Abstract
Angiogenesis is required in normal physiological processes, but is also involved in tumor growth, progression and metastasis. Vascular endothelial growth factor (VEGF), a primary mediator of angiogenesis in normal physiology and in disease, and other VEGF family members and their receptors provide targets that have been explored extensively for cancer therapy. Small molecule inhibitors and antibody/protein-based strategies that target the VEGF pathway have been studied in multiple types of cancer. This review will focus on VEGF pathway targeting antibodies that are currently being evaluated in pre-clinical and clinical studies.
Introduction
Angiogenesis is a tightly regulated process responsible for the development of new blood vessels from a pre-existing vascular network. During development and normal physiological processes such as wound healing and the menstrual cycle, angiogenesis is regulated by endogenous activators and inhibitors. Within adult animals, the levels of endogenous mediators are balanced and endothelial cells (ECs) are largely quiescent.Citation1,Citation2 In pathological settings, such as age-related macular degeneration, rheumatoid arthritis, diabetic retinopathy and tumor growth and metastasis, angiogenesis is critical for disease progression.Citation2,Citation3 A key step in tumor development, the ‘angiogenic switch’ occurs when endogenous activators of angiogenesis outweigh endogenous inhibitors, thereby shifting the balance of angiogenic mediators and stimulating angiogenesis. This results in increased blood vessel formation, which supplies growing tumors with necessary oxygen and nutrients for sustained growth;Citation4 however the resulting vasculature is disorganized and poorly structured, leading to chaotic blood flow and leaky blood vessels.Citation5,Citation6 Although dysfunctional when compared to the hierarchical, well-structured vascular network found in normal tissue,Citation7,Citation8 tumor vasculature is nonetheless essential for continued tumor growth. In the absence of a blood vascular network, tumors are restrained in size due to limits in the diffusion of oxygen.Citation9 In 1971, Judah Folkman was the first to hypothesize the potential therapeutic benefit of targeting tumor angiogenesis.Citation9 The vascular endothelial growth factor (VEGF) family of proteins are key regulators of normal and tumor angiogenesis and so provide attractive targets for anti-cancer therapies. This review will focus on antibody-based strategies to target the VEGF pathway in tumors.
The VEGF Family
There are five members of the human VEGF family: VEGF-A (referred to in this review as VEGF), VEGF-B, VEGF-C, VEGF-D and placental growth factor (PlGF).Citation2,Citation3 In addition, multiple isoforms of VEGF, VEGF-B and PlGF are generated through alternative splicing of pre-mRNA.Citation1 The VEGF family ligands interact with the receptor tyrosine kinases VEGF receptor-1 (VEGFR1), VEGFR2 and VEGFR3. VEGF family interaction with VEGFRs is also regulated by the non-enzymatic co-receptors neuropilin (Nrp)-1 and Nrp2.Citation1
The VEGF gene contains eight exons and seven introns.Citation10,Citation11 VEGF binds to VEGFR1, VEGFR2, Nrp1 and Nrp2.Citation1 VEGF induces vascular permeabilityCitation12 and also functions as an EC mitogen and survival factor,Citation13–Citation15 and an inducer of EC cell and monocyte migration.Citation16,Citation17 Alternative splicing of VEGF yields nine different isoforms in total and four major isoforms: VEGF121, 165, 189 and 206.Citation18 The bioavailability of the different VEGF isoforms is mediated by their expression of heparin sulfate proteoglycan (HSP)-binding domains, encoded on exons 6a, 6b and 7.Citation19,Citation20 These domains have strong affinity for proteoglycans found on cell plasma membranes or within the extracellular matrix (ECM), thereby restricting the diffusion of larger isoforms of VEGF.Citation21 Release of VEGF from the ECM and cell membrane allows for VEGF-mediated activity and signaling. The proteolytic release of VEGF is mediated by the extracellular proteases plasmin,Citation22 urokinase type of plasminogen activator (uPA)Citation23 and matrix metalloproteinases.Citation24–Citation26 Proteolytic release of VEGF is induced by remodeling and microenvironment cues elicited during physiological and pathologic angiogenesis.Citation27
The VEGF-B gene contains seven exons that undergo alternative splicing to produce two isoforms, VEGF-B167 and VEGFB186.Citation28 VEGF-B binds to both VEGFR1 and Nrp1.Citation1 The overall function of VEGF-B remains unclear, with suggested roles in heart function in adults, but not in developmental angiogenesis or cardiovascular development since VEGF-B null mice are viable despite some abnormalities in cardiac conduction.Citation29
The VEGF-C gene is made up of eight exons, but does not undergo alternative splicing. Mature VEGF-C binds to VEGFR2 and VEGFR3 and is involved in developmental lymphangiogenesis and the maintenance of adult lymphatic vasculature.Citation30 VEGF-C null mice are embryonic lethal and heterozygous VEGF-C loss is characterized by lymphedema from defective development of the lymphatic vasculature.Citation31 Interestingly, VEGF-C is not required for blood vessel development since vessels appeared normal in VEGF-C null animals.Citation31
VEGF-D is composed of seven exons and is found on the X chromosome.Citation32 Mature VEGF-D binds to both VEGFR2 and VEGFR3 as a non-covalent homodimer.Citation33 Knock out studies in mice suggest that VEGF-C, and perhaps other growth factors, are capable of substituting for VEGF-D function, as VEGF-D null mice are viable and have a normal lymphatic vasculature during development and in the adult.Citation34
The last member of the human VEGF family is PlGF. The PlGF gene contains seven exons that generate four different isoforms by alternative splicing.Citation35–Citation37 These isoforms are primarily expressed in the placenta, but are also found within the heart, retina, skin and skeletal muscle.Citation1 There is reduced vascularization of the corpus luteum and retina in PlGF null mice, but these animals are viable.Citation38
The VEGF Receptors
There are three receptor tyrosine kinases that mediate the angiogenic functions of VEGF family members: VEGFR1, VEGFR2 and VEGFR3. Although these receptors potentiate diverse downstream functions, they are structurally very similar. The VEGF receptors each contain a seven member immunoglobulinlike domain extracellular region, a single transmembrane domain segment, a juxtamembrane segment, a split intracellular proteintyrosine kinase domain, and a carboxyterminal tail.Citation1
VEGFR1, also known as fms-like tyrosyl kinase-1 (Flt-1), binds VEGF, VEGF-B and PlGF.Citation39–Citation42 Alternative splicing of VEGFR1 produces a soluble form of the receptor (sVEGFR1) that contains the first six of the seven immunoglobulin domains, and binds to and inhibits the function of VEGF.Citation43 VEGFR1 can function as a decoy receptor, utilizing its strong affinity for VEGF (approximately 10 times stronger than that of VEGFR2 for VEGF) to sequester the ligand, preventing it from signaling through other receptors.Citation17 Despite the strong binding affinity of VEGFR1 to VEGF, the kinase activity of this receptor is weak making it difficult to evaluate levels of VEGFR1 auto-phosphorylation in cells that have not been engineered to express high levels of the receptor.Citation17 VEGFR1 is essential during development. VEGFR1 null animals are embryonic lethal, characterized by ECs that do not form a structured, organized vascular network.Citation44 Interestingly, mice that do not express the tyrosine kinase domain of VEGFR1 but retain the ligand-binding extracellular domains and the transmembrane segment (VEGFR1-TK−/−) are viable, emphasizing the importance of ligand sequestration in VEGFR1 function.Citation45 The mutant phenotype resulting from VEGFR1 loss in embryonic stem cell-derived blood vessels can be rescued with VEGFR2 small molecule inhibitors.Citation46 Although VEGFR1 signaling remains unclear, there is support for the involvement of the receptor in hematopoiesis,Citation47,Citation48 the migration of monocytes and the recruitment of bone marrow-derived progenitor cells.Citation16,Citation49 VEGFR1 has also been implicated in the paracrine release of growth factors from ECsCitation50 and inducing VEGF-B-mediated EC expression of matrix metalloproteinase-9, uPA and plasminogen activator inhibitor-1, molecules important for ECM degradation that can potentiate VEGF release and cell migration.Citation41 In addition, VEGF binding to VEGFR1 has been shown to induce SHP-1 phosphatase activity that in turn reduced VEGFR2 phosphorylation levels.Citation51 These data support VEGFR1 functioning to negatively regulate activity of VEGFR2, which could have important implications for targeting the VEGF pathway within tumors.
VEGFR2 [Fetal liver kinase-1 (Flk-1)/Kinase Domaincontaining Receptor (KDR)] is the predominant mediator of VEGF-induced angiogenic signaling.Citation52–Citation54 Functions of VEGFR2 include EC survival, migration, proliferation and vascular permeability.Citation1,Citation55,Citation56 VEGFR2 binds all VEGF isoforms, VEGF-C and VEGF-D. Although VEGR2 has a lower affinity for VEGF than VEGFR1, it has a stronger kinase activity. When VEGF binds VEGFR2, it induces receptor dimerization and trans auto-phosphorylation.Citation1 The predominant phosphorylation sites on VEGFR2 occur on tyrosine 1175 and 1214, inducing signaling cascades through PI3K, AKT, PLCγ, p38 MAPK and p42/44 MAPK.Citation1,Citation55,Citation57 Ebos et al. identified a soluble, circulating form of VEGFR2, and Albuquerque et al. recently found that this soluble receptor is a distinct splice variant that inhibits lymphangiogenesis.Citation58,Citation59 Recently, VEGFR2 expression by macrophages has been demonstrated to mediate macrophage infiltration in tumor bearing animals.Citation60 VEGFR2 null mice are embryonic lethal between day E8.5–9.5. These animals have severe defects in endothelial and hematopoietic cell development with no organized blood vessel found at any point within the developing embryo or the yolk sac.Citation61
VEGFR-3 (Flt-4) binds both VEGF-C and VEGF-D and functions in the remodeling of embryonic primary capillary plexus, with sustained roles in adult angiogenesis and lymphangiogenesis.Citation62–Citation64 VEGFR3 null mice are embryonic lethal at day E9.5 and display cardiovascular failure as a result of the abnormal structure and organization of large vessels that leads to defective vessel lumens and an accumulation of fluid within the pericardial cavity.Citation65 In humans, inactivating mutations within the catalytic loop of the VEGFR3 kinase domain cause Milroy disease, a hereditary form of lymphedema where defective lymphatic vessels cause chronic swelling of the extremities.Citation66 The lymphatic abnormalities of Milroy disease and the phenotype of VEGFR3 null mice, suggest that VEGFR3 functions first in the development of the cardiovascular system and later in the lymphatic vasculature in adults.Citation1
The two non-enzymatic co-receptors for the VEGF family, Nrp1 and Nrp2 were first identified as receptors for semaphorins, which function during neurogenesis.Citation1,Citation67,Citation68 Structurally, the Nrps have a large extracellular region, a transmembrane segment and a short intracellular domain that apparently does not function catalytically, but may serve as a binding site for other coreceptors or downstream signaling molecules.Citation69 Nrp1 binds VEGF isoforms with HSP-binding domains, PlGF and both VEGF-B isoforms and potentiates signaling through VEGFR2.Citation70–Citation72 Nrp1 functions during vascular development and in angiogenesis as demonstrated by genetic modifications in mouse models. Nrp1 overexpressing mice are embryonic lethal due to hemorrhaging and excessive capillary and blood vessel formation.Citation73 Nrp1 null mice are embryonic lethal between days E10.5–12.5 resulting from nervous system and cardiovascular abnormalities.Citation74 Nrp2 binds to VEGF145,165, VEGF-C and PlGF.Citation75,Citation76 Nrp2 functions during lymphatic development as indicated by Nrp2 null mice that are normal and display phenotypically normal vasculature but have a severe reduction in small lymphatic vessels.Citation77,Citation78 Additional work evaluating the role of Nrp1 and Nrp2 by Takashima et al. demonstrated that Nrp1 Nrp2 double null mice have avascular yolk sacs and are embryonic lethal at day E8.5. Further, Nrp1+/− Nrp2−/− or Nrp1−/− Nrp2+/− mice are embryonic lethal at day E10-0.5 with severe angiogenic abnormalities in both the yolk sac and the embryo and an overall reduction in embryo size.Citation79 These mutant embryos had a similar phenotype to VEGF or VEGFR2 null mice, highlighting the importance of the Nrps in embryonic blood vessel development.
Monoclonal Antibodies as Therapeutic Agents
The clinical use of monoclonal antibodies (mAbs) to treat cancer and other diseases is well established. There are currently more than 20 mAbs approved by the Food and Drug Administration (FDA) for therapeutic use.Citation80 Rituximab (Rituxan®, Genentech, Inc.), a chimeric mAb directed against CD20 used to treat patients with non-Hodgkin lymphoma, was the first anticancer mAb to gain FDA approval. Since then, mAbs targeting key pathways in tumor survival have been developed and successfully used clinically to treat patients including trastuzumab (Herceptin®, Genentech, Inc.) which recognizes the HER2/neu cell surface receptor expressed in 15–20% of breast cancers, and cetuximab (Erbitux®, ImClone Systems) which binds epidermal growth factor receptor (EGFR) and is approved for the treatment of metastatic colorectal cancer and head and neck cancer.Citation80
Tumor angiogenesis is a hallmark of cancer, and is required for continued growth and metastasis.Citation81 VEGF is a major mediator of angiogenesis in normal physiology and in cancer. There is an upregulation of VEGF family members and the VEGF receptors in many different tumors,Citation1 providing a target for cancer therapy. This target has been utilized by investigators, leading to the development of anti-angiogenic small molecule tyrosine kinase inhibitors such as sorafenib (Nexavar®, BAY 43-9006, Bayer Pharmaceuticals Corp.) and sunitinib (Sutent®, SU11248, Pfizer, Inc.,),Citation1 and a number of mAbs against VEGF ligands and receptors. The latter strategy will be the focus of the rest of this review (refer to and for a summary of the anti-VEGF pathway and the mAbs targeting this pathway discussed here).
Antibodies and Fusion Proteins Targeting VEGF
Bevacizumab (Avastin®, Genentech, Inc.).
In 1993, the Ferrara et al. introduced a mouse anti-human VEGF mAb (A.4.6.1) that inhibited the growth of A673 rhabdomyosarcoma, G55 glioblastoma and SK-LMS-1 leyomiosarcoma tumor cell line xenograft models in vivo but not in vitro,Citation82 illustrating an indirect role of VEGF signaling in tumor survival. The efficacy of A.4.6.1 in controlling the growth of a number of tumor cell lines in xenograft models in immunocompromised mice was described subsequently in a number of publications.Citation83–Citation85 A.4.6.1 underwent site-directed mutagenesis resulting in the humanized mAb bevacizumab (Avastin®, Genentech Inc.) that binds human VEGF with an affinity similar to A.4.6.1 (Kd ≈ 0.5 nM).Citation86 Structural analysis of VEGF bound to bevacizumab Fab gave insight to the specificity and mechanism of the mAb. Gly88 of human VEGF is required for bevacizumab to binding. In mouse and rat VEGF this residue is replaced by a serine, disrupting the binding of bevacizumab to rodent VEGF.Citation87 Further, bevacizumab Fab bound to VEGF does not induce structural conformational changes in the ligand, suggesting that bevacizumab is effective by sterically disrupting the ability of VEGF to interact with its receptors.Citation87–Citation89 Preclinical safety evaluations of bevacizumab were performed in Macaca fascicularis. Some treatment-induced changes were observed, including suppressed angiogenesis within the female reproductive tract, but all adverse effects were reversible with the cessation of bevacizumab treatment.Citation90
In 1997, bevacizumab entered Phase 1 clinical trials and was found to be relatively non-toxic and well-tolerated, without exacerbating toxicities related to patient chemotherapy treatment.Citation91,Citation92 In subsequent Phase 2 and 3 trials, the primary toxicities induced following bevacizumab therapy were hypertension, proteinuria and gastrointestinal perforations, which occurred more frequently in patients with colorectal cancer or metastases within the gastrointestinal tract. Bleeding events are also a concern with bevacizumab therapy and can be fatal with some tumor histologies, such as in squamous non-small cell lung cancer.Citation93 These Phase 2 and 3 clinical trials led to the approval of bevacizumab, the first anti-angiogenic strategy approved for cancer therapy, for the treatment of five different cancers to date. In metastatic colorectal cancer (mCRC), bevacizumab in combination with IFL chemotherapy [ironotecan, 5-fluorouracil (5-FU) and leucovorin (LV)] significantly increased median duration of patient survival by 4.7 months as compared to IFL treatment alone, leading to the approval of this regimen in 2004 as a first-line treatment for mCRC.Citation94 Approval of bevacizumab as a second-line treatment for mCRC in 2006 was the result of the eastern Cooperative Oncology Group E3200 (ECOG E3200) study where bevacizumab plus FOLFOX4 (oxaliplatin, 5-FU and LV) significantly increased patient overall survival (OS) and progression-free survival (PFS) by 2.1 and 2.6 months, respectively as compared to FOLFOX4 treatment alone.Citation95 In 2006, bevacizumab was also approved as a first-line treatment for non-small cell lung cancer (NSCLC) based on the findings of the ECOG E4599 study where bevacizumab in combination with paclitaxel and carboplatin versus chemotherapy alone significantly increased OS by 2 months and PFS by 1.7 months.Citation96 The results of the ECOG E2100 study lead to the approval of bevacizumab in 2008 as a first-line therapy for HER2-negative metastatic breast cancer (mBC) where bevacizumab plus paclitaxel failed to increase OS versus paclitaxel alone treatment, but did significantly increase PFS by 5.9 months.Citation97 Bevacizumab is not currently recommended for second- or third-line treatment of mBC that has progressed following anthracycline and taxane chemotherapy. This decision was made based on the findings of the AVF2119 study where mBC patients that had previously been treated with anthracycline and taxane had no increased in PFS or OS with bevacizumab in combination with capecitabine versus capecitabine alone.Citation98 FDA approval of single agent bevacizumab therapy for second-line treatment of glioblastoma in May 2009 was the result of AVF3708g and NCI 06-C-0064E trial that demonstrated durable objective response rates.Citation99,Citation100 In July 2009, bevacizumab was approved for the treatment of metastatic renal cell carcinoma (mRCC) based on the Hoffmann-La-Roche BO17705 trial where bevacizumab plus interferon (IFN) alfa-2a resulted in a statistically significant 4.8 month increase in PFS versus IFN alfa-2a treatment alone,Citation101 and the Cancer and Leukemia Group B (CALGB) 90206 trial that demonstrated a statistically significant 3.3 month increase in PFS with combination bevacizumab plus IFN alfa therapy versus IFN alfa alone.Citation102 Bevacizumab has paved the way for subsequent antiangiogenic therapies in cancer. A more detailed account of bevacizumab’s progress through clinical trials in a number of tumor types is provided in a thorough review by Grothey and Galanis.Citation93 However, the survival benefits of bevacizumab therapy, although reaching statistical significance, are modest and as mentioned above, are measured in months. These somewhat disappointing results of bevacizumab’s clinical efficacy in combination with bevacizumab-mediated toxicity highlight the need to re-evaluate how to best utilize anti-angiogenic therapies in the clinic.
2C3 and r84 (AT001, affitech AS).
2C3 is a murine monoclonal IgG2a,κ antibody that binds human VEGF and inhibits VEGF from interacting with VEGFR2, but not VEGFR1. 2C3-mediated blockade of VEGFR2 signaling blocks VEGF-mediated EC growth, VEGFR2 phosphorylation, vascular permeability and inhibits the growth of established human tumor xenografts in immunocompromised mice.Citation103,Citation104 In addition, 2C3 localizes to pools of VEGF in the tumor stroma and within the perivascular connective tissue of solid human tumors.Citation103 Since its initial identification, 2C3 has been characterized in numerous tumor xenograft models as an effective inhibitor of tumor growth and angiogenesis and as a modulator of macrophage and immune cell infiltration.Citation60,Citation105–Citation108
The success of 2C3 in preclinical models lead to the development of a phenotypically similar, fully human mAb, r84 (AT001, Affitech AS) that was generated by screening a human anti-VEGF single chain variable fragment library for specific 2C3-like properties. 2C3 and r84 cross-block each other in VEGF ELISA assays indicating that the epitope each mAb binds is very similar; however, the exact epitope on VEGF bound by each mAb has not been determined. Efforts are currently underway to solve the structure of VEGF bound by the Fab of r84, which is anticipated to uncover the epitope for r84. r84 binds mouse and human VEGF, but not other VEGF family members, and specifically inhibits VEGF from binding to VEGFR2, but allows VEGF to bind and activate VEGFR1. Through specific VEGF blockade, r84 inhibits VEGFR2-mediated endothelial cell migration and phosphorylation cascades in vitro and controls tumor growth, angiogenesis and immune cell infiltration in vivo.Citation108–Citation110 Research with 2C3 and r84 demonstrate that inhibition of VEGF-mediated VEGFR2 activation is sufficient to control tumor growth and calls into question the pro-tumorigenic function of VEGFR1. In addition, because r84 binds mouse and human VEGF, it facilitates evaluation of the effects of blocking tumor-derived (human) and stromal (mouse) VEGF in preclinical xenografts models. Therefore r84 should more closely mirror the activity of selective VEGF inhibition in human patients. A mouse chimeric version of r84 (mcr84) has been generated and is a useful tool for studying angiogenesis in immunocompetent animals and in syngenic tumor models.Citation109 To date, treatment of tumor-bearing mice with r84 has not induced anti-VEGF-related toxicities that have been characterized in other preclinical models using different inhibitors of the VEGF pathway.Citation111–Citation113 Therefore, the potential efficacy of r84 as a cancer therapeutic for treating cancer patients is highly anticipated.
VEGF-trap (aflibercept, Regeneron Pharmaceuticals, Inc.).
The precursor of VEGF-Trap (aflibercept, Regeneron Pharmaceuticals, Inc.), the therapeutic drug currently in Phase 2 and 3 clinical trials, was first developed by trying to harness the strong affinity of VEGFR1 for VEGF to inhibit VEGF-mediated angiogenesis within tumors. A soluble decoy receptor was engineered by fusing the first three immunoglobulin domains of VEGFR1 to the Fc constant region of human IgG1 antibody, creating a forced homodimer, mFlt(1–3)-IgG that bound VEGF and PlGF with high affinity;Citation114,Citation115 however, this fusion protein had poor pharmacokinetic properties in vivo that required large, frequent doses for efficacy.Citation116 To improve the half-life without losing affinity, mFlt(1–3)-IgG was modified to contain the second immunoglobulin domain of VEGFR1 and the third immunoglobulin domain of VEGFR2 fused to human IgG1 Fc region, creating the fusion protein known as VEGF-Trap. This fusion protein had improved pharmacokinetics and affinity for VEGF (approximately 1 pM) as compared to the parental mFlt(1–3)-IgG, and effectively inhibited tumor growth and angiogenesis in xenograft models.Citation117 Pre-clinical trials demonstrated the efficacy of VEGF-Trap in suppressing tumor growth and angiogenesis through its ability to bind both mouse and human VEGF,Citation118–Citation120 and supported its entry into clinical trials were it is being currently evaluated in a number of cancers and in age-related macular degeneration.
Antibodies Targeting the VEGF Receptors
MF1/IMC-18F1 (ImClone Systems).
The rat anti-mouse VEGFR1 IgG1 mAb, MF1 (ImClone Systems) was first shown to suppress tumor and ischemic retinal angiogenesis, as well as inflammation in an autoimmune arthritis model.Citation49 Kaplan et al. used MF1 to demonstrate VEGFR1 participates in the premetastatic niche in animal models, and that blockade of VEGFR1 with MF1 more effectively blocked tumor metastases than did inhibition of VEGFR2.Citation121 These studies led to the development of a fully human mAb directed against human VEGFR1 (mAb 6.12, IMC-18F1, ImClone Systems) that blocked VEGFR1 signaling in VEGFR1-expressing breast cancer cells lines in vitro, and inhibited tumor growth in vivo.Citation122 The final characterization of IMC-18F1 came in 2006. IMC-18F1 bound to VEGFR1 with high-affinity and was able to block VEGFR1 from interacting with VEGF, VEGF-B and PlGF, preventing downstream VEGFR1 signaling and breast cancer cell line growth in vitro and in vivo. Targeting human and mouse VEGFR1 with IMC-18F1 and MF1, respectively, more effectively controlled tumor growth than either agent alone.Citation123 The safety and dosing of IMC-18F1 is currently being evaluated in a Phase 1 clinical trial.
DC101/IMC-1C11 (ImClone Systems).
DC101 is a rat antimouse VEGFR2 (Flk-1) mAb that inhibits ligand-induced activation of VEGFR2.Citation124 In vivo syngenic and xenograft tumor models in mice demonstrated the ability of DC101 to control tumor growth and reduce tumor angiogenesis by targeting ECor tumor-expressed VEGFR2.Citation125–Citation129 Since DC101 recognizes only mouse VEGFR2 and therefore is not a candidate for clinical trials, a single chain variable fragment with human VEGFR2 reactivity that was also able to block in vitro VEGFR2 signaling was isolated from a single chain antibody phage display library.Citation130 This fragment became IMC-1C11 (ImClone Systems), a chimeric mAb that blocked tumor growth and angiogenesis in tumor xenografts and was tested in a Phase 1 clinical trial in colorectal carcinoma patients with liver metastases.Citation131,Citation132 Although treatment with IMC-1C11 did not induce grade 3 or 4 toxicities, 50% of treated patients developed anti-chimeric antibodies that impeded the future progression of this antibody in the clinic.Citation132
IMC-1121B (ramucirumab, ImClone Systems).
To develop an anti-human VEGFR2 mAb that would not be immunogenic in clinical trials, ImClone Systems used a human antibody phage display library to isolate VEGFR2-specific human Fab fragments. The resulting best Fab bound to human VEGFR2 with high affinity, and inhibited ligand-induced VEGFR2 activation in endothelial cells.Citation133,Citation134 Affinity maturation of the best Fab clone and subsequent synthesis of a full length antibody yielded IMC-1121B, a fully human IgG1 mAb with higher affinity for VEGFR2 that was a more potent inhibitor of VEGF-induced VEGFR2 signaling and EC migration in vitro. IMC-1121B also increased the survival of murine xenograft models in vivo more effectively than other anti-VEGFR2 antibodies, including IMC-1C11.Citation135,Citation136 There are 13 current Phase 1 or 2 clinical trials with IMC-1121B to assess the safety and efficacy of this mAb.
Future Directions
The development of anti-angiogenic therapies was highly anticipated. This therapeutic strategy was hypothesized to avoid the tumor resistance pathways of traditional anti-cancer drugs by targeting the vasculature as opposed to the genetically instable and highly mutagenic tumor cell population. The pre-clinical success of targeting the VEGF pathway using mAb-based therapy further bolstered this hypothesis. However, clinical studies of anti- VEGF strategies in cancer patients have not delivered the level of efficacy anticipated. To date, bevacizumab is the most developed anti-VEGF pathway mAb. Bevacizumab is currently indicated as a first- or second-line treatment in five different tumor types, and is being evaluated in many clinical trials. Therefore, experience with bevacizumab in the clinic provides a working model for the benefits and pitfalls of anti-angiogenic mAb therapies, as well as a benchmark for other anti-angiogenic mAb discussed in this article.
As discussed previously, the results of bevacizumab in Phase 2 and 3 clinical trials have been modest when compared to the success of anti-VEGF therapy in pre-clinical models.Citation90 In the clinic, responses with bevacizumab as a single agent therapy (in glioblastoma) or in combination with standard chemotherapy (in nonsmall cell lung cancer, metastatic colorectal cancer, breast cancer and renal cell carcinoma) is measured in a few months correlating with small, albeit statistically significant increases in PFS, and rarely in OS.Citation93 This differs from some anti-angiogenic small molecule tyrosine kinase inhibitors (TKIs) that do not display improved efficacy when combined with chemotherapy.Citation137 The differences between anti-angiogenic TKIs and mAbs may be due to functional changes occurring within the tumor in response to the different drugs. Treating tumors with bevacizumab, or other anti-VEGF pathway mAbs counteracts the inherent disorganization and abnormalities of the tumor vasculature. This process has been termed “normalization.”Citation138 The pruned, normalized tumor vasculature achieved with anti-angiogenic therapy has increased pericyte coverage and stability and a reduction in vessel leakiness and interstitial fluid pressure, which in combination improves the subsequent delivery of chemotherapy and other drugs. This process allows for cytostatic anti-angiogenic therapy (e.g., bevacizumab and other anti-VEGF pathway mAbs) combined with cytotoxic chemotherapy to have improved clinical response as compared to either agent alone. This absence of synergy with some VEGFR TKIs and chemotherapy is perhaps due to the targeting of other tyrosine kinase receptors such as PDGFR that can disrupt the normalization process.Citation137 A better understanding of the normalization process among tumor types would allow for optimization of anti-angiogenic and chemotherapeutic drug delivery schedules, perhaps improving the overall efficacy of these drugs in the clinic.
Bevacizumab therapy is associated with several adverse effects. The most common toxicities are hypertension, proteinuria and bleeding events that result from a loss of homeostatic VEGF signaling and vascular maintenance.Citation139 Certain histologies, such as squamous non-small cell lung cancer, were more prone to fatal bleeding events, leading to the exclusion of these patients from future studies.Citation93 In addition, perforations were more frequent in patients with metastatic colorectal cancer, ovarian cancer or metastases within the gastrointestinal tract. Inhibition of VEGFR signaling with sorafenib and sunitinib therapy is also associated with hypertension, proteinuria and bleeding events similar to treatment with bevacizumab. However, there are a host of off-target adverse effects with sorafenib and sunitinib therapy, including skin reactions, hand-foot syndrome, fatigue and diarrhea resulting from TKI inhibition of targets other than the VEGFRs.Citation139 Based on these patterns, specific blockade of VEGF, PlGF, VEGFR1 or VEGFR2 with r84, VEGF-Trap, IMC-18F1 or IMC-1121B might induce toxicities more similar to bevacizumab rather than sorafenib and sunitinib. It is also possible that VEGFR1 signaling is important for maintaining the homeostatic function of VEGF, and thus therapies allowing for continued VEGFR1 signaling such as r84 and IMC-1121B may provide a less severe toxicity profile than bevacizumab. In support of this, pre-clinical studies in our laboratory with extended (12 week) treatment of tumor-bearing and non-tumor bearing mice with r84 failed to induce toxicity (unpublished data). Results from IMC- 1121B on-going clinical trials and future studies with r84 in the clinic will ultimately answer these questions about differences in toxicities between the VEGF pathway antibodies. Alternatively, the severity or frequency of toxicities between mAbs targeting the VEGF pathway may depend more on the relative affinity of the drug for its target. In pre-clinical studies with mice engineered to express human VEGF, anti-VEGF antibodies of increasing affinity had a greater toxicity induction.Citation111 Additionally, chemotherapy regimens such as carboplatin and paclitaxel, in combination with bevacizumab or VEGFR TKIs can exacerbate the toxicities of these targeted therapies.Citation140 Therefore, carefully assessing both drug affinity for its target and chemotherapy doses and regimens is required to control toxicity in patients receiving drugs targeting the VEGF pathway. The distribution of toxicities within mAb strategy and patient groups should be taken into consideration as future anti-VEGF pathway therapies are introduced into the clinic to minimize adverse effects and to monitor for new patterns of toxicity.
With the expanding use of anti-angiogenic therapy in the clinic, it has become more apparent that not all tumors respond or will remain responsive to this treatment option. The inherent non-responsiveness of certain tumors and the development of acquired resistance to therapy following an initial response, has been termed intrinsic and evasive resistance, respectively.Citation141 Pre-clinical research from a number of investigators has identified several mechanisms mediating resistance to anti-angiogenic therapy. This includes a switch from a primary reliance on VEGF to an alternative growth factor such as fibroblast growth factor, interleukin-8 and ephrins, increased stabilization of existing vessels by improved pericyte coverage, stimulating infiltration of pro-angiogenic immune cells and the co-option of normal vasculature through enhanced tumor invasiveness and metastasis.Citation141 Patients with intrinsically resistant tumors would have no clinical benefit from anti-angiogenic therapy and therefore should be excluded from these treatments; however, we currently lack effective biomarkers that would allow for the stratification of intrinsically resistant or sensitive tumors, or for the monitoring of tumor response and the development of resistance to therapy.Citation142
The identification of biomarkers of response to anti-angiogenic drugs is being actively pursued. In mBC, baseline levels of circulating VEGF have shown promise as a biomarker of response to bevacizumab therapy, but this has not translated to other tumor types such as mCRC or NSCLC.Citation143 In addition, the utility of baseline VEGF levels as a biomarker of response varies depending on tumor type and therapy regimen (mAb or VEGFR TKI), bringing into question the universal applicability of this marker. Genetic polymorphisms of VEGF and interleukin-8 have been indicated as predictive biomarkers of response to bevacizumab in mBC and ovarian cancer, respectively; however these markers have yet to be validated in other tumor types or in relation to other anti-VEGF pathway therapies.Citation143 In addition, there are increased levels of circulating PlGF following anti-VEGF therapy that have been positively correlated with improved outcome in bevacizumab-treated rectal cancer patients, although it remains to be seen if this increase in PlGF is predictive or prognostic of response to therapy.Citation143 A systemic increase in blood pressure has potential as a pharmacodynamic biomarker of the effectiveness of VEGF blockade by bevacizumab and VEGFR TKIs. Recent studies have implicated the degree of hypertension in patients as a diagnostic biomarker of response to VEGF targeted therapy, although this has yet to be validated in large studies.Citation143
Although there are several candidates awaiting validation as pharmacodynamic or prognostic biomarkers of anti-angiogenic therapy, including circulating levels of VEGF and PlGF, genetic polymorphisms of angiogenic factors and the degree of hypertension following therapy, we still lack biomarkers predictive of response to therapy that exist for other targeted drugs such as the overexpression of human epidermal growth factor receptor 2 (HER2/neu) in BC as a predictive marker of response to trastuzumab therapy. Similar to HER2/neu overexpression, predictive markers of response to anti-angiogenic therapy may be tumor-dependent. Our laboratory and others are currently assessing tumor-specific factors in xenograft models while others are evaluating patient samples for potential biomarkers of response. Further studies are required to identify and validate predictive versus prognostic biomarkers and may require carefully designed clinical trials and more frequent collection of patient tumor and serum samples throughout treatment. A better understanding of what dictates patient response and how to monitor resistance could lead to improved efficacy of mAbs targeting the VEGF pathway.
The opportunity to better understand the function of individual components of the VEGF pathway in the tumor microenvironment is afforded by evaluation of the efficacy and biology of the anti-VEGF strategies outlined in this review. As there are very few studies that directly compare anti-VEGF pathway mAbs, it is difficult to say with certainty that one strategy is best or will work for every patient. All of the mAbs discussed in this review target the VEGF pathway; however the specificity of the different mAbs affects the function of these therapies within the tumor microenvironment and provides clues to the potential advantages and disadvantages among the different therapies. It is of particular interest to compare the strategies that are specific to VEGF (bevacizumab and r84) to VEGF-Trap and those that inhibit VEGFR1 (MF1, IMC-18F1) or VEGFR2 (DC101, IMC-1C11, IMC-1121B) directly.
Bevacizumab and r84 are both highly specific for VEGF-A and do not directly interfere with other VEGF family members. r84 is even more selective than bevacizumab due to the fact that it only inhibits VEGFR2 activation, leaving intact VEGFR1 signaling. In endothelial cells, VEGFR1 primarily functions as a negative inhibitor of VEGFR2 signaling by acting as a decoy receptor for VEGF and preventing VEGF from binding to and inducing angiogenesis through VEGFR2. This idea is supported by previously mentioned genetic experiments where loss of VEGFR1 leads to embryonic death due to too many endothelial cells, but mice expressing only the extracellular domain of VEGFR1 are viable.Citation44,Citation45 Additionally, VEGF binding to VEGFR1 can stimulate SHP-1 phosphatase to actively reduce levels of VEGFR2 phosphorylation.Citation51 Further, whereas VEGF activation of VEGFR1 does not alter gene expression, PlGF binding to VEGFR1 in vitro changes the gene expression of more than 50 genes.Citation144 PlGF may also provide an escape mechanism for anti-VEGF targeted therapy and blocking PlGF directly has been demonstrated to have anti-tumor effects.Citation145 Treatment with r84 would allow for regulatory signaling through VEGFR1 and could reduce PlGF activation of VEGFR1 as a result of competition with PlGF for VEGFR1 binding. Therefore, VEGF binding to VEGFR1 may be an important negative regulator of tumor angiogenesis that could be harnessed with r84, but not with bevacizumab. VEGFTrap blocks VEGF from binding to VEGFR1 and VEGFR2 and blocks PlGF from binding to VEGFR1, thereby preventing negative regulation of VEGFR2 activity by VEGFR1 similar to bevacizumab and uniquely controlling VEGFR1 activation by PlGF. Thus the in vivo mechanisms of action of VEGF-Trap may fall somewhere between that of r84 and bevacizumab.
Alternatively, blocking VEGFR1 activity with mAbs has been very effective in VEGFR1 expressing tumors.Citation122 VEGFR1 activity has also been linked to tumor metastasis and blocking VEGFR1 with MF1/IMC-18F1 reduces tumor growth, demonstrating the potential importance of this receptor in tumor progression and the need to inhibit its function in patients. Despite these data, the overall functions of VEGFR1 remain unclear and the full effects of VEGFR1 blockade are uncertain. However, there is still a strong possibility that VEGFR1 functions as a negative regulator of VEGFR2 and direct targeting of VEGFR1 with MF1/IMC-18F1 may be in effect, inhibiting an inhibitor of angiogenesis, which may be therapeutically counterproductive. Therefore, the results of on-going IMC-18F1 and IMC-1121B clinical trials and entry of r84 into the clinical arena are highly anticipated to elucidate the importance of VEGFR1 signaling in tumor angiogenesis and progression.
As previously mentioned, VEGFR2 is the predominant mediator of VEGF-induced angiogenesis and consequently, blocking functional signaling of this receptor with neutralizing antibodies such as DC101, IMC-1C11 and IMC-1121B is effective at reducing angiogenesis and tumor growth. There is increased expression of VEGFR2 in the tumor microenvironment, which in turn increases the potential of anti-VEGFR2 therapies to specifically target the tumor and not normal tissues. Directly targeting VEGFR2 also prevents receptor activation by other VEGF family members (e.g., VEGF-C, -D), which are not blocked by bevacizumab, r84, or VEGF-Trap. Additionally, the anti-VEGFR2 mAbs do not inhibit VEGFR1 negative regulation of VEGFR2, potentially enhancing blockade of VEGFR2 function. However, the anti-VEGFR2 mAbs will also block soluble VEGFR2, a natural inhibitor of lymphangiogenesis.Citation59 This effect is a potential disadvantage of directly targeting VEGFR2, given the importance of the lymphatic vasculature in metastasis.Citation146 Infiltration of immune cells such as macrophages within tumors can promote tumor survival and progression.Citation147 Macrophages in tumor bearing animals express VEGFR2, and blockade of this receptor has been demonstrated to reduce macrophage migration and infiltration in tumors.Citation60,Citation109 Therefore, targeting VEGFR2 directly can negatively affect tumor growth and metastasis by reducing the population of tumor-promoting immune cells within the tumor microenvironment.
The anti-VEGFR antibodies have a broader specificity profile given that these agents interfere with signaling pathways stimulated by multiple members of the VEGF family. Thus far, it is unclear if a broader specificity anti-VEGF agent is more effective than bevacizumab or r84. In fact, a recent direct comparison of mouse chimeric r84 to sunitinib and a peptoid that binds and inhibits VEGFR1 and VEGFR2 demonstrated that r84 was as or more effective in controlling tumor growth in two models of breast cancer in immunocompetent mice.Citation109 This study also evaluated the immunological phenotype of tumors under therapy and found that in general, treatment of r84 resulted in fewer immunosuppressive cells (e.g., myeloid-derived suppressor cells, Treg, immature dendritic cells) in the tumor microenvironment. These cell based changes are likely the result of an altered cytokine profile after therapy.Citation109 To our knowledge similar studies have not been performed with the other anti-VEGF agents discussed here. Species specificity issues and other inherent challenges with pharmaceutical-based novel therapies preclude a head-to-head test of these leading anti-VEGF strategies in pre-clinical models. Thus we are forced to make assumptions regarding the potential superiority of one agent over another based on the efficacy observed in similar models and the biology of the therapy employed.
In reality, arguing the benefits and shortcomings of the individual mAb-based strategies available for targeting the VEGF pathway in cancer may be short-sighted. Selectively targeting angiogenesis in patients will most likely require an arsenal of therapeutics and the best strategy may very well depend upon the tumor type, stage, histology or may be entirely patient specific. This again highlights the need for biomarkers that can predict a patient’s response to anti-angiogenic therapy, as well as the need for an array of selective therapies to improve patient survival by inhibiting tumor angiogenesis.
Declaration of Interest
R.A.B. is a consultant for Peregrine Pharmaceuticals, which is developing the r84 antibody.
Abbreviations
| VEGF | = | vascular endothelial growth factor |
| VPF | = | vascular permeability factor |
| EC | = | endothelial cell |
| Nrp | = | neuropilin |
| VEGFR | = | vascular endothelial growth factor receptor |
| HSP | = | heparin-sulfate proteoglycan |
| ECM | = | extracellular matrix |
| uPA | = | urokinase type of plasminogen activator |
| Flt | = | fms-like tyrosyl kinase |
| Flk | = | fetal liver kinase |
| 5-FU | = | 5-fluorouracil |
| LV | = | leucovorin |
| ECOG | = | eastern cooperative oncology group |
| OS | = | overall survival |
| PFS | = | progression-free survival |
| CALGB | = | cancer and leukemia group B |
| Fab | = | fragment antigen binding |
Figures and Tables
Figure 1 Blockade of the VEGF pathway with mAbs. The specificity of the VEGF family ligands for the VEGF receptors and coreceptors are shown. The clinically-relevant mAbs targeting the anti-VEGF pathway discussed in this review are placed based on their blockade of VEGF ligand or receptor. The ligand-binding antibodies bevacizumab (bev), r84, and VEGF-Trap inhibit ligand binding to the indicated receptor. IMC-18F1 and IMC-1121B bind VEGFR1 and VEGFR2 respectively, and prevent ligand binding.
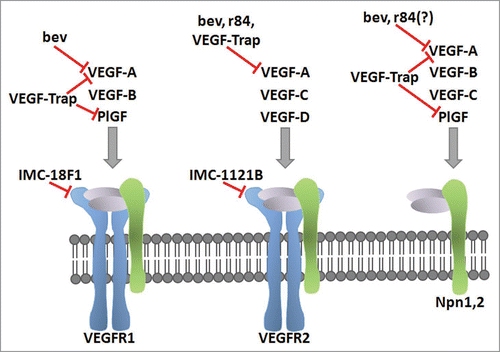
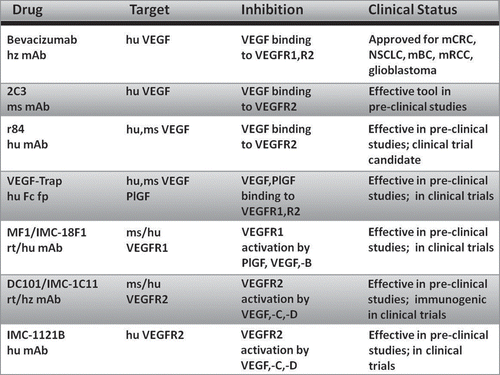
Figure 2 Current anti-angiogenic mAbs with applications in cancer therapy. hz, humanized; hu, human; ms, mouse; rt, rat; fp, fusion protein; mCRC, metastatic colorectal cancer; NSCLC, non-small cell lung cancer; mBC, metastatic breast cancer; mRCC, metastatic renal cell carcinoma.
Acknowledgements
This work was supported in part by the Randall Bridwell Lung Cancer Research Grant from Uniting Against Lung Cancer, and the Effie Marie Cain Scholarship in Angiogenesis Research (to R. Brekken).
References
- Roskoski R Jr. Vascular endothelial growth factor (VEGF) signaling in tumor progression. Crit Rev in Oncol Hematol 2007; 62:179 - 213
- Ferrara N. Vascular endothelial growth factor: basic science and clinical progress. Endocr Rev 2004; 25:581 - 611
- Hoeben A, Landuyt B, Highley MS, Wildiers H, Van Oosterom AT, De Bruijn EA. Vascular endothelial growth factor and angiogenesis. Pharmacol Rev 2004; 56:549 - 580
- Hanahan D, Folkman J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell 1996; 86:353 - 364
- Gerwins P, Skoldenberg E, Claesson-Welsh L. Function of fibroblast growth factors and vascular endothelial growth factors and their receptors in angiogenesis. Crit Rev Oncol Hematol 2000; 34:185 - 194
- Jain RK. Molecular regulation of vessel maturation. Nat Med 2003; 9:685 - 693
- Dvorak HF. Angiogenesis: update 2005. J Thromb Haemost 2005; 3:1835 - 1842
- Less JR, Skalak TC, Sevick EM, Jain RK. Microvascular architecture in a mammary carcinoma: branching patterns and vessel dimensions. Cancer Res 1991; 51:265 - 273
- Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med 1971; 285:1182 - 1186
- Houck KA, Ferrara N, Winer J, Cachianes G, Li B, Leung DW. The vascular endothelial growth factor family: identification of a fourth molecular species and characterization of alternative splicing of RNA. Mol Endocrinol 1991; 5:1806 - 1814
- Tischer E, Mitchell R, Hartman T, Silva M, Gospodarowicz D, Fiddes JC, et al. The human gene for vascular endothelial growth factor. Multiple protein forms are encoded through alternative exon splicing. J Biol Chem 1991; 266:11947 - 11954
- Senger DR, Connolly DT, Van de Water L, Feder J, Dvorak HF. Purification and NH2-terminal amino acid sequence of guinea pig tumor-secreted vascular permeability factor. Cancer Res 1990; 50:1774 - 1778
- Alon T, Hemo I, Itin A, Pe’er J, Stone J, Keshet E. Vascular endothelial growth factor acts as a survival factor for newly formed retinal vessels and has implications for retinopathy of prematurity. Nat Med 1995; 1:1024 - 1028
- Ferrara N, Henzel WJ. Pituitary follicular cells secrete a novel heparin-binding growth factor specific for vascular endothelial cells. Biochem Biophys Res Commun 1989; 161:851 - 858
- Gerber HP, McMurtrey A, Kowalski J, Yan M, Keyt BA, Dixit V, et al. Vascular endothelial growth factor regulates endothelial cell survival through the phosphatidylinositol 3′-kinase/Akt signal transduction pathway. Requirement for Flk-1/KDR activation. J Biol Chem 1998; 273:30336 - 30343
- Barleon B, Sozzani S, Zhou D, Weich HA, Mantovani A, Marme D. Migration of human monocytes in response to vascular endothelial growth factor (VEGF) is mediated via the VEGF receptor flt-1. Blood 1996; 87:3336 - 3343
- Waltenberger J, Claesson-Welsh L, Siegbahn A, Shibuya M, Heldin CH. Different signal transduction properties of KDR and Flt1, two receptors for vascular endothelial growth factor. J Biol Chem 1994; 269:26988 - 26995
- Leung DW, Cachianes G, Kuang WJ, Goeddel DV, Ferrara N. Vascular endothelial growth factor is a secreted angiogenic mitogen. Science 1989; 246:1306 - 1309
- Houck KA, Leung DW, Rowland AM, Winer J, Ferrara N. Dual regulation of vascular endothelial growth factor bioavailability by genetic and proteolytic mechanisms. J Biol Chem 1992; 267:26031 - 26037
- Park JE, Keller GA, Ferrara N. The vascular endothelial growth factor (VEGF) isoforms: differential deposition into the subepithelial extracellular matrix and bioactivity of extracellular matrix-bound VEGF. Mol Biol Cell 1993; 4:1317 - 1326
- Robinson CJ, Stringer SE. The splice variants of vascular endothelial growth factor (VEGF) and their receptors. J Cell Sci 2001; 114:853 - 865
- Keyt BA, Berleau LT, Nguyen HV, Chen H, Heinsohn H, Vandlen R, Ferrara N. The carboxyl-terminal domain (111–165) of Vascular Endothelial Growth Factor is critical for its mitogenic potency. J Biol Chem 1996; 271:7788 - 7795
- Flaumenhaft R, Rifkin DB. The extracellular regulation of growth factor action. Mol Biol Cell 1992; 3:1057 - 1065
- Bergers G, Brekken R, McMahon G, Vu TH, Itoh T, Tamaki K, et al. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat Cell Biol 2000; 2:737 - 744
- Mignatti P, Rifkin DB. Biology and biochemistry of proteinases in tumor invasion. Physiol Rev 1993; 73:161 - 195
- Lee S, Jilani SM, Nikolova GV, Carpizo D, Iruela-Arispe ML. Processing of VEGF-A by matrix metalloproteinases regulated bioavailability and vascular patterning in tumors. J Cell Biol 2005; 169:681 - 691
- Pepper MS. Extracellular proteolysis and angiogenesis. Thromb Haemost 2001; 86:346 - 355
- Olofsson B, Pajusola K, von Euler G, Chilov D, Alitalo K, Eriksson U. Genomic organization of the mouse and human genes for vascular endothelial growth factor B (VEGF-B) and characterization of a second splice isoform. J Biol Chem 1996; 271:19310 - 19317
- Aase K, von Euler G, Li X, Ponten A, Thoren P, Cao R, et al. Vascular endothelial growth factor-B-deficient mice display an atrial conduction defect. Circulation 2001; 104:358 - 364
- Lymboussaki A, Olofsson B, Eriksson U, Alitalo K. Vascular endothelial growth factor (VEGF) and VEGF-C show overlapping binding sites in embryonic endothelia and distinct sites in differentiated adult endothelia. Circ Res 1999; 85:992 - 999
- Karkkainen MJ, Haiko P, Sainio K, Partanen J, Taipale J, Petrova TV, et al. Vascular endothelial growth factor C is required for sprouting of the first lymphatic vessels from embryonic veins. Nat Immunol 2004; 5:74 - 80
- Rocchigiani M, Lestingi M, Luddi A, Orlandini M, Franco B, Rossi E, et al. Human FIGF: cloning, gene structure, and mapping to chromosome Xp22.1 between the PIGA and the GRPR genes. Genomics 1998; 47:207 - 216
- Stacker SA, Stenvers K, Caesar C, Vitali A, Domagala T, Nice E, et al. Biosynthesis of vascular endothelial growth factor-D involves proteolytic processing which generates non-covalent homodimers. J Biol Chem 1999; 274:32127 - 32136
- Baldwin ME, Halford MM, Roufail S, Williams RA, Hibbs ML, Grail D, et al. Vascular endothelial growth factor D is dispensable for development of the lymphatic system. Mol Cell Biol 2005; 25:2441 - 2449
- Cao Y, Ji WR, Qi P, Rosin A. Placenta growth factor: identification and characterization of a novel isoform generated by RNA alternative splicing. Biochem Biophys Res Commun 1997; 235:493 - 498
- Maglione D, Guerriero V, Viglietto G, Delli-Bovi P, Persico MG. Isolation of a human placenta cDNA coding for a protein related to the vascular permeability factor. Proc Natl Acad Sci USA 1991; 88:9267 - 9271
- Yang W, Ahn H, Hinrichs M, Torry RJ, Torry DS. Evidence of a novel isoform of placenta growth factor (PlGF-4) expressed in human trophoblast and endothelial cells. J Reprod Immunol 2003; 60:53 - 60
- Carmeliet P, Moons L, Luttun A, Vincenti V, Compernolle V, De Mol M, et al. Synergism between vascular endothelial growth factor and placental growth factor contributes to angiogenesis and plasma extravasation in pathological conditions. Nat Med 2001; 7:575 - 583
- Shibuya M, Yamaguchi S, Yamane A, Ikeda T, Tojo A, Matsushime H, et al. Nucleotide sequence and expression of a novel human receptor-type tyrosine kinase gene (flt) closely related to the fms family. Oncogene 1990; 5:519 - 524
- de Vries C, Escobedo JA, Ueno H, Houck K, Ferrara N, Williams LT. The fms-like tyrosine kinase, a receptor for vascular endothelial growth factor. Science 1992; 255:989 - 991
- Olofsson B, Korpelainen E, Pepper MS, Mandriota SJ, Aase K, Kumar V, et al. Vascular endothelial growth factor B (VEGF-B) binds to VEGF receptor-1 and regulates plasminogen activator activity in endothelial cells. Proc Natl Acad Sci USA 1998; 95:11709 - 11714
- Park JE, Chen HH, Winer J, Houck KA, Ferrara N. Placenta growth factor. Potentiation of vascular endothelial growth factor bioactivity, in vitro and in vivo, and high affinity binding to Flt-1 but not to Flk-1/KDR. J Biol Chem 1994; 269:25646 - 25654
- Kendall RL, Thomas KA. Inhibition of vascular endothelial cell growth factor activity by an endogenously encoded soluble receptor. Proc Natl Acad Sci USA 1993; 90:10705 - 10709
- Fong GH, Rossant J, Gertsenstein M, Breitman ML. Role of the Flt-1 receptor tyrosine kinase in regulating the assemble of vascular endothelium. Nature 1995; 376:66 - 70
- Hiratsuka S, Minowa O, Kuno J, Noda T, Shibuya M. Flt-1 lacking the tyrosine kinase domain is sufficient for normal development and angiogenesis in mice. Proc Natl Acad Sci USA 1998; 95:9349 - 9354
- Roberts DM, Kearney JB, Johnson JH, Rosenberg MP, Kumar R, Bautch VL. The Vascular Endothelial Growth Factor (VEGF) Receptor Flt-1 (VEGFR-1) Modulates Flk-1 (VEGFR-2) Signaling During Blood Vessel Formation. Am J Pathol 2004; 164:1531 - 1535
- Gerber HP, Malik AK, Solar GP, Sherman D, Liang XH, Meng G, et al. VEGF regulates haematopoietic stem cell survival by an internal autocrine loop mechanism. Nature 2002; 417:954 - 958
- Hattori K, Heissig B, Wu Y, Dias S, Tejada R, Ferris B, et al. Placental growth factor reconstitutes hematopoiesis by recruiting VEGFR1(+) stem cells from bonemarrow microenvironment. Nat Med 2002; 8:841 - 849
- Luttun A, Tjwa M, Moons L, Wu Y, Angelillo-Scherrer A, Liao F, et al. Revascularization of ischemic tissues by PlGF treatment, and inhibition of tumor angiogenesis, arthritis and atherosclerosis by anti-Flt1. Nat Med 2002; 8:831 - 840
- LeCouter J, Moritz DR, Li B, Phillips GL, Liang XH, Gerber HP, et al. Angiogenesis-independent endothelial protection of liver: role of VEGFR-1. Science 2003; 299:890 - 893
- Nozaki M, Sakurai E, Raisler BJ, Baffi JZ, Witta J, Ogura Y, et al. Loss of SPARC-mediated VEGFR-1 suppression after injury reveals a novel antiangiogenic activity of VEGF-A. J Clin Invest 2006; 116:422 - 429
- Terman BI, Dougher-Vermazen M, Carrion ME, Dimitrov D, Armellino DC, Gospodarowicz D, et al. Identification of the KDR tyrosine kinase as a receptor for vascular endothelial cell growth factor. Biochem Biophys Res Commun 1992; 187:1579 - 1586
- Millauer B, Wizigmann-Voos S, Schnurch H, Martinez R, Moller NP, Risau W, et al. High affinity VEGF binding and developmental expression suggest Flk-1 as a major regulator of vasculogenesis and angiogenesis. Cell 1993; 72:835 - 846
- Quinn TP, Peters KG, de Vries C, Ferrara N, Williams LT. Fetal liver kinase 1 is a receptor for vascular endothelial growth factor and is selectively expressed in vascular endothelium. Proc Natl Acad Sci USA 1993; 90:7533 - 7537
- Bernatchez PN, Soker S, Sirois MG. Vascular endothelial growth factor effect on endothelial cell proliferation, migration and platelet-activating factor synthesis is Flk-1-dependent. J Biol Chem 1999; 274:31047 - 31054
- Takahashi T, Yamaguchi S, Chida K, Shibuya M. A single autophosphorylation site on KDR/Flk-1 is essential for VEGF-A-dependent activation of PLC-gamma and DNA synthesis in vascular endothelial cells. EMBO J 2001; 20:2768 - 2778
- Shibuya M, Claesson-Welsh L. Signal transduction by VEGF receptors in regulation of angiogenesis and lymphangiogenesis. Exp Cell Res 2006; 312:549 - 560
- Ebos JM, Bocci G, Man S, Thorpe PE, Hicklin DJ, Zhou D, et al. A naturally occurring soluble form of vascular endothelial growth factor receptor 2 detected in mouse and human plasma. Mol Cancer Res 2004; 2:315 - 326
- Albuquerque RJ, Hayashi T, Cho WG, Kleinman ME, Dridi S, Takeda A, et al. Alternatively spliced vascular endothelial growth factor receptor-2 is an essential endogenous inhibitor of lymphatic vessel growth. Nat Med 2009; 15:1023 - 1030
- Dineen SP, Lynn KD, Holloway SE, Miller AF, Sullivan JP, Shames DS, et al. Vascular endothelial growth factor receptor 2 mediates macrophage infiltration into orthotopic pancreatic tumors in mice. Cancer Res 2008; 68:4340 - 4346
- Shalaby F, Rossant J, Yamaguchi TP, Gertsenstein M, Wu XF, Breitman ML, et al. Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature 1995; 376:62 - 66
- Pajusola K, Aprelikova O, Korhonen J, Kaipainen A, Pertovaara L, Alitalo R, et al. FLT4 receptor tyrosine kinase contains seven immunoglobulin-like loops and is expressed in multiple human tissues and cell lines. Cancer Res 1992; 52:5738 - 5743
- Galland F, Karamysheva A, Pebusque MJ, Borg JP, Rottapel R, Dubreuil P, et al. The FLT4 gene encodes a transmembrane tyrosine kinase related to the vascular endothelial growth factor receptor. Oncogene 1993; 8:1233 - 1240
- Kaipainen A, Korhonen J, Mustonen T, van Hinsbergh VW, Fang GH, Dumont D, et al. Expression of the fms-like tyrosine kinase 4 gene becomes restricted to lymphatic endothelium during development. Proc Natl Acad Sci USA 1995; 92:3566 - 3570
- Dumont DJ, Jussila L, Taipale J, Lymboussaki A, Mustonen T, Pajusola K, et al. Cardiovascular failure in mouse embryos deficient in VEGF receptor-3. Science 1998; 282:946 - 949
- Olsson A-K, Dimberg A, Kreuger J, Claesson-Welsh L. VEGF receptor signalling-in control of vascular function. Nat Rev Mol Cell Biol 2006; 7:359 - 371
- Takagi S, Kasuya Y, Shimizu M, Matsuura T, Tsuboi M, Kawakami A, et al. Expression of a cell adhesion molecule, neuropilin, in the developing chick nervous system. Dev Biol 1995; 170:207 - 222
- Chen H, Chedotal A, He Z, Goodman CS, Tessier-Lavigne M. Neuropilin-2, a novel member of the neuropilin family, is a high affinity receptor for the semaphorins Sema E and Sema IV but not Sema III. Neuron 1997; 19:547 - 559
- Mamluk R, Gechtman Z, Kutcher ME, Gasiunas N, Gallagher J, Klagsbrun M. Neuropilin-1 binds vascular endothelial growth factor 165, placenta growth factor-2, and heparin via its b1b2 domain. J Biol Chem 2002; 277:24818 - 24825
- Soker S, Takashima S, Miao HQ, Neufeld G, Klagsbrun M. Neuropilin-1 is expressed by endothelial and tumor cells as an isoform-specific receptor for vascular endothelial growth factor. Cell 1998; 92:735 - 745
- Migdal M, Huppertz B, Tessler S, Comforti A, Shibuya M, Reich R, et al. Neuropilin-1 is a placenta growth factor-2 receptor. J Biol Chem 1998; 273:22272 - 22278
- Makinen T, Olofsson B, Karpanen T, Hellman U, Soker S, Klagsbrun M, et al. Differential binding of vascular endothelial growth factor B splice and proteolytic isoforms to neuropilin-1. J Biol Chem 1999; 274:21217 - 21222
- Kitsukawa T, Shimono A, Kawakami A, Kondoh H, Fujisawa H. Overexpression of a membrane protein, neuropilin, in chimeric mice causes anomalies in the cardiovascular system, nervous system and limbs. Development 1995; 121:4309 - 4318
- Kitsukawa T, Shimizu M, Sanbo M, Hirata T, Taniguchi M, Bekku Y, et al. Neuropilin-semaphorin III/Dmediated chemorepulsive signals play a crucial role in peripheral nerve projection in mice. Neuron 1997; 19:995 - 1005
- Gluzman-Poltorak Z, Cohen T, Herzog Y, Neufeld G. Neuropilin-2 is a receptor for the vascular endothelial growth factor (VEGF) forms VEGF-145 and VEGF-165. J Biol Chem 2000; 275:18040 - 18045
- Karkkainen MJ, Saaristo A, Jussila L, Karila KA, Lawrence EC, Pajusola K, et al. A model for gene therapy of human hereditary lymphedema. Proc Natl Acad Sci USA 2001; 98:12677 - 12682
- Giger RJ, Cloutier JF, Sahay A, Prinjha RK, Levengood DV, Moore SE, et al. Neuropilin-2 is required in vivo for selective axon guidance responses to secreted semaphorins. Neuron 2000; 25:29 - 41
- Yuan L, Moyon D, Pardanaud L, Breant C, Karkkainen MJ, Alitalo K, et al. Abnormal lymphatic vessel development in neuropilin 2 mutant mice. Development 2002; 129:4797 - 4806
- Takashima S, Kitakaze M, Asakura M, Asanuma H, Sanada S, Tashiro F, et al. Targeting of both mouse neuropilin-1 and neuropilin-2 genes severely impairs developmental yolk sac and embryonic angiogenesis. Proc Natl Acad Sci USA 2002; 99:3657 - 3662
- Scolnik PA. mAbs A business perspective. mAbs 2009; 1:179 - 184
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000; 100:57 - 70
- Kim KJ, Li B, Winer J, Armanini M, Gillett N, Phillips HS, et al. Inhibition of vascular endothelial growth factor-induced angiogenesis suppresses tumour growth in vivo. Nature 1993; 362:841 - 844
- Warren RS, Yuan H, Matli MR, Gillett NA, Ferrara N. Regulation by vascular endothelial growth factor of human colon cancer tumorigenesis in a mouse model of experimental liver metastasis. J Clin Invest 1995; 95:1789 - 1797
- Melnyk O, Shuman MA, Kim KJ. Vascular endothelial growth factor promotes tumor dissemination by a mechanism distinct from its effect on primary tumor growth. Cancer Res 1996; 56:921 - 924
- Borgstrom P, Bourdon MA, Hillan KJ, Sriramarao P, Ferrara N. Neutralizing anti-vascular endothelial growth factor antibody completely inhibits angiogenesis and growth of human prostate carcinoma micro tumors in vivo. Prostate 1998; 35:1 - 10
- Presta LG, Chen H, O’Connor SJ, Chisholm V, Meng YG, Krummen L, et al. Humanization of an Anti-Vascular Endothelial Growth Factor Monoclonal Antibody for the Therapy of Solid Tumors and Other Disorders. Cancer Res 1997; 57:4593 - 4599
- Muller YA, Chen Y, Christinger HW, Li B, Cunningham BC, Lowman HB, et al. VEGF and the Fab fragment of a humanized neutralizing antibody: crystal structure of the complex at 2.4 A resolution and mutational analysis of the interface. Structure 1998; 6:1153 - 1167
- Muller YA, Li B, Christinger HW, Wells JA, Cunningham BC, de Vos AM. Vascular endothelial growth factor: crystal structure and functional mapping of the kinase domain receptor binding site. Proc Natl Acad Sci USA 1997; 94:7192 - 7197
- Wiesmann C, Fuh G, Christinger HW, Eigenbrot C, Wells JA, de Vos AM. Crystal structure at 1.7 A resolution of VEGF in complex with domain 2 of the Flt-1 receptor. Cell 1997; 91:695 - 704
- Ferrara N, Hillan KJ, Gerber HP, Novotny W. Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nat Rev Drug Discov 2004; 3:391 - 400
- Gordon MS, Margolin K, Talpaz M, Sledge GW Jr, Holmgren E, Benjamin R, et al. Phase I safety and pharmacokinetic study of recombinant human anti-vascular endothelial growth factor in patients with advanced cancer. J Clin Oncol 2001; 19:843 - 850
- Margolin K, Gordon MS, Holmgren E, Gaudreault J, Novotny W, Fyfe G, et al. Phase Ib trial of intravenous recombinant humanized monoclonal antibody to vascular endothelial growth factor in combination with chemotherapy in patients with advanced cancer: pharmacologic and long-term safety data. J Clin Oncol 2001; 19:851 - 856
- Grothey A, Galanis E. Targeting angiogenesis: progress with anti-VEGF treatment with large molecules. Nat Rev Clin Oncol 2009; 6:507 - 517
- Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W, et al. Bevacizumab plus irinotecan, fluorouracil and leucovorin for metastatic colorectal cancer. N Engl J Med 2004; 350:2335 - 2342
- Giantonio BJ, Catalano PJ, Meropol NJ, O’Dwyer PJ, Mitchell EP, Alberts SR, et al. Bevacizumab in Combination With Oxaliplatin, Fluorouracil and Leucovorin (FOLFOX4) for Previously Treated Metastatic Colorectal Cancer: Results From the eastern Cooperative Oncology Group Study E3200. J Clin Oncol 2007; 25:1539 - 1544
- Sandler A, Gray R, Perry MC, Brahmer J, Schiller JH, Dowlati A, et al. Paclitaxel-carboplatin alone or with bevacizumab for non-small cell lung cancer. N Engl J Med 2006; 355:2542 - 2550
- Miller K, Wang M, Gralow J, Dickler M, Cobleigh M, Perez EA, et al. Paclitaxel plus Bevacizumab versus Paclitaxel Alone for Metastatic Breast Cancer. N Engl J Med 2007; 357:2666 - 2676
- Miller KD, Chap LI, Holmes FA, Cobleigh MA, Marcom PK, Fehrenbacher L, et al. Randomized phase III trial of capecitabine compared with bevacizumab plus capecitabine in patients with previously treated metastatic breast cancer. J Clin Oncol 2005; 23:792 - 799
- Friedman HS, Prados MD, Wen PY, Mikkelsen T, Schiff D, Abrey LE, et al. Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J Clin Oncol 2009; 27:4733 - 4740
- Norden AD, Young GS, Setayesh K, Muzikansky A, Klufas R, Ross GL, et al. Bevacizumab for recurrent malignant gliomas: efficacy, toxicity and patterns of recurrence. Neurology 2008; 70:779 - 787
- Escudier B, Pluzanska A, Koralewski P, Ravaud A, Bracarda S, Szczylik C, et al. Bevacizumab plus interferon alfa-2a for treatment of metastatic renal cell carcinoma: a randomised, double-blind phase III trial. Lancet 2007; 370:2103 - 2111
- Rini BI, Halabi S, Rosenberg JE, Stadler WM, Vaena DA, Ou SS, et al. Bevacizumab plus interferon alfa compared with interferon alfa monotherapy in patients with metastatic renal cell carcinoma: CALGB 90206. J Clin Oncol 2008; 26:5422 - 5428
- Brekken RA, Huang X, King SW, Thorpe PE. Vascular Endothelial Growth Factor as a Marker of Tumor Endothelium. Cancer Res 1998; 58:1952 - 1959
- Brekken RA, Overholser JP, Stastny VA, Waltenberger J, Minna JD, Thorpe PE. Selective Inhibition of Vascular Endothelial Growth Factor (VEGF) Receptor 2 (KDR/Flk-1) Activity by a Monoclonal Anti-VEGF Antibody Blocks Tumor Growth in Mice. Cancer Res 2000; 60:5117 - 5124
- Stephan S, Datta K, Wang E, Li J, Brekken RA, Parangi S, et al. Effect of rapamycin alone and in combination with antiangiogenesis therapy in an orthotopic model of human pancreatic cancer. Clin Cancer Res 2004; 10:6993 - 7000
- Liang Y, Brekken RA, Hyder SM. Vascular endothelial growth factor induces proliferation of breast cancer cells and inhibits the anti-proliferative activity of antihormones. Endocr Relat Cancer 2006; 13:905 - 919
- Holloway SE, Beck AW, Shivakumar L, Shih J, Fleming JB, Brekken RA. Selective blockade of vascular endothelial growth factor receptor 2 with an antibody against tumor-derived vascular endothelial growth factor controls the growth of human pancreatic adenocarcinoma xenografts. Ann Surg Oncol 2006; 13:1145 - 1155
- Roland CL, Dineen SP, Lynn KD, Sullivan LA, Dellinger MT, Sadegh L, et al. Inhibition of vascular endothelial growth factor reduces angiogenesis and modulates immune cell infiltration of orthotopic breast cancer xenografts. Mol Cancer Ther 2009; 8:1761 - 1771
- Roland CL, Lynn KD, Toombs JE, Dineen SP, Udugamasooriya DG, Brekken RA. Cytokine levels correlate with immune cell infiltration after anti-VEGF therapy in preclinical mouse models of breast cancer. PLoS ONE 2009; 4:7669
- Sullivan L, Carbon JG, Roland CL, Toombs JE, Kavlie A, Schlunegger K, et al. Selective inhibition of vascular endothelial growth factor receptor 2 with a fully human anti-vascular endothelial growth factor monoclonal antibody. Mol Cancer Ther 2009; In review.
- Gerber H-P, Wu X, Yu L, Wiesmann C, Liang XH, Lee CV, et al. Mice expressing a humanized form of VEGF-A may provide insights into the safety and efficacy of anti-VEGF antibodies. PNAS 2007; 104:3478 - 3484
- Kamba T, Tam BYY, Hashizume H, Haskell A, Sennino B, Mancuso MR, et al. VEGF-dependent plasticity of fenestrated capillaries in the normal adult microvasculature. Am J Physiol Heart Circ Physiol 2006; 290:560 - 576
- Kamba T, McDonald DM. Mechanisms of adverse effects of anti-VEGF therapy for cancer. British Journal of Cancer 2007; 96:1788 - 1795
- Ferrara N, Chen H, Davis-Smyth T, Gerber HP, Nguyen TN, Peers D, et al. Vascular endothelial growth factor is essential for corpus luteum angiogenesis. Nat Med 1998; 4:336 - 340
- Gerber HP, Vu TH, Ryan AM, Kowalski J, Werb Z, Ferrara N. VEGF couples hypertrophic cartilage remodeling, ossification and angiogenesis during endochondral bone formation. Nat Med 1999; 5:623 - 628
- Gerber HP, Kowalski J, Sherman D, Eberhard DA, Ferrara N. Complete inhibition of rhabdomyosarcoma xenograft growth and neovascularization requires blockade of both tumor and host vascular endothelial growth factor. Cancer Res 2000; 60:6253 - 6258
- Holash J, Davis S, Papadopoulos N, Croll SD, Ho L, Russell M, et al. VEGF-Trap: a VEGF blocker with potent antitumor effects. Proc Natl Acad Sci USA 2002; 99:11393 - 11398
- Huang J, Frischer JS, Serur A, Kadenhe A, Yokoi A, McCrudden KW, et al. Regression of established tumors and metastases by potent vascular endothelial growth factor blockade. Proc Natl Acad Sci USA 2003; 100:7785 - 7790
- Fukasawa M, Korc M. Vascular endothelial growth factor-trap suppresses tumorigenicity of multiple pancreatic cancer cell lines. Clin Cancer Res 2004; 10:3327 - 3332
- Hu L, Hofmann J, Holash J, Yancopoulos GD, Sood AK, Jaffe RB. Vascular endothelial growth factor trap combined with paclitaxel strikingly inhibits tumor and ascites, prolonging survival in a human ovarian cancer model. Clin Cancer Res 2005; 11:6966 - 6971
- Kaplan RN, Riba RD, Zacharoulis S, Bramley AH, Vincent L, Costa C, et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the premetastatic niche. Nature 2005; 438:820 - 827
- Wu Y, Hooper AT, Zhong Z, Witte L, Bohlen P, Rafii S, Hicklin DJ. The vascular endothelial growth factor receptor (VEGFR-1) supports growth and survival of human breast carcinoma. Int J Cancer 2006; 119:1519 - 1529
- Wu Y, Zhong Z, Huber J, Bassi R, Finnerty B, Corcoran E, et al. Anti-vascular endothelial growth factor receptor-1 antagonist antibody as a therapeutic agent for cancer. Clin Cancer Res 2006; 12:6573 - 6584
- Rockwell P, Neufeld G, Glassman A, Caron D, Goldstein N. In vitro neutralization of vascular endothelial growth factor activation of Flk-1 by a monoclonal antibody. Mol Cell Differ 1995; 3:91 - 109
- Skobe M, Rockwell P, Goldstein N, Vosseler S, Fusenig NE. Halting angiogenesis suppresses carcinoma cell invasion. Nat Med 1997; 3:1222 - 1227
- Prewett M, Huber J, Li Y, Santiago A, O’Connor W, King K, et al. Antivascular endothelial growth factor receptor (fetal liver kinase 1) monoclonal antibody inhibits tumor angiogenesis and growth of several mouse and human tumors. Cancer Res 1999; 59:5209 - 5218
- Shaheen RM, Tseng WW, Vellagas R, Liu W, Ahmad SA, Jung YD, et al. Effects of an antibody to vascular endothelial growth factor receptor-2 on survival, tumor vascularity and apoptosis in a murine model of colon carcinomatosis. Int J Oncol 2001; 18:221 - 226
- Zhang L, Yu D, Hicklin DJ, Hannay JA, Ellis LM, Pollock RE. Combined anti-fetal liver kinase 1 monoclonal antibody and continuous low-dose doxorubicin inhibits angiogenesis and growth of human soft tissue sarcoma xenografts by induction of endothelial cell apoptosis. Cancer Res 2002; 62:2034 - 2042
- Bruns CJ, Shrader M, Harbison MT, Portera C, Solorzano CC, Jauch KW, et al. Effect of the vascular endothelial growth factor receptor-2 antibody DC101 plus gemcitabine on growth, metastasis and angiogenesis of human pancreatic cancer growing orthotopically in nude mice. Int J Cancer 2002; 102:101 - 108
- Zhu Z, Rockwell P, Lu D, Kotanides H, Pytowski B, Hicklin DJ, et al. Inhibition of vascular endothelial growth factor-induced receptor activation with antikinase insert domain-containing receptor single-chain antibodies from a phage display library. Cancer Res 1998; 58:3209 - 3214
- Zhu Z, Witte L. Inhibition of tumor growth and metastasis by targeting tumor-associated angiogenesis with antagonists to the receptors of vascular endothelial growth factor. Invest New Drugs 1999; 17:195 - 212
- Posey JA, Ng TC, Yang B, Khazaeli MB, Carpenter MD, Fox F, et al. A phase I study of anti-kinase insert domain-containing receptor antibody, IMC-1C11, in patients with liver metastases from colorectal carcinoma. Clin Cancer Res 2003; 9:1323 - 1332
- Lu D, Jimenez X, Zhang H, Bohlen P, Witte L, Zhu Z. Selection of high affinity human neutralizing antibodies to VEGFR2 from a large antibody phage display library for antiangiogenesis therapy. Int J Cancer 2002; 97:393 - 399
- Lu D, Shen J, Vil MD, Zhang H, Jimenez X, Bohlen P, et al. Tailoring in vitro selection for a picomolar affinity human antibody directed against vascular endothelial growth factor receptor 2 for enhanced neutralizing activity. J Biol Chem 2003; 278:43496 - 43507
- Zhu Z, Hattori K, Zhang H, Jimenez X, Ludwig DL, Dias S, et al. Inhibition of human leukemia in an animal model with human antibodies directed against vascular endothelial growth factor receptor 2. Correlation between antibody affinity and biological activity. Leukemia 2003; 17:604 - 611
- Miao HQ, Hu K, Jimenez X, Navarro E, Zhang H, Lu D, et al. Potent neutralization of VEGF biological activities with a fully human antibody Fab fragment directed against VEGF receptor 2. Biochem Biophys Res Commun 2006; 345:438 - 445
- Kerbel RS. Antiangiogenic therapy: a universal chemosensitization strategy for cancer?. Science 2006; 312:1171 - 1175
- Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science 2005; 307:58 - 62
- Roodhart JM, Langenberg MH, Witteveen E, Voest EE. The Molecular Basis of Class Side Effects Due to Treatment with Inhibitors of the VEGF/VEGFR Pathway. Curr Clin Pharmacol 2008; 3:132 - 143
- Chen HX, Cleck JN. Adverse effects of anticancer agents that target the VEGF pathway. Nat Rev Clin Oncol 2009; 6:465 - 477
- Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer 2008; 8:592 - 603
- Sessa C, Guibal A, Del Conte G, Ruegg C. Biomarkers of angiogenesis for the development of antiangiogenic therapies in oncology: tools or decorations?. Nat Clin Pract Oncol 2008; 5:378 - 391
- Jain RK, Duda DG, Willett CG, Sahani DV, Zhu AX, Loeffler JS, et al. Biomarkers of response and resistance to antiangiogenic therapy. Nat Rev Clin Oncol 2009; 6:327 - 338
- Autiero M, Waltenberger J, Communi D, Kranz A, Moons L, Lambrechts D, et al. Role of PlGF in the intra- and intermolecular cross talk between the VEGF receptors Flt1 and Flk1. Nat Med 2003; 9:936 - 943
- Fischer C, Jonckx B, Mazzone M, Zacchigna S, Loges S, Pattarini L, et al. Anti-PlGF Inhibits Growth of VEGF(R)-Inhibitor-Resistant Tumors without Affecting Healthy Vessels. Cell 2007; 131:463 - 475
- Saharinen P, Tammela T, Karkkainen MJ, Alitalo K. Lymphatic vasculature: development, molecular regulation and role in tumor metastasis and inflammation. Trends Immunol 2004; 25:387 - 395
- Murdoch C, Muthana M, Coffelt SB, Lewis CE. The role of myeloid cells in the promotion of tumour angiogenesis. Nat Rev Cancer 2008; 8:618 - 631