Abstract
Neutralizing antibody responses to the surface glycoproteins of enveloped viruses play an important role in immunity. Many of these glycoproteins, including the severe acute respiratory syndrome-coronavirus (SARS-CoV) spike (S) protein form trimeric units in the membrane of the native virion. There is substantial experimental and pre-clinical evidence showing that the S protein is a promising lead for vaccines and therapeutics. Previously we generated a panel of monoclonal antibodies (mAbs) to whole inactivated SARS-CoV which neutralize the virus in vitro. 1,2 Here, we define their specificity and affinity, map several of their epitopes and lastly characterise chimeric versions of them. Our data show that the neutralizing mAbs bind to the angiotensin-converting enzyme 2 (ACE2) receptor-binding domain (RBD) of the SARS S protein. Three of the chimeric mAbs retain their binding specificity while one conformational mAb, F26G19, lost its ability to bind the S protein despite high level expression. The affinity for recombinant S is maintained in all of the functional chimeric versions of the parental mAbs. Both parental mAb F26G18 and the chimeric version neutralize the TOR2 strain of SARS-CoV with essentially identical titres (2.07 and 2.47 nM, respectively). Lastly, a comparison with other neutralizing mAbs to SARS-CoV clearly shows that the dominance of a 33 amino acid residue loop of the SARS-CoV RBD is independent of repertoire, species, quaternary structure, and importantly, the technology used to derive the mAbs. In cases like this, the dominance of a compact RBD antigenic domain and the central role of the S protein in pathogenesis may inherently create immunoselection pressure on viruses to evolve more complex evasion strategies or die out of a host species. The apparent simplicity of the mechanism of SARS-CoV neutralization is in stark contrast to the complexity shown by other enveloped viruses.
Introduction
The rapid extension of severe acute respiratory syndrome-coronavirus (SARS-CoV) virus from animals into the human population in 2003 illustrates the iterative pressure pathogens place on hosts while developing new niches.Citation3 Conversely, the rapid departure of the SARS-CoV from humans and the lack of a chronic state indicate that the virus was not ready to make a permanent jump into humans. This also implies that the host defence mechanisms were capable of repelling the virus, or at least were not quite suitable for sustained SARS-CoV viral pathogenesis. However, as an emerging infectious disease, the virus remains as a model system and a warning for vigilance.
It is clear that SARS-CoV is actively undergoing antigenic variation in its membrane glycoprotein spike (S) protein. The S protein mediates attachment to host cells, and causes fusion of the host cell and viral membranes. The S protein exists in vivo as a trimer composed of monomers that contain globular heads, formed by the S1 region, which contains the receptor binding domain (RBD; amino acids 318–510).Citation4,Citation5 Each monomer also contains an S2 region that forms the stalk and possesses two heptad repeat motifs of the coiled-coil structure important in membrane fusion.Citation6 Host cell receptors that bind S protein have been identified. Angiotensin-converting enzyme 2 (ACE2), a membrane-bound host protein, binds S protein and mediates virion entry, making it the primary receptor for the virus.Citation7 The RBD alone has been shown to be sufficient to mediate binding to ACE2.Citation8 The Spike protein is quite divergent among coronaviruses and suffers point mutational changes. The X-ray crystal structures of the RBD in complex with ACE-2 and monoclonal antibodies (mAbs) reveal critical contact residues that provide a molecular explanation for virus neutralization and species-specific receptor binding differences.Citation4,Citation9–Citation11
Membrane glycoproteins represent some of the most challenging targets for antibody discovery. The ability to engender broad and potent neutralizing antibodies to viral membrane proteins is generally thought to be limited by our ability to generate properly folded and oligomerized forms of these membrane proteins in vaccine preparations. Moreover, RNA viruses as a group have evolved a spectrum of antigenic variation systems, including point mutation, recombination, cryptic fusion domains/epitopes, glycan shields and multimeric assemblies.Citation12–Citation15 For these reasons, recombinant monomeric fragments of the envelope proteins of RNA viruses like HIV-1, Ebola and influenza A (gp120 or HA subunit proteins) are poorly immunogenic and elicit extreme type-specific protective antibody.Citation16–Citation19 This may be due to a lack of native structure, high epitope mobility, incorrect or absent glycosylation or response to cryptic non-neutralizing or narrow spectrum epitopes.
Immunity to SARS-CoV has been established in several animal models and is mediated via humoral responses to the S protein. Vaccines are often predicated upon the findings of passive vaccines whereby the protective antigens are identified.Citation20 Subunit delivery of the S protein using live virus,Citation21 DNA delivery,Citation22 recombinant S protein,Citation23 or even the minimalist RBDCitation24–Citation26 provides protection against infection by eliciting antibodies that neutralize SARS virus in vitro or in vivo. This suggests that dominant protective epitopes are contained in the S protein, and that these key epitopes are readily recapitulated in a variety of expression systems and formats. Higher neutralizing antibody responses to the S protein were correlated with a favourable disease outcome in SARS patients.Citation27 Active immunization with whole virus induces S protein specific antibodies that neutralize the virusCitation1,Citation28 and block receptor binding.Citation28 Previously, we assisted in demonstrating that polyclonal serum antibody responses elicited by whole SARS-CoV virus inoculation in six different species, map to an extended loop (residues 424–494) that mediates all contact with ACE2. This strongly suggests that most protective antibody should map to the RBD.
When administered as prophylaxis, neutralizing antibodies to viruses can serve as a type of viral entry inhibitor drug. Prophylaxis is effective in a variety of viral infections, including varicella, hepatitis A, hepatitis B, rabies and respiratory syncytial virus (RSV).Citation30 During the SARS epidemic, hyperimmune sera from convalescent patients were used as treatment with no adverse side effects.Citation31 An assessment of the efficacy of this treatment is hard to complete due to the small sample numbers available, though it did seem to shorten hospitalisation time. Soon after the isolation of SARS-CoV, mAbs were developed in mice by hybridoma techniques,Citation1 and from memory B cells of convalescent patients by phage displayCitation32,Citation33 and Epstein-Barr virus transformation.Citation34 In addition to providing essential reagents for diagnostics and screening, many potent neutralizing antibodies have been isolated and characterized.
Many improved strategies are being used to develop mAbs to infectious agents. These include high-throughput screening for antigenic variants,Citation35 direct cloning of B cells,Citation36,Citation37 more accurate portrayal of viral membrane proteins using VLPs or live virusCitation38–Citation43 or altering immunodominance through re-focussing antibody responses to epitopes.Citation44–Citation50 Our understanding of antibody responses is a direct reflection of the techniques used to derive the antibody (immune, synthetic or naive antibody systems), which are each presumed to be affected by the structural characteristics of the immunogen or antigen used to derive the mAb.
Native virions represent the most natural immunogen or antigen for use in immunization or mAb discovery, respectively.Citation13,Citation14 Ideally, to understand natural human immunity to a pathogen, the epitopes targeted through the human B cell response to whole virus must be obtained; conversely, the epitopes targeted by mAbs made to recombinant proteins, would not necessarily be expected to target the same epitopes as those in human infections. In some cases the use of whole virus as an antigen is not feasible, and recombinant protein was used as a simple and safe surrogate for screening. While there are multiple immune, synthetic and naive mAb discovery methods, the process is still empirical,Citation51 and the method used to produce a mAb clearly can bias characteristics of the mAbs produced. It is generally recognized that these methods are capable of collectively providing unique mAbs to diverse epitopes if applied to the same targets.
mAbs further enable the specific dissection of the fine specificity of host protective immune responses.Citation52–Citation54 The identification of protective epitopes reveals important functional domains that might lead to therapies that may prevent viral escape. We previously generated murine mAbs to whole inactivated SARS-CoV virus and the most potent of these neutralizing mAbs were F26G9, F26G10, F26G18 and F26G19.Citation1 Molecular characterization of these neutralizing mAbs revealed a relatively conserved immunoglobulin structure suggesting a pauciclonal B cell response, likely due to structural properties of the S-protein that result in a stringent B cell selection.Citation2
Here, we characterize the epitopes of parental murine and chimeric versions of mAbs generated to the whole SARS-CoV. Recently, neutralizing mAb F26G19 was co-crystallized in complex with the RBD and the structure clearly reveals the contact sites critical to this interaction.Citation10 Further characterisation of these antibodies and an additional non-neutralizing S protein specific monoclonal antibody (mAb G8) was performed to define their epitopes and affinities. Epitope competition using labelled mAbs was performed on whole virus and peptide mapping revealed the identity of two linear epitopes for mAbs F26G18 and F26G8. Despite having attributes of conformational epitope binding, F26G18 and its chimera map to a discrete sequential epitope (460FSPDGKPCT PPALNCYW476) in the RBD and neutralize live virus in vitro. Four mAbs (F26G9, F26G10, F26G18 and F26G19) utilize related VH genes and they previously demonstrated high neutralization titres in vitro against wild-type virus. However, chain swapping experiments performed here revealed the importance of the natural pairing of these related VH genes and showed that the light chain partners are important in maintaining epitope specific binding. These data suggest that these mAbs have potential use in antiviral therapy, but, more importantly, clearly show that blocking the RBD is the mechanism for neutralization by SARS-CoV neutralizing antibodies.
Results
Determination of S-protein and RBD specificity for neutralizing mAbs raised to whole virus using ELISA assays
All five of the F26 series mAbs tested (F26G8, F26G9, F26G10, F26G18, F26G19) were verified to bind to full-length S protein (rFS) using recombinant protein. In contrast, among these only the neutralizing mAbs (F26G9, F26G10, F26G18, F26G19) bind a fragment (rACE2BDS) spanning the receptor binding domain (RBD) from amino acids (aa) 318–510, compared to non-neutralizing spike mAbs F26G8 which does not bind (). Additionally, the four neutralizing mAbs bind to a deglycosylated version of the 318–510 fragment (dgrACE2BDS) as shown by the ability of the deglycosylated protein to inhibit binding to the rFS by increasing concentrations of dgrACE2BDS (). F26G8 binding, on the other hand, is unaffected by soluble 318–510 fragment; this shows that the epitopes of F26G9, F2610, F2618 and F2619 are not composed of glycosylations, but are found in the 318–510 region. The F26 series mAbs do not cross-react with the irrelevant control protein anthrax toxin (PA), which is bound by F20G7-5 anti-PA toxin mAb. Furthermore, the neutralizing mAbs F26G9, F2610, F2618 and F2619 all recognize mammalian expressed RBD 318–510 fragment in ELISA () and also in western immunoblot (data not shown).
A competition ELISA (cELISA) performed between mAbs on whole virus reveals several major overlapping groups (“bins”) of epitopes. Three spike specific mAbs were biotinylated for direct detection in immunoassay: biot-F26G6 (immunofluorescence [IFA] positive, non-neutralizing mAb), biot-F26G9 (IFA positive, neutralizing mAb), biot-F26G18 (IFA positive, highly potent neutralizing mAb). A concentration of labelled mAb which produced a half maximal signal (OD 405 nm) in ELISA was mixed 1:1 with the other mAb supernatants from cell culture.
The neutralizing and non-neutralizing mAbs to SARS-CoV essentially fall into four main bins as depicted in . There is a non-neutralizing epitope bin () where F26G8 and competes with biot-F26G6 to greater than a 90% reduction in binding; this is consistent with F26G8 being unable to bind to the RBD fragment (residues 318–510) and both being non-neutralizing mAbs. Next is the biot-F26G9 neutralizing mAb bin () which overlaps with all neutralizing mAbs (F26G7, F26G10, F26G18, F26G19; all IFA positive, neutralizing mAbs to the RBD 318–510 fragment), except F26G3 and non-neutralizing F26G4. Interestingly, the non-neutralizing mAb F26G8 competes better with this bin than neutralizing mAb F26G18. This suggests that critical contact residues are key to neutralization, and that some mAbs compete by simple steric interference. The biot-F26G18 bin () overlaps by about 90% with F26G9 bin, although there is less than 50% overlap with F26G19; this is consistent with the pepscan data that shows the critical contacts of F26G18 are adjacent to, but do not overlap with, the linear stretch of F26G19 epitope as determined in X-ray co-crystal.Citation10 Lastly, F26G3 (IFA positive, neutralizing mAb) and F26G4 (IFA negative, non-neutralizing mAb) have essentially unique non-overlapping epitopes with the other mAbs. In general, the neutralizing mAbs tend to cluster within one bin, but there are some surprisingly low overlaps. For example, F26G18 and F26G19 show less than 50% overlap using biot-F26G18. There is also a 50% reduction of the non-neutralizing biot-F26G6 signal by both the F26G18 and F26G19, which is likely due to steric hindrance. Interestingly, the steric hindrance appears to be one-way as the biotin-G18 does not show the same phenomenon with unbiotinylated G6. F26G8 and F26G6 both have very short VH-CDR3 regions which distinguish them from the neutralizing mAbs.Citation2
Chimeric mAbs were developed for these genetically related mAbs whereby human constant regions were fused in frame to the murine variable region gene cDNA. The human-mouse chimeras of F26G9, F26G10, F26G18 and F26G19 were named 9H9L, 10H10L, 18H18L and 19H19L, respectively. This also allowed simple naming of chain swapped pairs and tracking back to the original F26 clone. Transfection of 293 cells followed by ELISA screening of the supernatant shows that three of the original VH and VL pairs (9H9L, 10H10L and 18H18L) and two of the uniquely assembled chain-swapped pairs (9H10L and 10H9L) produce antibody that binds to rFS (). The rationale for chain swapping was that many of these VH and VL genes were highly related and may be promiscuous binders. Of note, when paired, 10H19L and 18H19L are the only constructs that seem to not produce significant antibody, although there is a slight increase in the gamma heavy and kappa light chain signal for 18H19L. This suggests an incompatibility between these two specific sets of immunoglobulin variable region genes. Interestingly, the plasmid parental V gene pair of 19H19L produces significant amount of antibody in the 293 cell line but the antibody does not bind rFS (data not shown). This is the only parental pairing where rFS binding is lost, and shows the changes to the constant domains through the chimerization process have a negative impact on the function of these V genes. We cannot discount the potential effects of the addition of a couple of two artificial amino acid residues on binding of 19H19L however these are not in the antigen contact regions and do not effect binding of the remaining mAbs. These modifications were instituted during the addition of a cloning site for the hinge region. Furthermore, this shows that V gene relatedness is not always a good predictor of promiscuity and that somatic mutations clearly impact functional pairings empirically. The binding of the chimeric mAbs to the Spike protein was evaluated in ELISA assay and cELISA. As can be seen from Figure 3A, many of the chimeric mAbs bind to rFS, rACE2BDS and dgrACE2BDS, although binding to rACE2BDS and the deglycosylated form is lower for 10H9L and 9H10L. A cELISA was then done to confirm these results (), which demonstrates that all of the chimeric antibodies are inhibited in their binding to rFS by increasing concentrations of dgrACE2BDS.
Western immunoblot.
Previous western blotting with whole viral lysate from Vero-E6 cells showed that the parental murine mAbs F26G8, F26G18 and F26G19 bound to a band corresponding to S protein via a linear epitope, while F26G9 and F26G10 did not.Citation1 When this experiment was repeated herein instead using the recombinant baculovirus expressed S protein, F26G8 and F26G18 bind rFS by western blot compared to F26G9, F26G10 and F26G19 which do not (data not shown); F26G19 was retested at several concentrations to verify this result. While it is not clear why F26G19 binds to viral expressed S protein but not baculovirus expressed Spike, it may be due to differences in conformation or lack of ability to standardise amounts of S protein in viral fractions versus pure recombinant protein. However, F26G18 binds to ACE2BDS, which clearly confirms the ELISA and cELISA data. Western immunoblotting was repeated for the most potent chimeric mAb 18H18L. Using anti-human Fc-gamma secondary reagents 18H18L shows the same binding pattern in western blot as parental murine F26G18, binding to both rFS and rACE2BDS and not rPA ( and inset).
Linear epitope mapping of F26G8 and F26G18.
Since western immunoblot shows F26G8 and F26G18 recognized linear epitopes in rFS, further epitope mapping was undertaken to minimize the binding area. The F26G18 epitope (neutralizing mAb) is clearly localized to a single pin-peptide at amino acid 460–476, with sequence 460FSPDGKPATPPALNAYW476 (). This novel linear epitope is located within the receptor binding domain region, as the ELISA and western blotting experiments predicted it would. Pepscan experiments show clear recognition of two pins for non neutralizing mAb F26G8, 604–620 and 612–628, suggesting that the epitope lies within the 9-mer overlap region with sequence ADQLTPAWR (). This epitope was also found as part of a motif recognized by F26G8 using random phage peptide library. Biopanning the commercial Ph.D-12-mer random library reveals a motif that matches pepscan (, inset). Consistency between pepscan and phage peptide libraries has been seen previously.Citation55 Some binding also seems to occur at amino acid residues 444–460, which can be discounted as background since C-ELISA with dgrACE2RBS shows no competition for binding to rFS for F26G8 (). Another signal was seen for F26G9 at pin corresponding to amino acid residues 540–556. Although it is most probably background, it is hard to discount immediately, since the sequence does lie outside of the 318–510 and in the region of S1 that it is known to bind.
Soluble peptides were also utilized to confirm epitope specificity. Based on pepscan information, four soluble peptides were synthesized; two were identical to the respective epitope regions, and two were scrambled versions of the epitopes. A direct ELISA on tethered soluble peptide showed that both murine antibodies recognized their respective peptide epitopes directly, and did not bind to the scrambled versions ( and inset). A cELISA was then performed with rFS coated to the plate and the soluble peptides used as binding competitors (). Both G8PEP and G18PEP compete in a concentration dependent manner with rFS for binding to F26G8 and F26G18, respectively. Compared to the scrambled peptides, G8PEPScr and G18PEPScr, did not affect binding. As for F26G18, peptide mapping was performed on its chimera, 18H18L, using 17-mer pin peptides with 9-mer overlaps and soluble G18PEP and G18PEPScr. Pepscan mapping, using the anti-human reagents, was unable to locate the epitope for the chimeric human constructs (); solid phase peptide ELISA were equally as inconclusive as18H18L did not bind to G18PEP stronger than to background (G18PEPScr, G8PEP, G8PEPScr) (). A cELISA using soluble G18PEP and G18PEPScr as competitors clearly showed that G18PEP inhibits 18H18L binding to rFS in a concentration dependent fashion (). G18PEPScr, on the other hand, produces no inhibitory effect. This demonstrates that 18H18L binds to the specific sequence in G18PEP and maintains the same epitope specificity as the parental murine version. While it is not clear why pepscan fails and soluble peptides succeeds in mapping chimeric F26G18 binding the authors suspect that the problems were typical of many other human antibodies which generally do not work well in pepscan (Berry JD, personal experience).
Surface plasmon affinity analysis.
Affinity analysis was performed using the BIACore 2000 surface plasmon surface resonance (SPR) technique with a CM5 chip coated with rFS. Varying concentrations of the murine F26 series and chimeric human mAbs were flowed over the chip for association to occur, then disassociation was measured by the drop in resonance over time when only buffer was flowing (data not shown). Calculation of the kinetic constants (ka and kd) and affinity constant (KD) was performed and the results are shown in . All spike specific antibodies have nM level affinities (consistent with an affinity matured T cell dependent response). Interestingly, the two mAbs with linear epitopes, the non-neutralizing mAb F26G8, and neutralizing mAb F26G18, binds rFS strongest with KD of 0.83 (±0.36) nM and 1.78 (±0.63) nM, respectively. The conformational mAbs, F26G9, F26G10 and F26G19 have similar values of 10.3 (±0.52), 7.49 (±0.40) and 4.05 (±1.01) nM, respectively. When affinity measurements were repeated using a full length S protein without the transmembrane region provided by the Biodefense and Emerging Infectious Pathogens Resources (BEI) Resources, the affinity measures were essentially identical, thereby illustrating the specificity for the ectodomain (data not shown). Finally, the chimeric forms had essentially the identical affinity when the original VH/VL pairing from the parental hybridomas was maintained and where there was measurable expression of functional mAb (). These VH and VL genes had previously been shown to be highly related, so more light chain promiscuity was expected.Citation2
Neutralization assay on chimeric antibodies.
Neutralization assays were performed on the chimeric antibodies, while running the original murine version (i.e., F26G18) side-by-side so as to limit errors due to different lab adapted viral stocks. All the antibodies successfully neutralized the wild-type TOR2 strain of SARS-CoV. The titres for the mAbs are shown in . Chimeric mAb18H18L had a neutralization titre of 0.37 µg/ml which matched the parental F26G18 mAb on the same viral stock.
Discussion
The majority of SARS-CoV neutralizing mAbs, regardless of the dependence on epitope conformation, function by binding epitopes within the RBD domain and preventing receptor binding. F26G18, arguably one of the most potent mAbs to SARS-CoV, primarily binds a linear epitope within the RBD (residues 460–476). F26G19, also generated to the whole virus, binds a discontinuous conformational epitope; however, a linear surface exposed loop (residues 486–492) within the epitope, is clearly a major determinant of binding.Citation10 There are many other examples of neutralizing mAbs that map to the RBD that were developed using alternative S1 protein targets and discovery platforms. Similar to F26G19, the neutralizing human mAb 80R binds via a conformational epitope in the RBD. Unlike the F26 mAbs, 80R was isolated as a single-chain variable fragment (scFv) from a non-immune human B cell (naïve) library screened against recombinant insect cell expressed S1 protein.Citation32,Citation57 The mAb 80R has potent in vitro neutralizing capability, high affinity for the RBD (1.59 nM),Citation32 and it reduced viral titres below detection when used prophylactically in a mouse model.Citation56 Viral escape from 80R has lead to the study of recombinant mAbs with broadened activity.Citation57 The crystal structure of 80R in complex with the RBD shows that all six complementary determining region (CDR) loops, as well as extensive framework (FR) residues, are involved in binding.Citation9 The dependence on FR residues for binding may be a result of the fact that 80R was selected from a naïve library rather than an immune individual. Similarly, Zhu et al. isolated m396 from a non-immune human scFv library through screening against a recombinant form of the RBD.Citation58 The IgG1 version of this antibody demonstrates an affinity of 4.6 pM and can neutralize the GD03T0013 isolate.Citation11,Citation58 Crystal structure analysis of Fab m396 in complex with the RBD S protein shows that m396 recognizes the epitope 482–491, using CDR loops H1, H2, H3 and L3.Citation58 Other examples include the use of transgenic mice with human immunoglobulin genes,Citation59 or semi-synthetic librariesCitation33 (). This supports the notion that the RBD must be relatively exposed on the viral surface.Citation60,Citation61 This is supported by the immunogenicity of this region in animalsCitation62 and humans.Citation63 Collectively it’s clear, that mAbs F26G18 and F26G19, and other neutralizing mAbs to SARS-CoV neutralize SARS-CoV primarily by preventing binding of S1 to the ACE-2 receptor.
The SARS-CoV S1 protein has a dominant neutralization region within the RBD consisting of a 33 residue segment. Despite the use of different host species and immunoglobulin repertoires (naive, immune, synthetic) different forms of antigen (monomeric, multimeric), and different glycoforms (mammalian, insectoid, bacterial) essentially all S1 mAbs neutralize via a common footprint. Indeed the critical contact residues involve the 33 amino acid segment consisting of residues 460FSPDGKPCTPPALNCYWPLNDYGFYTTTGIGYQ492 (), and various neutralizing mAbs recognize both common overlapping sets of residues and unique contact residues relative to that recognized by the ACE-2 receptor.Citation58 This region is exposed regardless of the glycoform, and has a critical role in attachment to the host ACE2 receptor. These factors combine to make this antigenic region an immunological “Achilles heel,” for the virus. The mAb F26G18 uses seventeen of these residues as a core of critical contacts as determined by pepscan, and conformational mAb F26G19 engages seven of these amino acid residues.
Chimeric mouse-human mAbs do not always retain functionality. Three out of four recombinant chimeras functioned as the parental murine versions, but one (19H19L) failed to bind its cognate antigen. While some studies have shown that changes in fine specificity can result from chimerization with alternative isotypes,Citation64 the process can clearly have more profound effects as 19H19L completely failed to function as a chimera. This may be due to somatic mutational changes of the germline V genes which are unique to the F26G19 pairing but incompatible with the human Fc regions in chimerization. The results of the ELISA assays confirm the specificity of our neutralizing mAbs to the S protein, specifically the RBD domain (residues 318–510). Previous analysis of these murine mAbs was performed using whole SARS CoV. The neutralizing mAbs F26G9 and F26G10 were western immunoblot negative and as such their protein target was unknown. These data confirm the conformational nature of the corresponding epitopes of F26G9 and F26G10 as they could not be mapped via pepscan and extend the findings in that we are able to show that they recognize both the SARS-CoV spike protein and the RBD in ELISA. We were able to measure SPR using the recombinant full length spike protein produced from BEI. The mAbs competition data showed that there are at least four easily discernable bins of mAbs. Moreover, multiple mAbs can bind to epitopes in the RBD with partial overlap. Our data on the binding of the neutralizing mAbs to dgrACE2BDS show that carbohydrates are not part of the epitopes for these mAbs.
Ideally, a chimeric mAb should retain the same immmunochemical parameters and biological activity as the parental mAb. The most potent murine mAb F26G18, was created as a chimera, 18H18L. Chimeric 18H18L binds to the full-length spike and the minimal ACE2BDS in western-immunoblot (). Furthermore, 18H18L has the same affinity for recombinant spike and it neutralizes live virus in vitro as well as the parental mAb does (). Epitope mapping of the binding specificity of 18H18L was performed using both pepscan and soluble peptides. The chimeric 18H18L mAb failed to perform in pepscan, which is likely due to a general incompatibility of secondary antibody reagents. The F26G18 and F26G19 epitope data from pepscan and X-ray crystallography,Citation10 respectively, showed that critical residues for F26G18 and F26G19 are closely located but not overlapping (), and this is consistent with the partial overlap of these mAbs seen in competition ELISA on the whole virus. Soluble peptide inhibition experiments with G18PEP containing the critical contact residues of F26G18 confirmed the peptide epitope by specifically competing for binding to rFS.
Recombinant integral membrane proteins are not always reliable immunogens for eliciting broadly protective anti-viral antibodies. Indeed, the immune response to recombinant fragments of the S protein would not be expected to accurately mimic a trimeric integral membrane protein. Moreover, the dominance of epitopes on different forms of the same immunogen might be perturbed when expressed as a monomer as most neutralizing antibodies bind to conformational epitopes and can, as in the case of Ebola, span subunits.Citation65 In this regard, a soluble recombinant monomeric S protein would be expected to be the least desirable immunogen. Similarly, the use of a monomeric S protein fragment as an antigen in phage biopanning would not be expected to necessarily select for the best virus neutralizing mAb as the presentation and prominence of a given epitope is not likely to be native. This has been the case for other enveloped viruses including HIV-1 where monomeric Env can fail to select for potent antibody responses to the virus.Citation66 Thus our initial attempts at antibody development for SARS-CoV made use of the most native form of the virus, that obtained by gentle chemical inactivation. Since that time, the collective mAb field has shown that mAbs of essentially equivalent potency can be derived to SARS-CoV S protein using technically distinct methods. While the virus does show the capacity to mutate residues to escape neutralization by individual mAbs, the extreme dominance of the region along with the ability of mAbs to redundantly recognize key critical residues likely contributed to the limited infection of humans.
Epitopes in a single region in the S-protein consistently elicit or select the majority of neutralizing mAbs for SARS-CoV. With the addition of our data to the body of the literature, we establish the following conclusion regarding the novel properties of the SARS-CoV spike RBD: dominance of the neutralizing subregion in the receptor binding domain of Severe Acute Respiratory Virus S1 is independent of antibody repertoire, host species, target quaternary structure and mAb technology (). This seems unlike what has been frequently observed with many other enveloped RNA viruses such as those found in the Filoviruses and Lentiviruses, where each of these factors has produced confounding obstacles that slow the development of useful and safe antibody therapeutics or vaccines. Indeed, SARS-CoV is relatively simple with the vast majority of antibody bioactivity mediated through one site critical to the virus pathogenesis, which thus represents a single fatal weakness. Other viruses have perhaps had more evolutionary time to evolve their membrane glycoproteins to avoid direct immune recognition of a single Achilles heel by gaining structural features to sequester the conserved receptor or co-receptor binding surfaces. Relative to even bacterial systems,Citation67 RNA viruses have a high level of gene expression noise, which makes them highly adaptable to environmental stress and immunological selection; however the fact that human naive mAb libraries provide high-affinity protective mAbs to SARS suggests that it was repelled from the human population because of relatively resistant people with pre-existing antibody to the protective domain. An important caveat is that we simply have not yet studied the virus in enough detail to observe antigenic variants and to understand the importance of other regions of the S protein.Citation68,Citation69 It will be important to continue to monitor the evolution of SARS-CoV for the emergence of escape variants that could confound current thinking on antibody-based vaccines.
Materials and Methods
Cells, hybridomas, constructs, proteins and antibodies
Monoclonal antibodies were generated by hybridoma fusion to whole inactivated SARS-CoV viral particles as the immunogen and the antigen as described previously.Citation1,Citation2 The mAbs examined in this paper are listed in . Several purified murine mAbs as indicated were labelled commercially (Rockland Immunochemicals) with biotin to perform mAb competition studies on whole SARS-CoV grown at the National Microbiology Laboratory under BL-3 conditions. Free-style 293 cells were obtained from Invitrogen. Recombinant S-protein were produced in mammalian and baculovirus expression systems and purified. Both full length spike protein (rFS) and transmembrane deficient Spike (rFS-TMless) were graciously provided by the Biodefence and Emerging Infectious Pathogens Resources (BEI). A mammalian expressed ACE-s minimal binding domain fragment (rACE2BDS) protein fragment (aa 318–510) was prepared as described previously.Citation62,Citation70 Purity was assessed by Coomassie blue staining following SDS-PAGE. A deglycosylated version of the rACE2BDS, termed (dgrACE2BDS), was prepared by pngase digestion. Sequence data on the variable regions (VH and VL) of the F26 mAbs have been published previously.Citation2 Chain promiscuity was assessed by interchanging Vh and VLs in chimeric pIgG constructs and the original pairings (i.e., 18H18L) and uniquely assembled pairings (i.e., 9H10L) were analyzed. While the original pairings represent chimeric leads, the uniquely built pairings might reveal information regarding the relative contributions of both heavy and light chain V genes to binding.
Cloning of VH and VL genes, construction and expression of chimeric mAbs.
Chimeric mAbs were produced in several versions of a mammalian expression vector with human IgG1 and kappa constant regions. Immunoglobulin variable region gene RT-PCR was performed essentially as describedCitation2,Citation71 with the following exceptions. Briefly, the GeneRacer™ System (Invitrogen) was used to perform RNA ligase-mediated rapid amplification of cDNA ends (RLM-RACE) when the blocking primer failed to prevent AbVk amplification.Citation72 Polymerase Chain Reaction (PCR) was then used to amplify the VL and VH using combinations primers published previously.Citation2,Citation71 Amplified VH and VL genes were cloned into the pIgG vectorCitation73,Citation74 and all clones were sequenced at each step. Sequence data obtained from commercial services or the NML DNA core were analyzed using DNAStar software. Variable region genes with an internal Sac I site in the VH region that would interfere with the digestion and ligation strategy was removed via site-directed mutagenesis using the QuikChange® II XL Site-Directed Mutagenesis Kit (Stratagene). The light chain variable region gene was fused to the human Ckappa constant region using overlap PCR. Primers were developed to link the murine VL to the human Cκ and provide the appropriate 5′ and 3′ RE sites (Hind III and Xba I respectively) for ligation in the pIgG vector. The pIGG-P14 plasmid clone and parental pIgG vector were obtained as a gift from Christoph Rader (NIH) and Carlos Barbas (TSRI).Citation74,Citation75 Following this the VH genes were inserted into the pIGG-containing the VLCκ. Individual clones were screened for inserts using restriction digests and by PCR and sequencing, followed by expression tests. The human-mouse IgG1 antibodies were expressed transiently in batches in free-style 293F cells. The 9H and 10H immunoglobulin variable region gene cDNAs from F26G9 and F26G10 parental hybridomas were paired with all four possible combinations of light chains (9L, 10L, 18L and 19L), while 18H and 19H were each paired with 18L and 19L only. This gave a total of 12 constructs, with four original pairings and eight uniquely assembled pairs. Eight clones of the ligation reactions were screened by sequencing, showing that all the constructs created were correct and in-frame (data not shown). The cells were transfected with plasmids using 293fectin™ according to the manufacture’s protocol (Invitrogen) and incubated at 37δC with 8% CO2 for six days prior to collection, screening in ELISA, concentration and finally protein A purification.
Enzyme-linked immunosorbent assays (ELISAs).
Monoclonal antibody were tested for binding to full-length baculovirus expressed recombinant spike protein (rFS; at 200 ng/well), rACE2BDS (100 ng/well), and recombinant protective antigen (rPA; 200 ng/well) (Cedarlane) coated to individual wells of a MaxiSorp 96 well plate (NUNC™). The plates were handled as described previously.Citation1,Citation2 Biotinylated F26G6, F26G9 and F26G18 were used to group epitopes on the whole inactivated SARS coated on plate and conjugated mAbs used at 2 × 1/2 maximum optical density were than mixed 1:1 with unlabelled competitor mAbs. A competition ELISA was performed on the mAbs in order to test for specificity for proteinaceous epitopes in the ACE-2 BDS protein. For the RBD competition ELISA, 200 ng of rFS was coated per well on MaxiSorp 96 well plates overnight at 4°C as above. Tissue culture supernatant from parental murine hybridomas were diluted with PBS to a dilution, determined previously, that yielded an absorbance unit of approximately OD = 1 at 405 nm versus rFS when the substrate is applied for an hour, and incubated for 30–40 minutes with twofold serial dilutions in PBS of deglycosylated (dgrACE2BDS), starting at 0.03625 mg/ml down to 1.77 × 10−5 mg/ml. Competitive ELISA using the soluble peptides was carried out, with the following modifications. Instead of dgrACE2BDS, the soluble peptides were mixed at twofold serial dilutions starting from 100 µg/ml, and ending at 0.097 µg/ml. Each antibody was tested against the peptide containing its putative binding domain and the corresponding scrambled peptide. The protocols outlined above were followed for characterisation of the chimeric antibody with the following modifications. In ELISA, cELISA and western blotting, the GαH IgG-γ HRP antibody was substituted for the anti-mouse secondary.
Western immunoblots.
Antibody binding specificity was determined using western immunoblotting as described previously.Citation1,Citation2 Diaminobenzidine (DAB)/Metal Substrate (Pierce, Rockford, IL) was applied to the membrane until substantial colour development could be seen, usually 0.5 to 4 minutes. Excess substrate was then washed away with water and the membrane allowed to dry prior to scanning to obtain a digital image.
Epitope mapping of F26G8 and F26G18 with pepscan.
Using the TOR2 strain S protein as a template, forty-nine 17-mers with 9aa overlap were developed, synthesised and attached via an extra C-terminal cysteine to pins (synthesis and attachment performed by PepScan Presto) spanning from 300–700 aa. Internal cysteines were replaced by alanine. For the assay, the pins were blocked for 2 hours and the mAb was diluted to 1 ug/mL in 0.1% skim milk and 0.1% Tween-20 in PBS and reacted with the pins overnight at 4°C. The pins were washed three times then incubated with GαM IgG-γ HRP (Southern Biotech) in dilution buffer for 1 hour. After washing again, 200 µl of ABTS substrate was applied to a MaxiSorp 96-well plate and the readings taken at timed intervals. The absorbance reading was then plotted versus the peptide sequence, and secondary antibody alone used as the negative control. The pin block was regenerated via sonication at 60°C for 1 hour in a phosphate solution with 1% SDS and 0.1% BME, followed by sonication for 30 minutes in water according to the manufacturer’s instructions.
Epitope mapping of F26G8 and F26G18 using soluble peptides
Based on the pin peptide analysis, soluble 17-mers of the putative binding sequence were commercially developed with an N-terminal biotin (United Biochemical Research). Epitope mapping was performed on the huG1-F26G18 chimeric monoclonal antibody using pin peptides and soluble peptides identical to the methods described, except substituting the GαH IgG-γ HRP antibody for the anti-mouse secondary. Oligopeptides containing the identical amino acid residues but in scrambled order were synthesized as controls. The soluble synthetic peptide names and sequences are as follows: G18PEP, N-Biotin-FSPDGKPCTPPALNCYW-COOH; G18PEPScr, N-Biotin-KGCPWAYLSPPCTDPNF-COOH; G8PEP, N-Biotin-TAIHADQLTPAWRIYST-COOH; G8PEPScr, N-Biotin-HITARQTPWAADLITYS-COOH. The mAbs were first tested for binding to the synthetic oligopeptides in an indirect ELISA using streptavidin (Zymed) coated on a MaxiSorp 96 well plate to capture the peptides. A competitive peptide ELISA was performed in order to confirm specificity for the minimal peptide epitopes. In this case the soluble peptides were used to inhibit mAb binding to rFS.
Surface plasmon resonance
Affinity analysis was performed on a BIACore 2000 essentially as described previously using CM5 chipsCitation75 coated with rFS. Briefly, surface plasmon resonance was done using 0.15 M NaCl, 0.005% Surfactant P20, 0.01 M HEPES pH 7.4 (HEPES-P) (BIACore) as the buffer with a flow rate of 5 µl/minute. The rFS was coated onto flow cell 1 in all cases of an activated commercial grade CM-5 chip. An increase in signal from baseline (after activation) to after coating indicated that sample had been successfully coated onto the chip. A minimum of five samples runs was used for each antibody.
Virus neutralization assay.
Vero-E6 cells were grown in a tissue culture 96 well plate to confluence in DMEM with 2% FBS (Gibco). Antibody was diluted tenfold in the medium and heat inactivated at 56°C for 30 minutes. The Vero-E6 cells and antibody were then taken into the Biosafety Level 3 Laboratory. Various dilutions of antibody were then mixed with a TCID50 = 100 of SARS-CoV TOR2 strain (uncloned) and incubated at 37°C for 1 hour. After incubation, 20 µl of the antibody/virus dilutions were applied to six wells per dilution to the plate and incubated for 1 hour to allow for infection. Virus titers of TCID50 = 10, 1 and 0.1 were used as a positive control. Next, 100 µl of medium was then added to each well and the plates incubated at 37°C for 3–4 days, then scored under a microscope for the presence of cytopathic effect (CPE).
Abbreviations
| SARS-CoV | = | human severe acute respiratory syndrome coronavirus |
| SDS-PAGE | = | sodium dodecyl sulfate-polyacrylamide gel electrophoresis |
| RBD | = | receptor binding domain, 318–510 |
Figures and Tables
Figure 1 ELISA Epitope characterisation of murine F26 series mAbs. All the F26 series mAbs tested bind to rFS and F26G8 is the only mAb that does not bind to the ACE2BD region, as demonstrated by lack of binding to either rACE2BDS or dgrACE2BDS (A). These results were confirmed by a competitive with rFS coating and dgrACE2BDS inhibition which clearly shows a reduction in signal with increasing dgrACE2BDS protein concentration for all mAbs except F26G8 (B).
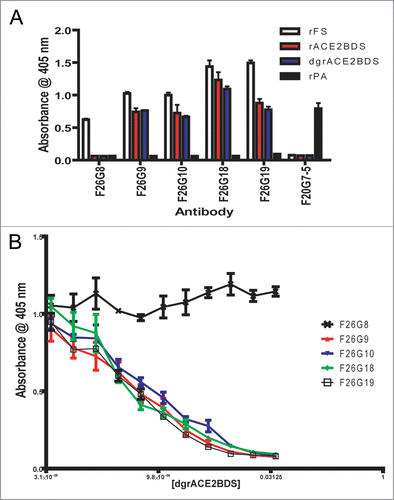
Figure 2 Competition ELISA with biotinylated mAbs to whole inactivated SARs-CoV. The biotinylated mAbs F26G6 (A), F26G9 (B) and F26G18 (C), were evaluated for the ability to bind in the presence of competing mAbs. signal was detected using goat anti-mouse Fc gamma hrp and normalized to 100% binding in the presence of PBS. White bars, less than 0–50% inhibition; Gray bars, around 50% inhibition of binding; Black bars, >75% inhibition of the biotinylated mAb.
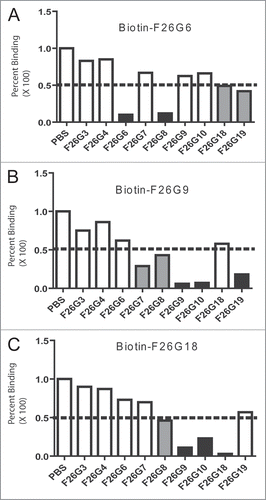
Figure 3 ELISA epitope characterisation of chimeric mAbs. Direct ELISA (A) shows that all the chimeric mAbs bind strongly to rFS, while rACE2BDS and rdgACE2BDS bind with varying degrees. Competitive ELISA (B) shows that rdgACE2BDS successfully competes with rFS for antibody binding for all the chimerics. B-inset shows western immunoblotting on rFS (2), rACE2BDS (3) and rPA (4) using chimeric antibody 18H18L. The chimeric antibody binds to both rFS and rACE2BDS, without binding rPA. Lane 1 is the molecular weight marker, with the values in kiloDaltons (kDa) on the left hand side.
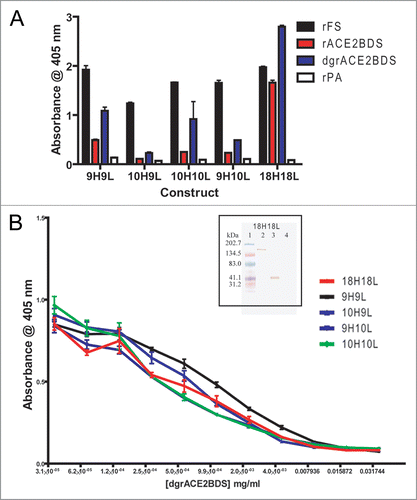
Figure 4 Epitope Mapping of murine mAbs F26G8 and F26G18. (A) Pepscan Mapping of murine mAbs F26G8 and F26G18 on overlapping pin peptides spanning the S1 region (A) inset, Random Phage peptide mapping of mAb F26G8. Mimotope identification of anti-SARS monoclonal antibody, F26G8 after the second selection from Ph.D-12-mer phage displayed random peptide library mapping. Above the clone sequences is shown the actual S protein motif sequence. (B) inset, Direct ELISA on tethered peptide in an ELISA and (B), a competitive soluble peptide ELISA on rFS which confirms that mAbs F26G8 and F26G18 bind S1 with critical contacts in regions aa612–620 and aa460–476, respectively.
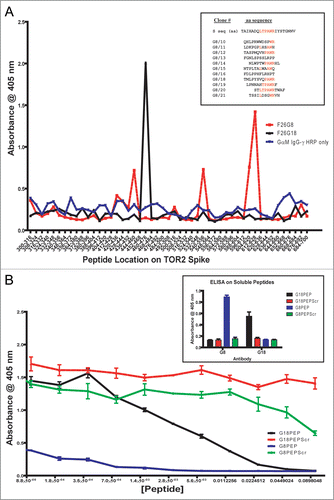
Figure 5 Peptide Mapping of 18H18L. Pin Peptide Mapping (A) and direct peptide binding ELISA (B) results are inconclusive compared to background (black bar, G18PEP; white bar, G18PEPScr; grey bar, G8PEP; hatched bar, G8PEPScr), while the competitive ELISA (C) demonstrates that 18H18L binds the aa460–476 peptide.
Figure 6 Depiction of the SARS-CoV Achilles heel. A schematic depicting the location of neutralizing epitopes and binding domains on the 193 amino acid receptor binding domain (RBD) of the S1 protein (Li et al. 2005). (A) Contact residues of ACE2 and mAbs which interrupt S1 binding to ACE2 are depicted with blue filled boxes. The DC-SIGN and mAbs which are known to block S1 binding to DC-SIGN are depicted with a pink filled box. The mAbs F26G18 and F26G19 were raised in immune response to native SARS-CoV spike protein (whole virus) and are outlined in red lined boxes. mAbs with linear epitopes are shown with solid lined boxes. mAbs with conformation epitopes have dashed lines on their boxes to indicate contact residues fall in the region as determined from co-crystal structure with S1 proteins. (B) The core 33 amino acid residues containing the known critical contacts of neutralizing mAbs and the ACE-2 receptor in the RBD (green dashed box). This immunodominant determinant corresponds to amino acid residues 460–492 of the S1 protein (green dashed box). This region is shown to illustrate the diversity of recognition of this compact region as well as the proximity of the contact residues of neutralizing mAbs in relation to the contact residues of the ACE2 host cellular receptor. Occlusion of the receptor is the main mechanisms of SARS virus neutralization for mAbs binding in this region. The boxes show the minimal epitope contact “footprint” in this region either from crystal structures and or other epitope mapping strategies for each of: ACE2 (red broken box); conformational mAb 80R (blue dotted box); conformational mAb F26G19 (black dashed box, 486–492); conformational mAb m396 main contact residues (brown box); and the linear epitope of mAb F26G18 (black box, 460–476). The known contact residues for ACE-2 are color coded red. Data has been extracted from this paper and other previously published articles.Citation4,Citation9,Citation11,Citation10,Citation56,Citation76
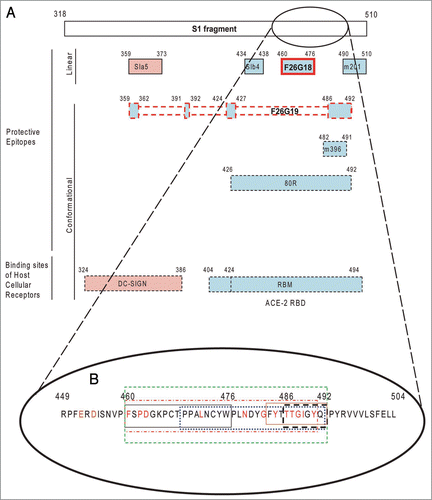
Table 1 Binding characteristics of murine and chimeric human mAbs on recombinant full length baculovirus spike protein and virus neutralisation titres
Table 2 Different methods and forms of S protein all generate or select neutralizing mAbs to the SARS-CoV RBD
Acknowledgements
The authors would like to thank the Critical Reagents Program, as well as BEI Resources (Biorepository for Emerging Infectious Agents), for providing reagents, Dr. Jack Nunberg (University of Montana) for providing advice on cell lines, and Drs. Christoph Rader (NCI-NIH) and Carlos F. Barbas, III (TSRI) for providing assistance with mammalian expression vectors and design. Funding for this work was partially provided by the Public Health Agency of Canada and the CBRN research technology initiative. K.H. is the recipient of a Canadian NSERC graduate fellowship stipend and the University of Manitoba Medical College Gold Medal entrance award.
References
- Berry JD, Jones S, Drebot MA, Andonov A, Sabara M, Yuan X, et al. Development and characterisation of neutralising monoclonal antibody to the SARS-coronavirus. J Virol Methods 2004; 120:87 - 96
- Gubbins MJ, Plummer FA, Yuan XY, Johnstone D, Drebot M, Andonova M, et al. Molecular characterization of a panel of murine monoclonal antibodies specific for the SARS-coronavirus. Mol Immunol 2005; 42:125 - 136
- Stadler K, Masignani V, Eickmann M, Becker S, Abrignani S, Klenk HD, et al. SARS—beginning to understand a new virus. Nat Rev Microbiol 2003; 1:209 - 218
- Li F, Li W, Farzan M, Harrison SC. Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science 2005; 309:1864 - 1868
- Chakraborti S, Prabakaran P, Xiao X, Dimitrov DS. The SARS coronavirus S glycoprotein receptor binding domain: fine mapping and functional characterization. Virol J 2005; 2:73
- Liu S, Xiao G, Chen Y, He Y, Niu J, Escalante CR, et al. Interaction between heptad repeat 1 and 2 regions in spike protein of SARS-associated coronavirus: implications for virus fusogenic mechanism and identification of fusion inhibitors. Lancet 2004; 363:938 - 947
- Li W, Moore MJ, Vasilieva N, Sui J, Wong SK, Berne MA, et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003; 426:450 - 454
- Wong SK, Li W, Moore MJ, Choe H, Farzan M. A 193-amino acid fragment of the SARS coronavirus S protein efficiently binds angiotensin-converting enzyme 2. J Biol Chem 2004; 279:3197 - 3201
- Hwang WC, Lin Y, Santelli E, Sui J, Jaroszewski L, Stec B, et al. Structural basis of neutralization by a human anti-severe acute respiratory syndrome spike protein antibody, 80R. J Biol Chem 2006; 281:34610 - 34616
- Pak JE, Sharon C, Satkunarajah M, Auperin TC, Cameron CM, Kelvin DJ, et al. Structural insights into immune recognition of the severe acute respiratory syndrome coronavirus S protein receptor binding domain. J Mol Biol 2009; 388:815 - 823
- Prabakaran P, Gan J, Feng Y, Zhu Z, Choudhry V, Xiao X, et al. Structure of severe acute respiratory syndrome coronavirus receptor-binding domain complexed with neutralizing antibody. J Biol Chem 2006; 281:15829 - 15836
- Forsell MN, Dey B, Morner A, Svehla K, O’dell S, Hogerkorp CM, et al. B cell recognition of the conserved HIV-1 co-receptor binding site is altered by endogenous primate CD4. PLoS Pathog 2008; 4:1000171
- Parren PW, Fisicaro P, Labrijn AF, Binley JM, Yang WP, Ditzel HJ, et al. In vitro antigen challenge of human antibody libraries for vaccine evaluation: the human immunodeficiency virus type 1 envelope. J Virol 1996; 70:9046 - 9050
- Parren PW, Burton DR, Sattentau QJ. HIV-1 antibody—debris or virion?. Nat Med 1997; 3:366 - 367
- Parren PW, Moore JP, Burton DR, Sattentau QJ. The neutralizing antibody response to HIV-1: viral evasion and escape from humoral immunity. AIDS 1999; 13:137 - 162
- King JC Jr, Cox MM, Reisinger K, Hedrick J, Graham I, Patriarca P. Evaluation of the safety, reactogenicity and immunogenicity of FluBlok((R)) trivalent recombinant baculovirus-expressed hemagglutinin influenza vaccine administered intramuscularly to healthy children aged 6–59 months. Vaccine 2009; 27:6589 - 6594
- Olsen CW, McGregor MW, Dybdahl-Sissoko N, Schram BR, Nelson KM, Lunn DP, et al. Immunogenicity and efficacy of baculovirus-expressed and DNA-based equine influenza virus hemagglutinin vaccines in mice. Vaccine 1997; 15:1149 - 1156
- Treanor JJ, Wilkinson BE, Masseoud F, Hu-Primmer J, Battaglia R, O'Brien D, et al. Safety and immunogenicity of a recombinant hemagglutinin vaccine for H5 influenza in humans. Vaccine 2001; 19:1732 - 1737
- Wei CJ, Xu L, Kong WP, Shi W, Canis K, Stevens J, et al. Comparative efficacy of neutralizing antibodies elicited by recombinant hemagglutinin proteins from avian H5N1 influenza virus. J Virol 2008; 82:6200 - 6208
- Plotkin SA, Mortimer EA. Vaccines 1994; 2nd Ed Philadelphia WB Saunders
- Du L, He Y, Wang Y, Zhang H, Ma S, Wong CK, et al. Recombinant adeno-associated virus expressing the receptor-binding domain of severe acute respiratory syndrome coronavirus S protein elicits neutralizing antibodies: Implication for developing SARS vaccines. Virology 2006; 353:6 - 16
- Yang ZY, Kong WP, Huang Y, Roberts A, Murphy BR, Subbarao K, et al. A DNA vaccine induces SARS coronavirus neutralization and protective immunity in mice. Nature 2004; 428:561 - 564
- Bisht H, Roberts A, Vogel B, Subbarao K, Moss B. Neutralizing antibody and protective immunity to SARS coronavirus infection of mice induced by a soluble recombinant polypeptide containing and N-terminal segment of the spike glycoprotein. Virology 2005; 334:160 - 165
- Du L, Zhao G, He Y, Guo Y, Zheng BJ, Jiang S, et al. Receptor-binding domain of SARS-CoV spike protein induces long-term protective immunity in an animal model. Vaccine 2007; 25:2832 - 2838
- He Y, Zhou Y, Liu S, Kou Z, Li W, Farzan M, et al. Receptor-binding domain of SARS-CoV spike protein induces highly potent neutralizing antibodies: implication for developing subunit vaccine. Biochem Biophys Res Commun 2004; 324:773 - 781
- He Y, Lu H, Siddiqui P, Zhou Y, Jiang S. Receptor-binding domain of severe acute respiratory syndrome coronavirus spike protein contains multiple conformation-dependent epitopes that induce highly potent neutralizing antibodies. J Immunol 2005; 174:4908 - 4915
- Zhang L, Zhang F, Yu W, He T, Yu J, Yi CE, et al. Antibody responses against SARS coronavirus are correlated with disease outcome of infected individuals. J Med Virol 2006; 78:1 - 8
- He Y, Zhou Y, Siddiqui P, Jiang S. Inactivated SARS-CoV vaccine elicits high titers of spike protein-specific antibodies that block receptor binding and virus entry. Biochem Biophys Res Commun 2004; 325:445 - 452
- Yu M, Stevens V, Berry JD, Crameri G, McEachern J, Tu C, et al. Determination and application of immunodominant regions of SARS coronavirus spike and nucleocapsid proteins recognized by sera from different animal species. J Immunol Methods 2007; 331:1 - 12
- Zhu Z, Dimitrov AS, Chakraborti S, Dimitrova D, Xiao X, Broder CC, et al. Development of human monoclonal antibodies against diseases caused by emerging and biodefense-related viruses. Expert Rev Anti Infect Ther 2006; 4:57 - 66
- Stockman LJ, Bellamy R, Garner P. SARS: systematic review of treatment effects. PLoS Med 2006; 3:343
- Sui J, Li W, Murakami A, Tamin A, Matthews LJ, Wong SK, et al. Potent neutralization of severe acute respiratory syndrome (SARS) coronavirus by a human mAb to S1 protein that blocks receptor association. Proc Natl Acad Sci USA 2004; 101:2536 - 2541
- van den Brink EN, ter Meulen J, Cox F, Jongeneelen MA, Thijsse A, Throsby M, et al. Molecular and biological characterization of human monoclonal antibodies binding to the spike and nucleocapsid proteins of severe acute respiratory syndrome coronavirus. J Virol 2005; 79:1635 - 1644
- Traggiai E, Becker S, Subbarao K, Kolesnikova L, Uematsu Y, Gismondo MR, et al. An efficient method to make human monoclonal antibodies from memory B cells: potent neutralization of SARS coronavirus. Nat Med 2004; 10:871 - 875
- Corbett CR, Elias MD, Simpson LL, Yuan XY, Cassan RR, Ballegeer E, et al. High-throughput homogeneous immunoassay readily identifies monoclonal antibody to serovariant clostridial neurotoxins. J Immunol Methods 2007; 328:128 - 138
- Babcook JS, Leslie KB, Olsen OA, Salmon RA, Schrader JW. A novel strategy for generating monoclonal antibodies from single, isolated lymphocytes producing antibodies of defined specificities. Proc Natl Acad Sci USA 1996; 93:7843 - 7848
- Smith K, Garman L, Wrammert J, Zheng NY, Capra JD, Ahmed R, et al. Rapid generation of fully human monoclonal antibodies specific to a vaccinating antigen. Nat Protoc 2009; 4:372 - 384
- Breathnach CC, Rudersdorf R, Lunn DP. Use of recombinant modified vaccinia Ankara viral vectors for equine influenza vaccination. Vet Immunol Immunopathol 2004; 98:127 - 136
- Galarza JM, Latham T, Cupo A. Virus-like particle (VLP) vaccine conferred complete protection against a lethal influenza virus challenge. Viral Immunol 2005; 18:244 - 251
- Mahmood K, Bright RA, Mytle N, Carter DM, Crevar CJ, Achenbach JE, et al. H5N1 VLP vaccine induced protection in ferrets against lethal challenge with highly pathogenic H5N1 influenza viruses. Vaccine 2008; 26:5393 - 5399
- Noad R, Roy P. Virus-like particles as immunogens. Trends Microbiol 2003; 11:438 - 444
- Pushko P, Tumpey TM, Van Hoeven N, Belser JA, Robinson R, Nathan M, et al. Evaluation of influenza virus-like particles and Novasome adjuvant as candidate vaccine for avian influenza. Vaccine 2007; 25:4283 - 4290
- Warfield KL, Swenson DL, Demmin G, Bavari S. Filovirus-like particles as vaccines and discovery tools. Expert Rev Vaccines 2005; 4:429 - 440
- Kwong PD, Doyle ML, Casper DJ, Cicala C, Leavitt SA, Majeed S, et al. HIV-1 evades antibody-mediated neutralization through conformational masking of receptor-binding sites. Nature 2002; 420:678 - 682
- Moulard M, Phogat SK, Shu Y, Labrijn AF, Xiao X, Binley JM, et al. Broadly cross-reactive HIV-1-neutralizing human monoclonal Fab selected for binding to gp120-CD4-CCR5 complexes. Proc Natl Acad Sci USA 2002; 99:6913 - 6918
- Pantophlet R, Wilson IA, Burton DR. Hyperglycosylated mutants of human immunodeficiency virus (HIV) type 1 monomeric gp120 as novel antigens for HIV vaccine design. J Virol 2003; 77:5889 - 5901
- Pantophlet R, Burton DR. Immunofocusing: antigen engineering to promote the induction of HIV-neutralizing antibodies. Trends Mol Med 2003; 9:468 - 473
- Pantophlet R, Wang M, guilar-Sino RO, Burton DR. The human immunodeficiency virus type 1 envelope spike of primary viruses can suppress antibody access to variable regions. J Virol 2009; 83:1649 - 1659
- Selvarajah S, Puffer B, Pantophlet R, Law M, Doms RW, Burton DR. Comparing antigenicity and immunogenicity of engineered gp120. J Virol 2005; 79:12148 - 12163
- Suguitan AL Jr, Marino MP, Desai PD, Chen LM, Matsuoka Y, Donis RO, et al. The influence of the multi-basic cleavage site of the H5 hemagglutinin on the attenuation, immunogenicity and efficacy of a live attenuated influenza A H5N1 cold-adapted vaccine virus. Virology 2009; 395:280 - 288
- Berry JD. Rational monoclonal antibody development to emerging pathogens, biothreat agents and agents of foreign animal disease: The antigen scale. Vet J 2005; 170:193 - 211
- Bui HH, Sidney J, Li W, Fusseder N, Sette A. Development of an epitope conservancy analysis tool to facilitate the design of epitope-based diagnostics and vaccines. BMC Bioinformatics 2007; 8:361
- Davies V, Vaughan K, Damle R, Peters B, Sette A. Classification of the universe of immune epitope literature: representation and knowledge gaps. PLoS ONE 2009; 4:6948
- Peters B, Sidney J, Bourne P, Bui HH, Buus S, Doh G, et al. The immune epitope database and analysis resource: from vision to blueprint. PLoS Biol 2005; 3:91
- Zhong G, Berry JD, Choukri S. Mapping epitopes of neutralizing monoclonal antibodies using phage random peptide libraries. J Ind Microbiol Biotechnol 1997; 19:71 - 76
- Sui J, Li W, Roberts A, Matthews LJ, Murakami A, Vogel L, et al. Evaluation of human monoclonal antibody 80R for immunoprophylaxis of severe acute respiratory syndrome by an animal study, epitope mapping and analysis of spike variants. J Virol 2005; 79:5900 - 5906
- Sui J, Aird DR, Tamin A, Murakami A, Yan M, Yammanuru A, et al. Broadening of neutralization activity to directly block a dominant antibody-driven SARS-coronavirus evolution pathway. PLoS Pathog 2008; 4:1000197
- Zhu Z, Chakraborti S, He Y, Roberts A, Sheahan T, Xiao X, et al. Potent cross-reactive neutralization of SARS coronavirus isolates by human monoclonal antibodies. Proc Natl Acad Sci USA 2007; 104:12123 - 12128
- Greenough TC, Babcock GJ, Roberts A, Hernandez HJ, Thomas WD Jr, Coccia JA, et al. Development and characterization of a severe acute respiratory syndrome-associated coronavirus-neutralizing human monoclonal antibody that provides effective immunoprophylaxis in mice. J Infect Dis 2005; 191:507 - 514
- Beniac DR, Andonov A, Grudeski E, Booth TF. Architecture of the SARS coronavirus prefusion spike. Nat Struct Mol Biol 2006; 13:751 - 752
- Beniac DR, deVarennes SL, Andonov A, He R, Booth TF. Conformational reorganization of the SARS coronavirus spike following receptor binding: implications for membrane fusion. PLoS ONE 2007; 2:1082
- Zakhartchouk AN, Sharon C, Satkunarajah M, Auperin T, Viswanathan S, Mutwiri G, et al. Immunogenicity of a receptor-binding domain of SARS coronavirus spike protein in mice: implications for a subunit vaccine. Vaccine 2007; 25:136 - 143
- Lu L, Manopo I, Leung BP, Chng HH, Ling AE, Chee LL, et al. Immunological characterization of the spike protein of the severe acute respiratory syndrome coronavirus. J Clin Microbiol 2004; 42:1570 - 1576
- Torres M, Casadevall A. The immunoglobulin constant region contributes to affinity and specificity. Trends in Immunol 2007; 29:91 - 97
- Lee JE, Fusco ML, Hessell AJ, Oswald WB, Burton DR, Saphire EO. Structure of the Ebola virus glycoprotein bound to an antibody from a human survivor. Nature 2008; 454:177 - 182
- Parren PW, Fisicaro P, Labrijn AF, Binley JM, Yang WP, Ditzel HJ, et al. In vitro antigen challenge of human antibody libraries for vaccine evaluation: the human immunodeficiency virus type 1 envelope. J Virol 1996; 70:9046 - 9050
- Fraser D, Kaern M. A chance at survival: gene expression noise and phenotypic diversification strategies. Mol Microbiol 2009; 71:1333 - 1340
- Coughlin MM, Babcook J, Prabhakar BS. Human monoclonal antibodies to SARS-coronavirus inhibit infection by different mechanisms. Virology 2009; 394:39 - 46
- Guo Y, Tisoncik J, McReynolds S, Farzan M, Prabhakar BS, Gallagher T, et al. Identification of a new region of SARS-CoV S protein critical for viral entry. J Mol Biol 2009; 394:600 - 605
- See RH, Zakhartchouk AN, Petric M, Lawrence DJ, Mok CP, Hogan RJ, et al. Comparative evaluation of two severe acute respiratory syndrome (SARS) vaccine candidates in mice challenged with SARS coronavirus. J Gen Virol 2006; 87:641 - 650
- Berry JD, Boese DJ, Law DK, Zollinger WD, Tsang RS. Molecular analysis of monoclonal antibodies to group variant capsular polysaccharide of Neisseria meningitidis: recurrent heavy chains and alternative light chain partners. Mol Immunol 2005; 42:335 - 344
- Yuan X, Gubbins MJ, Berry JD. A simple and rapid protocol for the sequence determination of functional kappa light chain cDNAs from aberrant-chain-positive murine hybridomas. J Immunol Methods 2004; 294:199 - 207
- Chung J, Rader C, Popkov M, Hur YM, Kim HK, Lee YJ, et al. Integrin alphaIIbbeta3-specific synthetic human monoclonal antibodies and HCDR3 peptides that potently inhibit platelet aggregation. FASEB J 2004; 18:361 - 363
- Hofer T, Tangkeangsirisin W, Kennedy MG, Mage RG, Raiker SJ, Venkatesh K, et al. Chimeric rabbit/human Fab and IgG specific for members of the Nogo-66 receptor family selected for species cross-reactivity with an improved phage display vector. J Immunol Methods 2007; 318:75 - 87
- Gubbins MJ, Berry JD, Corbett CR, Mogridge J, Yuan XY, Schmidt L, et al. Production and characterization of neutralizing monoclonal antibodies that recognize an epitope in domain 2 of Bacillus anthracis protective antigen. FEMS Immunol Med Microbiol 2006; 47:436 - 443
- Shih YP, Chen CY, Liu SJ, Chen KH, Lee YM, Chao YC, et al. Identifying epitopes responsible for neutralizing antibody and DC-SIGN binding on the spike glycoprotein of the severe acute respiratory syndrome coronavirus. J Virol 2006; 80:10315 - 10324
- Tripp RA, Haynes LM, Moore D, Anderson B, Tamin A, Harcourt BH, et al. Monoclonal antibodies to SARS-associated coronavirus (SARS-CoV): identification of neutralizing and antibodies reactive to S, N, M and E viral proteins. J Virol Methods 2005; 128:21 - 28
- Zhou T, Wang H, Luo D, Rowe T, Wang Z, Hogan RJ, et al. An exposed domain in the severe acute respiratory syndrome coronavirus spike protein induces neutralizing antibodies. J Virol 2004; 78:7217 - 7226