Abstract
In therapeutic or diagnostic antibody discovery, affinity maturation is frequently required to optimize binding properties. In some cases, achieving very high affinity is challenging using the display-based optimization technologies. Here we present an approach that begins with the creation and clonal, quantitative analysis of soluble Fab libraries with complete diversification in adjacent residue pairs encompassing every complementarity-determining region position. This was followed by alternative recombination approaches and high throughput screening to co-optimize large sets of the found improving mutations. We applied this approach to the affinity maturation of the anti-tumor necrosis factor antibody adalimumab and achieved ~500-fold affinity improvement, resulting in femtomolar binding. To our knowledge, this is the first report of the in vitro engineering of a femtomolar affinity antibody against a protein target without display screening. We compare our findings to a previous report that employed extensive mutagenesis and recombination libraries with yeast display screening. The present approach is widely applicable to the most challenging of affinity maturation efforts.
Introduction
Monoclonal antibodies continue to gain importance in the treatment of a number of diseases and this trend shows no signs of abating.Citation1 Protein engineering methods are often required to improve antibody characteristics for clinical development and manufacturing.Citation2 Properties that may need optimization include affinity, stability, lack of aggregation, selectivity, solubility, cross-reactivity to homologous antigens of relevant animal species, immunogenicity potential, expression yield or effector functions. For a given antibody, only a subset of these characteristics typically needs optimization. However, after isolation of antibodies by display-based selection from even very large naïve or synthetic libraries, and even from immunized animals, affinity maturation is often required. Indeed, several hundred or even a thousand-fold tighter binding may be required for selected applications, such as therapeutic antibodies against soluble targets that interfere with high affinity protein-protein interactions, or for diagnostic antibodies. In many cases, improved antigen binding affinity increases potency and lowers dosing requirements, contributing to enhanced efficacy, safety and lowered costs. These advantages, however, must be balanced by the resulting increase in non-germline substitutions, which can increase the risk of immunogenicity or affect other antibody properties such as stability or expression. It is thus critical to use the minimum of mutations to reach the needed increase in potency.
In the peer-review scientific literature, the engineering of antibodies for femtomolar binding affinities to protein targets has been described in a very few cases, and as yet only using display methods.Citation3,Citation4 Indeed, antibody display methods have substantial popularity in affinity enhancement efforts.Citation5 The widespread use of phage and yeast display can be attributed largely to the fact that, when well implemented, very complex libraries can be screened relatively easily by panning or cell sorting, respectively. However, the “single-pot” phage display panning approach of large libraries has well-documented drawbacks with respect to selection bias toward variants that are better displayed or that allow faster growth of the expressing host cell.Citation6-Citation9 The effects are magnified over the multiple rounds of selection and amplification, so that useful candidates may be lost, if they do not also compete well in these therapeutically irrelevant selection criteria. In addition, it is impractical to generate and screen by display methods many dozens of mutagenic libraries simultaneously but individually, an approach that proved important in the present work. Finally, display methods generally have insufficient dynamic range to allow reliable isolation of the tightest protein target binders when starting with an already tightly-binding antibody.
As an alternative to display methods, antibody variants can be tested in a clonal, well-based, high-throughput screening process with quantitative determination of properties to ensure selection of candidates with the best mix of desired characteristics.
In the present report, we effected the optimization of an antibody toward low picomolar/femtomolar binding constants, the extreme limit of resolvability for tightness of antibody binding. In contrast to two published reports of optimization of antibodies to the femtomolar affinity range,Citation3,Citation4 we chose soluble Fab (fragment antigen-binding) expression of our library variants, well-based screening for selection and limited our library diversification to complementarity-determining regions (CDRs) only. In addition, we compare our results to a study that applied yeast display and extensive library generation approaches to the same antibody and optimization parameters, namely, the antibody adalimumab.
Adalimumab (Humira®),Citation10 binds the inflammatory mediator, tumor necrosis factor (TNF), with ~100 pM affinity.Citation11 While it has not been demonstrated that increased affinity would increase the therapeutic efficacy of adalimumab, the molecule has already been affinity-matured antibody and thus serves as a good starting point for studying the engineering of very tight binding by different methods. With similar reasoning, adalimumab was recently subjected to affinity maturation using a single chain variable fragment (scFv) yeast display approach. In their paper, Rajpal et al.Citation12 describe an elegant and generic method to improve antibody affinities that included diversity generation at every CDR residue position and recombination of the beneficial mutations.
In the approach described here, we employed clonal, quantitative, well-based high-throughput screening of soluble Fab fragments. In doing so, we sought an approach that could efficiently identify the largest affinity benefits using the minimum number of needed CDR mutations while also avoiding undesired framework mutations. In principal, our multistep optimization approach includes a first “mutation gathering” round of library generation and screening, followed by rounds in which the gathered mutations are recombined by differing approaches.
For mutation gathering, we generated a first set of libraries that together covered all CDR residues. In each library, two neighboring residues were fully diversified, thus allowing comprehensive identification of 1) the best single substitutions at each CDR position that can contribute to affinity improvements, as well as 2) many adjacent residue pair combinations that might synergistically contribute to affinity improvements. The mutation pairs in each screening hit were deconvoluted to identify contributions from each individual residue change. Subsequently, the found improving substitutions (the “gathered mutations”) were subjected to alternative recombination strategies to select the best combinations of additive or synergistic mutations. In one strategy, we focused on the co-optimization of only positions that had conferred the greatest increase in affinity in the mutation gathering round (“anchor point” positions), and included multiple amino acid substitutions at each position in the recombination library. This was intended to capture the maximum benefit from the co-optimization of the positions that had conferred the highest benefit individually. This anchor point-optimization was followed by recombination of multiple 2-fold degenerate positions to capture improving contributions from improving residues dispersed over a larger number of positions.
In an alternative recombination approach, only the latter “dispersed binary” strategy was used, but with some co-optimized residue pairs from the mutation gathering round treated as single, linked substitutions. The output of both approaches was a substantial number of hits that met or exceeded the desired sub-picomolar affinity improvement, and, therefore, could have allowed further lead selection based on additional properties such as stability, selectivity, species cross-reactivity, expression yield or effector functions.
Results
Double and single NNK saturation mutagenesis library construction
The coding sequence for adalimumab Fab and scFv used was as published.Citation13 In each double NNK library, two adjacent amino acid positions of the parent antibody were diversified by saturation mutagenesis, resulting in a genotypic complexity of 1024 variants per library. In total, 33 double-NNK libraries were generated, encompassing all 62 CDR positions of adalimumab based on the Kabat CDR definition.Citation14 We also constructed 10 single NNK control libraries covering all five amino acids of CDR-H1 and five consecutive amino acids of CDR-L2 for comparative purposes. In total, all generated NNK libraries comprised ca. 34,000 genotypes.
Isolation of variants with decreased off-rate and synergistic effects
The single and double NNK libraries were screened for decreased off-rates using a competition ELISA format (), in which the amount of detected, remaining Fab scales with decreased off-rate (). This competition ELISA format allowed the analysis of > 20,000 Fab variants per day. In total, we screened 140,000 wells with oversampling to reduce the risk of missing underrepresented variants. Subsequently, ca. 1,000 hits from the primary screen were subjected to confirmatory assays applying two competition formats, time-resolved fluorescence resonance energy transfer (TR-FRET) and enzyme-linked immunosorbent assay (ELISA) assays in 384-well microtiter plates. In the confirmatory assays, 175 improved binding clones were identified and sequencing revealed 64 different genotypes: 1 triple mutant (resulting from an unintended oligonucleotide synthesis artifact), 39 double and 24 single mutant variants. The triple and double mutants were then deconvoluted into their corresponding single substitutions and these, along with all obtained single, double and triple substitution variants, 111 in total, were reevaluated and ranked according to their dissociation kinetics (koff). The “anchor points,” or three loci that contributed to the strongest affinity improvements were: (CDR-H1, 31–32); (CDR-L2: 53–54) and (CDR-L3:92–94).
Figure 1. Clonal well-based quantitative Fab library screening: (A) An example affinity histogram from five “co-optimized residue pair” CDR-L3 libraries. the histogram shows affinity distribution for 17,500 clones in off-rate limited ELISA format. Red line indicates distribution of adalimumab Fab koff levels; dashed line median koff of adalimumab Fab; blue circles mark double mutants with slowest dissociation rates. (B) Diagram of ratiometric Time-Resolved Fluorescence Resonance Energy Transfer (TR-FRET) competition assay of hits from the Anchor-Point Cooptimization (APC) library. Red diamonds, Ratiometric measurement of adalimumab Fab; green diamonds, Best single-substitution variant; blue diamonds, APC recombination library clones; blue circles, Confirmed hits with slowest dissociation kinetics. Direction of arrow indicates decreasing off-rates.
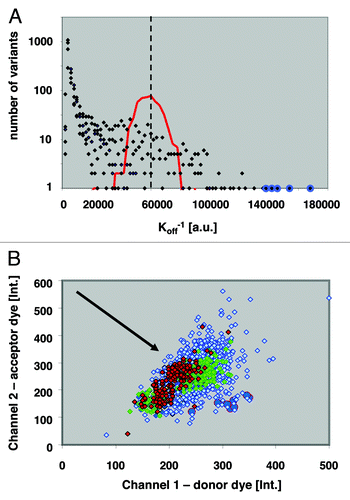
As part of the screening campaign, we also compared the retrieval of hits from single vs. double NNK libraries covering all amino acid positions of CDR-H1. The double mutation at position 31/32 (D31Q/Y32F) displayed slower dissociation compared with the best single mutant in position 31 (D31Q), and no positive substitution in position 32 was identified by screening. We then produced the Y32F variant as a single mutation and confirmed no detectable improvement in the above assay. These results are consistent with the potential for synergism involving positions 31 and 32. Similarly, two CDR-L1 variants containing mutation pairs, G28R/I29V or A25T/Q27M, displayed slower dissociation compared with adalimumab Fab, while remarkably each of the corresponding single mutants, G28R, I29V, A25T and Q27M, displayed faster dissociation (weaker binding) in the conditions tested. The above data suggest synergism, but would need confirmation by determination of kinetic constants for each variant before true synergism can be claimed. Nevertheless, the above data are consistent with the intention that the twin substitution strategy explore potentially synergistic affinity improvements that would have been missed in a single-residue-per-library approach.
Recombination strategy 1, round 1: anchor-point co-optimization (APC)
In first round of recombination, we focused on simultaneously co-optimizing the anchor points where adjacent residues best contributed to increased affinity. These comprised residue positions 31+32 in CDR-H1, positions 52+53 in CDR-L2 and 92–94 in CDR-L3. The diversity at each of these seven positions was 5, 4, 4, 2, 3, 4 and 2, respectively, for a total combinatorial complexity of 3840 possible unique variants (). In this manner, a high degree of co-optimization was performed within and among the three narrowly focused anchor points. To ensure a comprehensive sampling of this library, 17,500 wells were screened using a TR-FRET competition assay, which was superior to the above competitive ELISA assay format for the resolution of high affinity binders (). From this TR-FRET primary screen, 240 hits were selected for confirmatory assays, the 45 best confirmed clones were sequenced, and the best four variants purified for affinity characterization by SPR. The best APC variant, 2.1b_9.0_1, displayed 10 pM affinity ().
Recombination strategy 1, round 2: dispersed binary recombination (DBR)
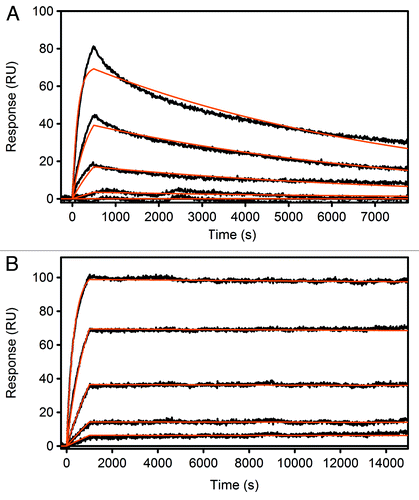
To explore the generation of still higher affinity clones, a second recombination library was generated using 2.1b_9.0_1 as starting scaffold. In this second round of recombination, 12 dispersed beneficial positions from CDRs H3, L1, L2 and L3 were co-optimized using only the original amino acid residue from adalimumab and the best single affinity-conferring substitution, hence 2-fold or “binary” degeneracy (). To ensure a comprehensive sampling of this library, 45,000 wells were screened using the TR-FRET competition assay (). Some 350 hits from the primary screen were further evaluated in confirmatory assays, 22 variants were sequenced, and three of these purified for SPR analysis. The best variant from this DBR step, 3.1d_6.1_2, carried the same number of substitutions (n = 10) as the best yeast-display variant, cb1–3, but had a 10-fold higher affinity (1 pM) in side-by-side analyses ().
Recombination strategy 1: optimizing contact-defined CDR residue positions
To take full advantage of all CDR residues in this antibody, we also altered the two additional CDR residue positions not included in the Kabat definition (). Chothia,Citation15 AbMCitation16 and ContactCitation17 define CDR residues based on available crystal structures of antibody-antigen complexes and slightly expand the Kabat definition to include additional amino acid positions that take part in antigen interactions. Using this definition, CDR-H1 and CDR-L2 each include one additional amino acid position, residues 30 and 49, respectively. For both sites, single-position NNK libraries were generated and all eight combinations of the highest affinity substitutions (D30G and Y49R, W, G) and the wild type residues were tested for affinity improvement by TR-FRET competition assay (). The best combination of residues at these two additional sites (CDR-H1: D30G; CDR-L2: Y49G or Y49W) increased the affinity of variant 3.1d_6.1_2 and 3.1f_12.1_1 to 400–500 fM according to SPR analysis (; ).
Figure 2. Surface Plasmon Resonance (SPR) measurements of adalimumab Fab (A) and the best hit after one round of APC and two rounds of DBR screening, 4.2a_6 (B). Adalimumab Fab was detected with a KD of 180 pM, 4.2a_6 with <1 pM.
Recombination strategy 2: single-round dispersed binary “locus” recombination
As an alternative to the multi-step sequential recombination described above, we compared a second approach in which up to 13 affinity-improving loci identified in the first round were co-optimized in a single recombination step (). We tested two sets that included only Kabat positions, or alternatively included the mentioned contact-defined CDR positions, 30 and 49, respectively. To maximize the number of positions in each library complexity, while still allowing screening of the complete combinatorial complexity, at each locus only the parental amino acids and the best substitution (or substitution pair) were recombined. The top 13 first round alterations comprised ten single and three double substitutions for both the Kabat-only and contact-defined CDR recombination experiments. The 13 alterations in each set were recombined in the adalimumab Fab scaffold, resulting in two libraries with a total combinatorial complexity of 8,192, each. To ensure complete sampling, for each library approximately 30,000 clones were examined by TR-FRET assay, approximately 150 were validated in confirmatory assays (TR-FRET / ELISA) and approximately 30 were sequenced. Finally, SPR measurements revealed two Kabat-defined CDR variants with an affinity of 2 pM, while the best contact-defined CDR variant displayed an equilibrium binding constant of 1 pM ().
Functional characterization of the affinity-enhanced adalimumab clones
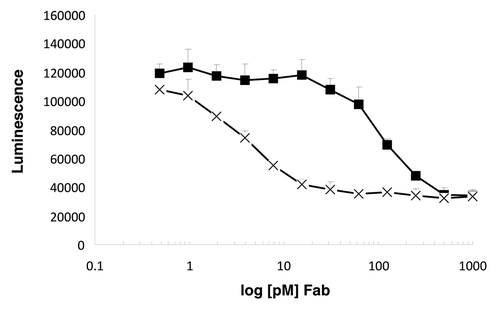
Adalimumab binds TNF and thereby prevents receptor activation. In a cell-based functional assay, the improved affinity of our variants correlated with improved TNF-neutralization activity (). The affinity-matured variants were determined to neutralize TNF-α-induced apoptosis of R1-Fas cellsCitation18 50-fold more effectively than adalimumab Fab.
Figure 3. The affinity-matured variant 3.1d_6.1_2 (X) neutralizes TNF-α-induced apoptosis of R1-Fas18 cells with a 50-fold increased efficacy in comparison to adalimumab Fab (■).
Discussion
Higher binding affinity, especially for soluble cytokines or growth factors, often correlates with improved potency and selectivity, resulting in lower dosage and potentially fewer or less severe adverse events (e.g., infusion reactions). We have also observed that a very tight protein:protein interaction in a disease mechanism may be disrupted only by antibodies with very low picomolar dissociation constants (data not shown). Diagnostic antibodies also generally require high affinities to efficiently detect low concentrations of analyte. Antibodies isolated by in vitro selection or hybridoma technologies, however, often have far weaker binding affinityCitation19,Citation20 than required for the above applications and thus are often candidates for extensive optimization.
Display methods have been extensively used to engineer high affinity antibodies,Citation3,Citation4,Citation21,Citation22 with less attention being given to compartmentalized screening methods. In our study we applied a compartmentalized well-based screening approach to the affinity maturation of the anti-TNF antibody, adalimumab. We compare the described approach to that of Rajpal, et al., who applied “walk-through mutagenesis” and extensive recombination strategies, employing a scFv yeast display selection format.Citation12
Both Rajpal, et al., and the current work included a first mutation gathering round followed by different recombination strategies. For mutation gathering, the two studies used libraries with diversification in all CDR positions of adalimumab based on the Kabat definition while we additionally included in certain cases two positions defined as CDR residues by Chothia, Contact and AbM. In contrast to our study, Rajpal et al. used nine of the 20 canonical amino acids at each position to test all biophysical classes, while allowing more complete simultaneous screening of a greater number of single-substitution libraries. Libraries were pooled and then screened by scFv yeast display.
The contributions of most affinity-increasing amino acid changes in antibody binding sites are largely independent and additive. For this reason, single-site mutagenesis at all positions, as applied by Rajpal et al., is able to identify many positive substitutions which when combined, yield highly improved binding. The present “adjacent pair” randomization approach was also able to identify all single improving mutations. However, we expected synergisms to occur most frequently at short-distances,Citation23 and thus chose to apply saturation mutagenesis to neighboring residues. Deconvolution of paired mutations in screening hits allowed determination of the contributions of individual residues, and at least 3 mutation-pairs were determined to be synergistic.
In a first round of APC recombination, we co-optimized what we call “anchor-points.” The three anchor points did not necessarily include contact residues, but were the loci where residue changes had the greatest contribution to increased affinity. We thus focused the first combinatorial co-optimization at only these positions to increase the chances of maximizing their benefit, and isolated variants with 10 pM binding constants. The subsequent more dispersed, binary recombination was then used to co-optimize contributions at a larger number of positions to further increase binding strength, and yielded 1 pM variants. By using the alternative Chothia CDR definition and thus including two additional positions, we obtained binders that were clearly sub-picomolar. In contrast, one round of dispersed binary recombination (DBR) alone enabled isolation of a 1 pM binder. Although a single round of dispersed binary recombination was remarkably efficient in the current example, it is expected to be less effective where any of the residues chosen at each anchor position clash with each other. Thus, the two approaches appear complementary, balancing reliability with speed and efficiency.
Rajpal et al. identified their best scFv adalimumab derivative, cb1–3, by five rounds of MACS and FACS screening of the initial set of libraries, followed by recombination, three rounds of sorting, a second recombination of the selected light and heavy chain variants and two final rounds of enrichment. We cloned and expressed an antibody Fab fragment with all 10 substitutions described for cb1–3 to allow head-to-head comparison with our hits. We measured a final affinity of 10 pM for the cb1–3, similar to variants we obtained after a single round of APC recombination (2.1b_9.0_1), but the latter possessed 4 fewer mutations. By contrast, in the present work we used 2 rounds of mutation generation and screening: The first was the adjacent-pair library generation/screening. The second was a single round of Dispersed Binary Recombination, in which we co-optimized 12 loci and were able to identify a 10-fold higher affinity improvement compared with cb1–3, but with the same number of substitutions as cb1–3.
The presented library strategies in combination with well-based quantitative measurements of individual clones thus compared favorably to the compared library approach with yeast-display screening in terms of affinity achieved, as well as a lower number of selection and screening rounds required.
The described library strategies combined with well-based screening, are applicable to any antibody. In comparison to Rajpal, et al., the described process resulted in fewer mutations for a given affinity increase, likely because all possible single and many synergistic CDR amino acid changes were exhaustively explored, and potentially because of the differing recombination strategies also with complete screening. Given the large flexibility in assay design for well-based screening, the presented approaches have clear applications to other antibody properties, such as stability, selectivity, species cross-reactivity, expression yield, receptor modulation or cellular responses and to the engineering of proteins beyond antibodies.
Materials and Methods
Reagents and E. coli strains
Synthetic oligonucleotides were obtained from Eurofins MWG Operon (Ebersberg, Germany). Restriction enzymes were obtained from New England Biolabs. KOD polymerase was obtained from Novagen. DNA purification was performed with kits from Qiagen. E.coli TOP 10F’ cells (Invitrogen) were used for standard cloning purposes, E.coli BL21 STAR (DE3) cells (Invitrogen) were used for library and selected Fab variant expression.
Adalimumab Fab construction
The nucleotide sequence of the adalimumab Fab fragment was derived from US patent Nr. US6090382A1. E. coli codon-optimized cDNA sequences of the adalimumab heavy and light chains were synthesized with N-terminal ompA or phoA secretion signal sequences by Invitrogen (Regensburg, Germany). In addition, the heavy chain carboxy-terminus was supplied with a 6xHis and a haemagglutinin (HA)-tag for improved affinity chromatography and immunodetection, respectively. Both chains were bicistronically expressed from vector pET21d or pET28a (Novagen). After expression in E.coli, Fabs were purified by Protein A sepharose affinity chromatography and subsequently subjected to mass-spectrometry (MS) and surface plasmon resonance (SPR) analysis for characterization.Citation18 Adalimumab Fab and scFv showed a KD of ca. 180 pM for TNF antigen (PeproTech or in-house expression), which is in line with the 100 pM KD for full length adalimumab previously published.Citation11,Citation24
Fab expression and purification
The Fab molecules were expressed in E. coli BL21 STAR (DE3) cells.Citation25 For protein expression, cells were first grown in 100 ml shaking flasks (10 ml LB, 30°C, over night, 160 rpm), transferred to 50 ml LB in 1 L shaking flasks to an inoculum of OD600 0.2 (LB, 30°C, 8 h, 160 rpm). From this solution 500 ml TBoNEX media (Novagen) were inoculated (OD600 0.1, 30°C, 50 h, 160 rpm). Cells were harvested by centrifugation (4°C, 20 min., 9000 rpm), resuspended in lysis buffer (50 mM NaH2PO4, 300 mM NaCl, 10 mM imidazole, pH 8.0, 30%; per 1 ml volume 20 µl protease inhibitor cocktail (Sigma P-8465) and 1 µl Benzonase Nuclease (Novagen) were added) and lysed by sonification (Branson Sonifier W-250D, head 0.5,” 70%, 5 x 1 min., 0.7 sec pulse/0.3 sec pause). Cell lysates were centrifuged at 20,000 rpm for 30 min at 4°C. Fabs were purified from cell lysates by absorption to Ni-NTA superflow resin (8 ml, Qiagen, buffer a: 50 mM NaH2PO4, 300 mM NaCl, 10 mM imidazole, pH 8; buffer b: 50 mM NaH2PO4, 300 mM NaCl, 250 mM Imidazole, pH 8; Gradient: 4% B 5 CV, 100% B 5 CV). Target fractions were dialysed (20 mM NaOAc, pH 5). Further purification was achieved by ion exchange chromatography (1 ml Resource S (GE), buffer a: 20 mM NaOAc, pH 5; buffer b: 20 mM NaOAc, 1 M NaCl, pH 5, step gradient). Buffer was changed to PBS for storage.
Construction of single / double NNK libraries
NNK (n = AGCT, K = G or T) randomizations at both individual (“NNK libraries”) and two adjacent codons (“double NNK libraries”) were generated by OE-PCR using synthetic oliogonucleotides and subcloned into vector pET21d. The sequencing of 224 random clones revealed that the libraries were highly diverse, with 87% possessing the expected double NNK mutants. The remaining 13% consisted of 1% wildtype clones, 4% with triple mutations and 8% with premature stop codons.
Construction of recombination libraries
After screening, beneficial mutations in the CDR regions of the variable domains were identified by sequencing. These mutations were then recombined. To this end, degenerate oligonucleotides were synthesized to effect the incorporation of the selected mutation(s) or the wild type amino acid at each selected position. Library construction was performed using sequential rounds of overlap extension PCR. The final PCR product was cut with XbaI/XhoI and ligated in pET21d.
Assay and screening
All methods described were performed on dedicated High-Throughput-Screening equipment in 1536-well format for Primary Screening and in 384-well plates for confirmative screens.
Library dispensing
Library stocks were reconstituted according to their titer in bacterial growth medium. Deposition of single cells into wells of the plates was performed by limited dilution, dispensing 4 ul per well, followed by approx. Thirty h growth period. Control plates were inoculated with 5 cells per well.
In confirmative screening, selected clones from primary screening were cultivated in 96-well plates with appropriate additional controls. Cultivation consisted of 2 precultures and one main culture.
Screening in competitive ELISA format
ELISA plates were prepared prior to screening by coating with antigen, incubation, washing and blocking. Plates were then stored at 10°C. Plates containing library and controls were replicated into fresh media for genotype backup, the remaining culture in the original plates were then lysed and supernatant transferred into ELISA plates (Perkin Elmer MaxiSorb). Assay was performed by incubation, washing and addition of antigen for competition, detection antibody and resorufin assay. Incubation times varied according to expected affinity of the variants. Finally the solution was transferred to reusable glass bottom plates (Bayer HealthCare) which can be substituted with standard disposable polystyrene ELISA plates when using a commercial readout system (i.e., Perkin Elmer Envision or comparable) and top readout. Fluorescence intensity (2-photon excitation 800 nm, emission 590 nm) was measured (Bayer HealthCare reader). The top performers (“hits”) on every plate were identified via automated online analysis and a work list for a single channel pipettor was generated for regeneration of clones from genotype backup plates into fresh media in a 96-well plate. In confirmative screening replicates of 4 were screened with measurements performed on a Tecan Genios Pro workstation.
Screening in HTRF format
Genotype backup and lysis was performed as described above, supernatant transferred to Perkin Elmer Optiplate. Antigen was added followed by incubation and addition of KF and competitor. Incubation time with the competitor varied according to the expected affinity of the variants. Readout was then done on Perkin Elmer EnVision, using the HTRF Laser option. Hit picking was performed automatically as described above. For confirmative screening replicates of 4 were screened.
Surface plasmon resonance analysis
TNF preparations were immobilized onto a GLC sensor chip within a ProteOn XPR biosensor (BioRad Laboratories) using standard amine coupling. The surfaces were activated for 5 min with sulf-NHS/EDC (0.01M/0.04 M), then the TNF was injected at 1 ug/ml in 10 mM sodium acetate, pH 4.5 for 5 min and finally the surfaces were blocked with a 5 min injection of 1 M ethanolamine pH 8.1. TNF immobilization levels were between 400 and 500 RU. Fabs were tested at 10 nM as the highest concentration in a 3-fold dilution series. The running buffer contained 10 mM HEPES, 150 mM NaCl, pH 7.4, with 0.005% tween-20 and 0.1 mg/ml BSA. Binding data were collected at 25°C. Binding kinetics were determined by globally fitting the response data to a simple 1:1 interaction model using Scrubber (Biologic Software Pty Ltd, Australia).
Cell-based assay
TNF-induced apoptosis was measured using immortalized TNFR1/2 double knockout mouse fibroblasts stably transfected with a TNFR1-FAS fusion construct (kind gift of P. Scheurich). Cells were seeded in 384-well plates at 6,000 cells per well the day before the assay in RPMI1640, 10% FCS, 2 mM L-Glut. Relevant Fabs at the indicated concentrations were preincubated with 1 p.m. TNF for 30 min at room temperature in cell culture medium. Cells were washed and Fab-TNF complexes were added. After 6 h caspase activity was quantified using caspase3/7 glo reagent (Promega) according to manufacturer’s instruction.
Acknowledgments
We thank Nick Lobrenz, Anne Geselbrecht, Susanne Steinig, Sabine Jach, Anja Brake, Astrid Rüter, Anne Heistermann, Mona Porschen, Nina Habrich, Sandra Geske, Alexander Korseska, Carsten Bäumchen, Kerstin Baral, Simone Brückner, Kornelia Kirchner, Danuta Cichon and Beya Hadidane for their important technical contributions.
Author contributions
C.V. coordinated execution of the project and coordinated analysis/interpretation of the data, C.V. and W.M.C. designed all library strategies. C.V. and H.P. developed homogeneous and heterogeneous assay systems for optimal resolution of high-affinity binders and C.V., H.P., O.H. and M.S. performed Fab rankings with the aforementioned assay systems, A.S., H.P. and C.V. optimized the Fab expression systems, A.S. and K.P. generated cloning strategies to realize library concepts, U.G. developed the TNF bioassay and contributed to ELISA assay development, M.S. and O.H. adjusted the high-throughput screening facility for running off-rate limited assays, D.M. performed and optimized SPR measurements for optimal resolution of high-affinity binders, P.S. and A.S. designed TNF expression constructs, P.S. coordinated Fab purification work, W.M.C. and U.H. suggested the comparative study with display optimization and decided for the adalimumab affinity maturation project with the goal of femtomolar affinity. C.V. and W.M.C assumed primary responsibility for writing and U.H. also contributed to the manuscript.
References
- Reichert JM. Metrics for antibody therapeutics development. MAbs 2010; 2:695 - 700; http://dx.doi.org/10.4161/mabs.2.6.13603; PMID: 20930555
- Lerner RA. Manufacturing immunity to disease in a test tube: the magic bullet realized. Angew Chem Int Ed Engl 2006; 45:8106 - 25; http://dx.doi.org/10.1002/anie.200603381; PMID: 17120282
- Boder ET, Midelfort KS, Wittrup KD. Directed evolution of antibody fragments with monovalent femtomolar antigen-binding affinity. Proc Natl Acad Sci U S A 2000; 97:10701 - 5; http://dx.doi.org/10.1073/pnas.170297297; PMID: 10984501
- Steidl S, Ratsch O, Brocks B, Dürr M, Thomassen-Wolf E. In vitro affinity maturation of human GM-CSF antibodies by targeted CDR-diversification. Mol Immunol 2008; 46:135 - 44; http://dx.doi.org/10.1016/j.molimm.2008.07.013; PMID: 18722015
- Hoogenboom HR. Selecting and screening recombinant antibody libraries. Nat Biotechnol 2005; 23:1105 - 16; http://dx.doi.org/10.1038/nbt1126; PMID: 16151404
- Kay BK, Adey NB, He YS, Manfredi JP, Mataragnon AH, Fowlkes DM. An M13 phage library displaying random 38-amino-acid peptides as a source of novel sequences with affinity to selected targets. Gene 1993; 128:59 - 65; http://dx.doi.org/10.1016/0378-1119(93)90153-T; PMID: 8508960
- Clackson T, Wells JA. In vitro selection from protein and peptide libraries. Trends Biotechnol 1994; 12:173 - 84; http://dx.doi.org/10.1016/0167-7799(94)90079-5; PMID: 7764900
- Derda R, Tang SK, Li SC, Ng S, Matochko W, Jafari MR. Diversity of phage-displayed libraries of peptides during panning and amplification. Molecules 2011; 16:1776 - 803; http://dx.doi.org/10.3390/molecules16021776; PMID: 21339712
- Krebber A, Burmester J, Plückthun A. Inclusion of an upstream transcriptional terminator in phage display vectors abolishes background expression of toxic fusions with coat protein g3p. Gene 1996; 178:71 - 4; http://dx.doi.org/10.1016/0378-1119(96)00337-X; PMID: 8921894
- Bain B, Brazil M. Adalimumab. Nat Rev Drug Discov 2003; 2:693 - 4; http://dx.doi.org/10.1038/nrd1182; PMID: 12953696
- Osbourn J, Groves M, Vaughan T. From rodent reagents to human therapeutics using antibody guided selection. Methods 2005; 36:61 - 8; http://dx.doi.org/10.1016/j.ymeth.2005.01.006; PMID: 15848075
- Rajpal A, Beyaz N, Haber L, Cappuccilli G, Yee H, Bhatt RR, et al. A general method for greatly improving the affinity of antibodies by using combinatorial libraries. Proc Natl Acad Sci U S A 2005; 102:8466 - 71; http://dx.doi.org/10.1073/pnas.0503543102; PMID: 15939870
- Salfeld JG, Allen DJ, Hoogenboom HR, Kaymakcalan Z, Labkovsky B, Mankovich JA, et al. Human antibodies that bind human TNFα. U.S. Patent 2003; 6,090,382.
- Kabat EA, Wu TT, Reid-Miller M, Perry H, Gottesman K. Sequences of Proteins of Immunological Interest, US Govt. Printing Off. 1987; Fourth Edition: 165-492.
- Al-Lazikani B, Lesk AM, Chothia C. Standard conformations for the canonical structures of immunoglobulins. J Mol Biol 1997; 273:927 - 48; http://dx.doi.org/10.1006/jmbi.1997.1354; PMID: 9367782
- Web Antibody Modeling. . http://antibody.bath.ac.uk
- MacCallum RM, Martin AC, Thornton JM. Antibody-antigen interactions: contact analysis and binding site topography. J Mol Biol 1996; 262:732 - 45; http://dx.doi.org/10.1006/jmbi.1996.0548; PMID: 8876650
- Jansson B, Uhlén M, Nygren PA. All individual domains of staphylococcal protein A show Fab binding. FEMS Immunol Med Microbiol 1998; 20:69 - 78; http://dx.doi.org/10.1016/S0928-8244(97)00108-9; PMID: 9514577
- Wark KL, Hudson PJ. Latest technologies for the enhancement of antibody affinity. Adv Drug Deliv Rev 2006; 58:657 - 70; http://dx.doi.org/10.1016/j.addr.2006.01.025; PMID: 16828920
- Foote J, Eisen HN. Kinetic and affinity limits on antibodies produced during immune responses. Proc Natl Acad Sci U S A 1995; 92:1254 - 6; http://dx.doi.org/10.1073/pnas.92.5.1254; PMID: 7877964
- Jermutus L, Honegger A, Schwesinger F, Hanes J, Plückthun A. Tailoring in vitro evolution for protein affinity or stability. Proc Natl Acad Sci U S A 2001; 98:75 - 80; http://dx.doi.org/10.1073/pnas.011311398; PMID: 11134506
- Hanes J, Plückthun A. In vitro selection and evolution of functional proteins by using ribosome display. Proc Natl Acad Sci U S A 1997; 94:4937 - 42; http://dx.doi.org/10.1073/pnas.94.10.4937; PMID: 9144168
- Wu H. Simultaneous humanization and affinity optimization of monoclonal antibodies. Methods Mol Biol 2003; 207:197 - 212; PMID: 12412476
- Santora LC, Kaymakcalan Z, Sakorafas P, Krull IS, Grant K. Characterization of noncovalent complexes of recombinant human monoclonal antibody and antigen using cation exchange, size exclusion chromatography, and BIAcore. Anal Biochem 2001; 299:119 - 29; http://dx.doi.org/10.1006/abio.2001.5380; PMID: 11730333
- Kwong KY, Rader C. E.coli Expression and Purification of Fab Antibody Fragments. Curr Protoc Protein Sci. 2009; 6: Unit 6.10.
- Krippner-Heidenreich A, Tübing F, Bryde S, Willi S, Zimmermann G, Scheurich P. Control of receptor-induced signaling complex formation by the kinetics of ligand/receptor interaction. J Biol Chem 2002; 277:44155 - 63; http://dx.doi.org/10.1074/jbc.M207399200; PMID: 12215450