Abstract
Genetically encoded fluorescent antibodies are desirable for many applications in biotechnology, proteomics, microscopy, cell biology and molecular diagnostics, although efficient production of fluorescent IgGs in mammalian cells has been hampered by different and mutually incompatible secretion- and folding-requirements of antibodies and green fluorescent protein-derived fluorescent entities. Here, we show that this hurdle can be overcome by generating whole antibody fusions with Citrine, a modified yellow fluorescent protein that folds properly in the endoplasmic reticulum of mammalian cells. Applying optimized connector sequences, one or more Citrine molecules can be fused to different positions of IgGs without interfering with folding, secretion or function of the fusion proteins. These proteins can be transiently expressed and purified to similar yields as unmodified antibodies using standard technologies. IgG-Citrine fusions fully retain binding specificity and affinity, and can be applied to assays that require labeled IgG. A particularly interesting feature is the pH-dependency of Citrine fluorescence. This makes IgG-Citrine fusion proteins a valuable tool to track antibody target binding, internalization and subsequent intracellular trafficking to acidic compartments.
Introduction
Recombinant antibodies have become widely used in research, treatment of diseases and many diagnostically relevant applications. If fluorescent antibodies are required, usually antibodies are chemically conjugated to fluorophores; however, this approach has numerous limitations: (1) the location of fluorophore attachment to the molecule can usually not be exactly determined and can ultimately interfere with antigen binding; (2) the stoichiometry of fluorophore/antibody can usually not be determined exactly, which means there is a heterogeneous mixture of different fluorophore/antibody complexes; and (3) green fluorescent protein (GFP)-derived proteins are more resistant to photobleaching than some fluorophores, e.g., fluorescein.Citation1–Citation5
One way to overcome the limitations inherent in chemical conjugates of fluorescent molecules to antibodies would be the use of fusions between fluorescent proteins and antibodies. This approach, however, is hampered by the different requirements of antibodies and GFP-related fluorescent proteins in terms of protein folding. Antibodies are hetero-tetrameric proteins stabilized and held together by disulfide bonds formed in the redox environment of the ER. In contrast, GFP-related fluorescent proteins are designed to be folded in the cytosol of eukaryotic jellyfish.Citation6
In recent years, multiple approaches have been undertaken to circumvent these problems and produce a fluorescent antibody fusion protein. Researchers have tried to overcome these problems by expression of either scFv or camelide VHH based fluorescent fusion proteins.Citation7–Citation10 These were expressed in E. coli either as cytosolic inclusion bodies or in the bacterial periplasm or Chinese hamster ovary cells. In general, these approaches were successful, but the yield usually was in the low µg range. Furthermore, the use of antibody fragments has various limitations such as reduced serum half-life when used for in vivo studies. On the other hand, the use of full-size antibody molecules allows use of well-established secondary detection methods and standard purification methods and the possibility of using Fc interacting molecules as downstream effectors.
In comparison to GFP, yellow fluorescent protein (YFP) exhibits higher photostability,Citation11 which is desirable for fluorescent antibodies used as tools for research and diagnostics. We therefore investigated the possibilities of combining an antibody with a version of YFP. Yellow fluorescent variants of GFP were generated by mutation of the Thr203 of GFP.Citation12,Citation13 These proteins were sensitive to pH and chloride interference, showed photo instability and were only poorly expressed at 37°C. These features interfere with the use of YFP as an attachment to an antibody; however, an expression screen in E. coli indentified the YFPQ69M mutant, Citrine, that showed improved expression in intracellular compartments such as the endoplasmic reticulum (ER), increased halide resistance and reduced, but still pronounced, pH dependent fluorescence.Citation14,Citation15
Expression of GFP-derived proteins inside the ER has been successfully carried out by many labs.Citation16 However, expression of sufficient protein to generate a fluorescent signal for microscopy is not comparable to the task of producing and purifying sufficient functional protein in quantitative amounts. Amounts below 1 µM of GFP are at the detection limit inside the mammalian cytosol, lower if the signal is within a defined cellular structure.Citation4,Citation17 This corresponds to several hundred molecules per cellular structure, with an estimated detection limit per cellular structure of about 50 molecules of GFP.Citation18 This detection limit obviously depends on the experimental setup used, as well as the structures of interest. Expression systems that load the cellular system to the maximum, e.g., the viral VSV-G reporter system, can yield up to several thousand molecules in large post-Golgi carriers.Citation19
With Citrine, we were able to express fluorescent antibody fusion proteins in the HEK293 system. We were able to purify the fluorescent antibodies using standard Protein A-based purification techniques with yields that were similar to those of standard IgG fusion proteins. The antibodies were capable of target binding comparable to non-fluorescent wild-type antibodies. Fluorescence properties, including the pH dependency of Citrine, were not altered by fusion to human IgG. This novel form of antibody has potential not only as a tool with multiple application in diagnostics and biotechnology, but also serves as a photostable, pH-sensitive tool to study cellular surface receptors.
Results
Design of antibody-Citrine fusion protein formats.
As antibodies are hetero-tetrameric proteins, numerous locations suitable for fusion to Citrine can be envisioned, e.g., the fluorescent protein could be attached to the N- or C-terminus of either antibody part; it could be fused to the heavy or light chains; advanced protein engineering might be used to target only one of the dimeric heavy or light chains. We thus generated a set of different formats to identify the candidate that could be expressed with the best yield, and generate molecules with either one or two Citrine moieties (). To facilitate the comparison, we used an antibody that targeted the digoxigenin (DIG) hapten for all the different formats.Citation20,Citation21
One antibody format had Citrine at the C-terminus of both light chains using a (G4S)2 linker. This approach resulted in an antibody carrying two fluorescent protein moieties. To analyze the influence of different connectors, we produced the same format with either no linker or with the sequence GIHRPVAT derived from the eGFP-N2 expression vector (BD Biosciences Clontech Cat.No 6081-1) ().
To generate antibody formats with only one fluorescent protein, we tagged the C-terminus of only one heavy chain with Citrine (). To produce this format, we made use of the “knobs into holes” technology.Citation22,Citation23 This technique allows the combination of two different heavy chains in one antibody. We generated a format that had one heavy chain with “hole” mutations, Y349C, T366S, L368A and Y407V, a corresponding heavy chain carrying the “knob” mutations S354C and T366W and, via a (G4S)2 linker, a single Citrine molecule.
Another approach to generate an antibody with only one fluorescent portion also uses the “knobs into holes” technique, but combines it with a published protein complementation assay (PCA) using two halves of Citrine.Citation24,Citation25 One heavy chain carries the “knob” mutation and the N-terminal half of Citrine connected via a (G4S)2 linker; the second half of Citrine is attached to a heavy chain with a “hole” mutation () via a (G4S)2 linker. When the antibody is folded in the ER and both heavy chains come in close contact, both halves of Citrine combine to form one fluorescent molecule.
To investigate whether our observations apply to antibodies of other binding specificity as well, we generated Citrine fusion antibodies specific for the IGF-1 receptor ().Citation26–Citation28 We also generated an antibody-Citrine fusion specific for the carbohydrate Lewis Y (LeY) antigen.Citation29,Citation30 Furthermore, antibodies with such binding specificities allow the analysis of the fluorescent fusion proteins on cells.
To further expand our inventory of fluorescent antibodies, we made use of the fact that antibodies can be expanded in their ability to bind antigen by the addition of additional binding sites, i.e., bispecific antibodies. We designed a full-size antibody that binds to the IGF-1 receptor and has additional disulfide stabilized scFvs specific for the DIG hapten,Citation31,Citation32 at the C-termini of both heavy chains () connected via a (G4S)2 linker. In this bispecific background, we used the same light chains used to generate the anti-IGF-1R mono-specific antibody with Citrine at the C-termini of both light chains.
To compare the full-size, Citrine fused antibodies to a smaller antibody-derived molecule with Citrine, we designed a format that has two disulfide stabilized scFvs,Citation31,Citation32 one that targeted the IGF-1 receptor and one that targeted the DIG hapten. The anti-IGF-1R scFv was attached to the N-terminus of Citrine via a (G4S)2 linker and the anti-DIG scFv was attached via the same linker to the C-terminus of Citrine (). To aid purification, this format included an additional 6x-His tag at its C-terminus.
To compare the ability of Citrine to form fluorescent antibodies with the ability of eGFP, we generated molecules in the aforementioned formats with eGFP to compare expression, folding and function of these fusion proteins ().
Purification of the fluorescent antibody formats.
We produced all antibody fusion proteins by transient expression in non-adherent human embryonic kidney (HEK) 293 cells. The proteins were secreted into the cell culture media in the same manner as standard recombinant antibodies. The supernatants were harvested 7 days after transfection, stored at −20°C, and thawed before purification. Freeze-thaw cycles did not cause any obvious reduction in the yield of proteins recovered from the supernatants. We obtained homogenous preparations of the antibody variants by applying Protein A affinity and size exclusion chromatography, except for the scFv based format, which was purified using Ni-NTA (GE Healthcare). This finding indicates that the Fc-region of the fluorescent antibody fusion proteins is fully accessible to the Protein A resin, and that the addition of extra protein entities does not interfere with the purification process. The antibody fusions showed the expected molecular mass by analytical SEC and SDS-PAGE.
An example of the results of the Protein A-based purification of an anti-DIG antibody with Citrine at the C-termini of the light chains is shown in . Remarkably, despite the low pH used for elution of the protein from the column, the eluted fractions were found to be yellow in daylight (data not shown) and the main peak fractions displayed fluorescence at 528 nM, which suggested that the Citrine portion of the purified antibodies was functional. shows an analytical SEC profile of the same protein after SEC purification. We used SEC purification to remove aggregated protein from all final products. Using this approach, we also selected for fusion proteins with properly folded Citrine moiety to ensure the expected ratio of fluorophore per antibody molecule.
An overview of the yields obtained after purification is given in . These data show that the addition of two Citrine molecules to the C-termini of the light chain did not significantly interfere with the production of the recombinant protein compared to the parental molecule without additional Citrine: purified protein recovered from 1 L HEK293 supernatants after transient expressions (without optimization of expression conditions) were 25 mg/L for the unmodified antibody and 23 mg/L for the Citrine fusions (L-chain). Addition of Citrine to one of the heavy chains reduced the yield, but it was still in the range observed for other IgG molecules. These data show that the addition of Citrine is compatible with the “knobs into holes” technology. The format with complemented Citrine attached to the heavy chains showed the lowest yield for a full size mono-specific antibody (3 mg/mL). Still, this yield is acceptable for in vitro analytics or microscopy. Comparable results were obtained using antibodies that targeted IGF-1R ().
When the linker connecting Citrine and the antibody was omitted and the Citrine directly fused to the antibody, a significant reduction of the produced material was observed (). Furthermore, more aggregates were observed after SEC purification. A similar result was observed when we used the GIHRPVAT linker that is present in the eGFP-N2 expression vector (). These findings indicate that a linker of sufficient length and flexibility is required for the proper, i.e., independent, folding of both the antibody and the Citrine moiety. A similar result was seen for formats with eGFP instead of Citrine as part of the whole antibody. While supernatants and cell lysates of cells expressing Citrine and eGFP fusion antibodies show comparable amounts of total protein mass (Sup. Fig. 1), fusions with eGFP show a significant reduction of the yield of properly folded active protein after purification (). Furthermore, eGFP was not compatible with the PCA-based approach using two halves of the fluorescent protein on the C-termini of the heavy chains. Despite the fact that some expression was observed in cell lysates and the supernatant (Sup. Fig. 1), no protein was purified. Generally, the Citrine fusion proteins showed a yield about 100% higher than the eGFP fusion proteins. This observation is consistent with published data that show superior folding of Citrine in mammalian cells at 37°C.Citation14
The absolute protein amount that was generated in expressions of small fusion molecules with both the scFv that targeted IGF-1R and DIG was lower than that obtained for the fusion proteins with whole IgGs (2 mg/L; ); however, considering that the molecular weight of such molecules is lower than that of IgG-fusions (60 kDa vs. 200 kD), the reduction in terms of molar concentration was not pronounced. The amounts obtained were still within ranges that allow easy purification of sufficient material for cellular assays, in vitro analytics or microscopy.
Taken together, the data show that Citrine fusion antibodies can be purified using standard purification technologies to yields comparable to standard IgG molecules. Furthermore, they offer the advantage of fluorescent eluted fractions, simplifying the process of determining the protein-containing fractions.
The data also show that Citrine offers the unique ability to generate fluorescent antibody in contrast to standard eGFP, which shows significant reduction of expression compared to parental antibody molecules. The reduced expression of formats without a linker or the reduced length linker derived from the eGFP-N2 vector highlight the importance of a flexible linker connecting the antibody and the fluorescent protein portion.
Antibody-Citrine fusions show affinities comparable to wild type antibodies.
To characterize the target binding properties of the Citrine fusion antibodies in comparison to the wild type antibodies we applied surface plasmon resonance (SPR).
The DIG-specific antibodies were captured onto the chip surface using a human Fc region specific antibody. Next, the binding to myoglobin carrying one DIG moiety was analyzed () and KD values were determined (). The analysis shows that the novel formats have comparable affinities to the wild type antibody. This finding was independent of the localization of Citrine in the fusion protein, indicating that Citrine forms a separate domain without any relevant interference with the function of the molecule. The affinities of the anti-DIG scFv were lower compared to the full-size anti-DIG antibodies, which was consistent with our earlier findings (data not shown).
When the anti-DIG scFv attached to the C-terminus of the heavy chains of the anti-IGF-1R maternal molecule was analyzed () and compared to the Citrine fusion protein, no significant difference in the ability of either molecule to bind mono-DIG- myoglobin was observed (). When the anti-IGF-1R Citrine fusion antibody was analyzed for its ability to bind to soluble IGF-1R, it showed a kinetic comparable to the wild type IgG (Sup. Fig. 2). The same was true for the bispecific molecule that targeted IGF-1R and DIG (data not shown).
Taken together, these data show that the addition of the Citrine moiety, independent of its localization in the fusion protein, does not interfere with the antigen binding capabilities of the IgG portion of the antibody.
Antibody-Citrine fusion proteins show pH-dependent fluorescence.
To analyze whether the fusion of Citrine to antibodies altered the pH-dependent fluorescence of the protein, we determined the fluorescence of the generated formats using an ELISA reader-based assay. For this assay, 50 nM of the antibody-Citrine fusion protein formats were incubated in PBS adjusted to the pH indicated. The sample was excited at 516 nm and the fluorescence was determined at 529 nm. Fluorescence was measured and normalized for all fusion proteins independently to generate a signal independent of the number of Citrine molecules. A DIG-specific IgG was used as a negative control.
The data () show that, independent of the localization of Citrine in the fusion protein, the fluorescence of the antibody fusion protein is pH-dependent. The format with two scFvs attached to the N- and C-terminus of Citrine showed a similar behavior. The fluorescence of Citrine reached its highest level at a pH above 7.5. The fluorescence was diminished as the pH was lowered, and followed a sigmoidal curve as previously described.Citation14 It reached a fluorescence of 50% of the maximal intensity at an approximate pH of 5.5. Even the complemented Citrine composed of two split portions attached separately to two H-chain C-termini showed the same result (). This indicates that the complemented molecule has the same properties as fusion proteins that contain the Citrine module as one complete polypeptide chain. Taken together these data show that the fluorescent properties of Citrine are not altered by fusion to an antibody, independent of the localization of the molecule in the fusion protein.
Antibody-Citrine fusion proteins can be used to stain fixed cells.
To analyze whether antibody-Citrine fusion proteins can be used on fixed cell samples, we made use of the fact that, upon binding to IGF-1R, antibodies not only block ligand, but also induce internalization of the receptor into the endocytic pathway.Citation33,Citation34 The antibody remains bound to its target and is co-internalized. A fluorescent antibody should also bind to cells and become internalized.
After application of the anti-IGF-1R antibody-Citrine fusion to IGR-1R positive cells at 4°C, we either fixed the cell samples immediately, or continued incubation of the cells with bound antibodies in standard growth medium overnight at 37°C. Such a prolonged incubation should lead to internalization of the majority of the bound antibody. After this incubation, the cells were fixed.
After a binding period of 1 hour on ice with subsequent fixation of the samples without elevation of the temperature, the fluorescent signal of the Citrine moiety observed in the FITC channel was found distributed equally over the cell surface (). When compared to a secondary antibody targeting the kappa light chain that shows the antibody in the Cy3 channel, both signals are found to co-localize.
When the cells bound to the Citrine-antibody fusion proteins were additionally incubated overnight at 37°C to allow internalization of the antibody bound to the IGF-1 receptor, the majority of the fluorescent signal of Citrine was found in intracellular vesicles (). Only a minor fraction was found on the cell surface, probably representing the re-cycling portion of the receptor. When compared to results from addition of a secondary antibody targeting the kappa light chain that shows the antibody in the Cy3 channel, both signals largely co-localize; however, a few membrane-bound structures with dominant green fluorescence can be observed ( and Arrow), probably reflecting subcellular structures containing degraded antibody and intact Citrine.
The data show that antibody-Citrine fusion proteins can be used to stain their targets on fixed cells. Furthermore, the fixation with PFA does not interfere with the fluorescent properties of Citrine when fused to an antibody. The antibody-Citrine fusion proteins were also not prone to rapid proteolysis once internalized into cells and exposed to the cellular interior for a prolonged time, as shown by the overnight incubation. The presence of some mainly green structures probably reflects the fact that Citrine is more resistant to degradation than the antibody moiety. This observation is consistent with our observations that, despite the complete degradation of the antibody, Citrine was still functional in excess papain (Sup. Fig. 3). Furthermore, internalization of the anti-IGF-1R-Citrine fusion antibody was comparable to that of an anti-IGF-1R antibody lacking the Citrine moiety (data not shown). This finding indicates that the Citrine moiety does not interfere with the antibody functionality.
Antibody-Citrine fusion proteins can be used to image living cells.
Next, we examined whether the novel antibody fusion proteins could be used to detect their respective targets on living cells. For this purpose we used FACS analysis. MCF7 cells were incubated in the presence of antibody-Citrine fusion proteins for 1 h at 4°C and immediately analyzed by FACS. When stained with a Cy5 labeled secondary antibody recognizing the Fc region of the antibodies, binding of the anti-IGF-1R antibody fused to Citrine on the C-terminus of the light chain was shown when analyzed in the APC-A channel (, left part). This finding shows that the antibody-Citrine fusion proteins are capable of antigen binding, and is consistent with the data obtained by SPR analysis and confocal microscopy.
When the same antibodies were analyzed in the FITC channel, the anti-IGF-1R antibody fused to Citrine on the C-terminus of the light chain gave rise to a signal, indicating direct labeling of the cells by the Citrine portion of the fusion proteins (, right part). It is important to keep in mind that the FITC channel is sub-optimal for this analysis because excitation with the 488 nm laser only leads to 35% of maximal Citrine excitation.
To correlate this finding to an antigen with a higher density than IGF-1R, we used a LeY-specific antibody with Citrine attached to the C-termini of the light chains. When this antibody was incubated with MCF7 cells, a stronger signal compared to that observed for the anti-IGF-1R antibody was obtained using secondary detection with an allophycoryanin (APC)-labeled anti-human antibody (, left part), illustrating the higher levels of antigen on the target cells. This Citrine-labeled antibody also gave rise to a signal in the FITC channel (, right part), again showing direct labeling of target positive cells with the Citrine moiety.
To compare these data to a monovalent small molecule, we used the format with Citrine between the anti-IGF-1R and anti-DIG scFvs. To show binding of this module to cells carrying the IGF-1 receptor, we used an APC-labeled antibody targeting the 6x His tag. The results show that the small module is capable of binding to target cells (, left part). When measured in the FITC channel, the small bispecific format showed a weak but significant signal (, right part), indicating direct labeling of target positive cells with the antibody-Citrine fusion proteins. Again, one has to keep in mind that the measurement is taken in a FITC optimized channel, and, in contrast to the full-size antibody formats, this format had only one Citrine molecule per protein.
These data show that Citrine fusion proteins secreted by mammalian cells are capable of binding to their cellular target as shown by staining with a secondary labeled antibody targeting the Fc portion of the fusion protein (), and that the fusion of Citrine to the C-termini of the antibody light chains does not interfere with standard secondary detection methods. Furthermore, we were able to detect the fluorescence of the Citrine-containing antibodies in the FITC channel and thus directly show binding of the antibodies to their cellular target without a secondary detection system.
Antibody-Citrine fusions can monitor intracellular acidification.
After having shown that the Citrine antibodies can be used to directly analyze living cells, we wondered whether it was possible to combine this feature of the fusion proteins with their pH-dependent fluorescence.
For these experiments, we relied on the fact that, upon binding, the anti-IGF-1R antibody induces internalization of the receptor into intracellular vesicles.Citation26,Citation34 As these vesicles are transported along the endocytic pathway, they become increasingly acidified, i.e., the initial neutral pH is reduced to approximately pH 6 to 5.5 in endosomal compartments and to a pH of approximately 5 in lysosomal compartments.Citation35 To interfere with this acidification, we made use of the fact that weak bases such as ammonia accumulate in acidic compartments and lead to an increase of the luminal pH.Citation36
For these experiments, one population of cells was treated with 50 mM NH4Cl for 1 h before starting the experiment while another population of cells was left untreated. Both populations were incubated with an anti-IGF-1R antibody fused to two Citrine molecules at the C-termini of the light chains for 1 h on ice. This incubation allows binding, but prevents internalization of the antibody. After this period, a fraction of both cell populations was immediately analyzed by FACS for antibody binding () by monitoring Citrine fluorescence in the FITC channel. Independent of the treatment with NH4Cl, both populations show a similar antibody binding to the cell surface. This finding shows that the NH4Cl incubation does not interfere with the binding of the antibody to the receptor and does not alter the receptor levels. After 2 h of incubation at 37°C, the antibodies are co-internalized with their receptor targets. At this time point, cells without NH4Cl treatment showed a reduction in Citrine fluorescence compared with cells treated with NH4Cl ().
This finding indicates that the fluorescence of the Citrine fusion protein is reduced in acidic compartments. Interfering with the acidification along the endocytic pathway by the weak base NH4Cl counteracts this effect and thus leads to more residual Citrine fluorescence. Taken together, these data show that antibody-Citrine fusion protein can be used to monitor the acidification upon receptor internalization.
Discussion
We have shown the successful design, production and analysis of a human antibody fused with a fluorescent GFP-derived protein. The different design variants we chose enabled us to generate formats with either one or two Citrine molecules per antibody. Interestingly, the addition of Citrine, especially to the C-termini of the light chains of antibodies, did not significantly interfere with the production of these molecules compared to the yields obtained from the expression of the respective wild-type proteins.
Furthermore, we demonstrated that these fluorescent antibodies are capable of binding their target at a level comparable to wild-type proteins. We showed this not only for our model anti-DIG antibodies, but also for antibodies targeting IGF-1R or the LeY carbohydrate, which suggests that the concept has general applicability. The formats we have chosen therefore seem to be applicable to multiple target specificities. This offers the possibility of generating a variety of fluorescent antibodies, with only exchange of the V regions to generate specific fluorescent antibodies.
When we analyzed alternate formats to generate antibodies with one or two fluorescent portions, we found that the Citrine fusions are compatible with the “knobs into holes” technology, which allows the generation of a multitude of fluorescent formats, including those with three or more Citrine moieties. This technology enables creation of formats with two identical half antibodies or two different half antibodies, and allows generation of antibodies carrying one or more fluorescent moieties. One can also use a split Citrine to generate a full-size antibody with a single fluorescent subunit that has the same biophysical properties as a single chain protein. Originally, fluorescent proteins that were split into two halves and generated fluorescent signals upon combining were devised for intracellular complementation assays.Citation24,Citation37 Such assays utilize PCA-mediated fluorescence signals to detect intracellular events in situ with high sensitivity. Our system expands the applicability of halved fluorescence proteins to the field of protein engineering. For example, if both half Citrine molecules are attached to the N-termini of light and heavy chain, one can distinguish association of correct pairs of different light and heavy chains that might be desired for various protein engineering applications.
Fluorescent antibodies have been derived mainly from small targeting modules such as scFvs or camelide VHH.Citation7–Citation10 We have shown the generation of a full-size antibody with a fluorescent protein portion, which has advantages for in vivo analysis. Due to their interaction with FcRn, full-size antibodies show long serum half-lives, which, when combined with the excellent stability of Citrine against proteolytic degradation, suggests that an antibody-Citrine fusion protein may have desirable PK properties. Alternatively, if a smaller molecule with targeting capabilities and a fluorescent label is desired, e.g., for improved tumor penetration, we have shown an additional format can be used, i.e., we have successfully shown the design, production and functional analysis of a small molecule that has Citrine in line between two scFvs of different specificity.
An auto-fluorescent antibody offers a number of advantages over the detection of an antigen by a primary and a secondary antibody. For example, the elimination of secondary detection leads to weaker signals, but fewer false positive signals. This results in less noise in the detection system and ultimately better signal-to-noise ratios. In addition, labeling of the primary antibody reduces the time required for staining, especially when used in high throughput systems. Furthermore, in contrast to antibodies that are labeled with a fluorescent dye in a conventional way, advantages conferred by antibodies fused with fluorescent proteins include: (1) exact definition of the positions where the fluorescent moieties are attached to the antibody; (2) precise stoichiometry of fluorescent entities attached to the antibody is known; and (3) the ability to make quantitative measurements based on this knowledge.
When using conventional labeling, which usually occurs at the epsilon amines of lysines, the exact residues of the antibody poly-peptide chain to which the dyes become attached are not known. This ultimately results in a heterogeneous population of labeled antibody, some of which do not bind to the antigen because of labeled lysine resides in one of the CDRs. Such problems are avoided when antibodies fused to fluorescent proteins are used.
We have shown two general formats with either one or two Citrine subunits. Using our formats, one can generate a module with three fluorescent subunits by simple combinatory methods. Thus, antibodies with a defined number of fluorescent subunits could be used to determine the concentration of antigen on cells in a fashion similar to published methods used to determine GFP tagged protein numbers and concentrations.Citation4,Citation18 One possible problem that might arise during expression of the antibody fusion proteins, and interfere with the predicted advantage of a precise stoichiometry of fluorophore to antibody, is the potential misfolding of the fluorophore. This problem can be minimized by SEC purification of the recombinant protein.
When we compared the production of antibodies with either Citrine or eGFP as a part of the polypeptide chain, we found that eGFP generally gave rise to substantially reduced yields. When we analyzed the general expression in cell lysates, no notable differences were observed, which suggests that eGFP fusions can be produced, but appear less functional as shown by reduced amounts of purified functional antibody fusion protein. This finding supports earlier publications that have shown superior folding abilities of Citrine in mammalian cells at 37°CCitation14 and gives rise to speculations about the general applicability of Citrine to secreted protein fusions other than antibodies. One can envision molecules comprising Citrine fused to secreted receptor ligands or extracellular portions of receptors that may have use in research and diagnostics. For example, a Citrine tagged receptor could be used to analyze binding of a cyan-labeled ligand by FRET analysis.
The reduction of the fluorescence of Citrine when exposed to lower pH suggests that this system could be used to track the intracellular trafficking of endocytosed receptors. We have shown that antibody-Citrine fusion proteins are co-internalized with the receptor that they target. Due to the fact that endosomes (pH ∼5.5) and lysosomes (pH ∼4.8) show a gradual reduction of the pH, the fluorescence of endocytosed antibody-Citrine fusions is lost gradually. One might therefore envision the use of an antibody-Citrine fusion protein to screen for factors that interfere with the acidification of endosomal compartments, or use of these antibodies to identify factors that interfere with the integrity of endosomal membranes, but one of the key characteristics of Citrine is its reduced sensitivity to pH. We think, however, that this feature goes hand in hand with its superior expression properties. In the light of the minor pH sensitivity and the potential applications, it would be worthwhile to investigate the expression properties of other, more pH-sensitive fluorescent proteins as antibody fusions.
Consistent with early publications on GFP, we have observed the exceptional stability of Citrine against proteolytic degradation (Sup. Fig. 3). When incubated in excess papain, the antibody was fully degraded while Citrine was still functional, as shown by its fluorescence. Furthermore, after an extended incubation of live cells with internalized antibody-Citrine proteins, we noted indications that the fluorescent signal of Citrine is still observed while the antibody starts to become degraded by intracellular proteases. Such stability offers advantages when combined with the targeting ability of an antibody. A cell that is targeted by the antibody is still stained by Citrine even if the antibody is already degraded by cellular proteases and can no longer be detected. This system might therefore function as a kind of cellular tagging system.
Materials and Methods
Cloning of antibody-Citrine fusion proteins.
DNA stretches of interest were synthesized (Geneart, Regensburg, Germany). Subsequently, the synthesized DNA stretches were subcloned into appropriate expression vectors and amplified in E. coli XL1Gold (Invitrogen). All gene segments encoding light and heavy chains of antibodies were synthesized with a 5′-end DNA sequence coding for a leader peptide (MGW SCI ILF LVA TAT GVH S) to target proteins for secretion in eukaryotic cells. Fragments were inserted into unique BamHI and XbaI restriction sites. Integrity of subcloned DNA sequences was confirmed using double-strand sequencing performed at Sequiserve GmbH (Vaterstetten, Germany). All proteins were coded as cDNA sequences. In addition to the antibody expression cassette, the vector contained an origin of replication, oriP, of Epstein-Barr virus (EBV), an origin of replication from the vector pUC18 which allows replication of this plasmid in E. coli, a beta-lactamase gene which confers ampicillin resistance in E. coli, the immediate early enhancer and promoter from the human cytomegalovirus (HCMV) and a poly-adenylation sequence.
Expression and purification of antibody-Citrine fusion proteins.
The antibody-Citrine fusion proteins were expressed by transient transfection of HEK293-F cells using the FreeStyle™ 293 Expression System according to the manufacturer's instruction (Invitrogen, USA). Light and heavy chains of the corresponding antibodies were constructed in expression vectors carrying pro- and eukaryotic selection markers. These plasmids were amplified in E. coli, purified and subsequently applied for transient transfections. The suspension FreeStyle™ 293-F cells were cultivated in FreeStyle™ 293 Expression medium at 37°C and 8% CO2; the cells were seeded in fresh medium at a density of 1–2 × 106 viable cells/ml on the day of transfection. The DNA-293fectin™ complexes were prepared in Opti-MEM medium (Invitrogen, USA) using 333 µl of 293 fectin™ (Invitrogen, Germany) and 250 µg of heavy and light chain plasmid DNA in a 1:1 molar ratio for a final transfection volume of 250 ml. The antibody-Citrine fusion protein containing cell culture supernatants were clarified 7 days after transfection by centrifugation at 14,000 g for 30 min and filtration through a sterile filter (0.22 µm). Supernatants were stored at −20°C until purification.
The recombinant proteins were purified from the supernatant in two steps by affinity chromatography using Protein A-Sepharose™ (GE Healthcare, Sweden) and Superdex 200 size exclusion chromatography. Briefly, the clarified culture supernatants were applied on a HiTrap 15 Protein A HP (5 ml) column equilibrated with PBS buffer (10 mM Na2HPO4, 1 mM KH2PO4, 137 mM NaCl and 2.7 mM KCl, pH 7.4). Unbound proteins were washed out with equilibration buffer. The antibodies were eluted with 0.1 M citrate buffer, pH 2.8, and the protein containing fractions were neutralized with 0.1 ml 1 M Tris, pH 8.5. Then, the eluted protein fractions were pooled, concentrated with an Amicon Ultra centrifugal filter device (MWCO: 30 20 K, Millipore) to a volume of 3 ml and loaded on a Superdex 200 HiLoad 120 ml 16/60 gel filtration column (GE Healthcare, Sweden) equilibrated with 20 mM histidine, 140 mM NaCl, pH 6.0. The protein concentration of purified antibodies and derivatives was determined by measuring the optical density (OD) at 280 nm with the OD at 320 nm as the background correction, using the molar extinction coefficient calculated on the basis of the amino acid sequence. Protein fractions were pooled, snap-frozen and stored at −80°C. The homogeneity of the recombinant proteins was confirmed by SDS-PAGE in the presence and absence of a reducing agent (5 mM 1,4-dithiothreitol) and staining with Coomassie brilliant blue. The NuPAGE® Pre-Cast gel system (Invitrogen, USA) was used according to the manufacturer's instruction (4–20% Tris-Glycine gels).
Surface plasmon resonance.
To analyse the binding properties of the recombinant antibody-Citrine fusion proteins, we used surface plasmon resonance (SPR) technology using a Biacore T100 or Biacore 3000 instrument (GE Healthcare Bio-Sciences AB, Uppsala). To perform the binding studies, a capturing anti-human IgG antibody was immobilized on the surface of a CM5 biosensor chip using amine coupling chemistry. Flow cells were activated with a 1:1 mixture of 0.1 M N-hydroxysuccinimide and 0.1 M 3-(N,N-dimethylamino)propyl-N-ethylcarbodiimide at a flow rate of 5 µl/min. The anti-human IgG antibody was injected in sodium acetate, pH 5.0 at 2 µg/ml, which resulted in a surface density of approximately 500 RU. A reference control flow cell was treated in the same way, but with vehicle buffers only instead of the capturing antibody. Surfaces were blocked with an injection of 1 M ethanolamine/HCl pH 8.5. To evaluate the functionality of the anti-DIG derivatives, binding to mono-digoxygenated myoglobin was analyzed. In order to measure binding affinities, digoxygenated myoglobin was injected at increasing concentrations. The regeneration was carried out by injecting 0.85% H3PO4 for 60 s at 5 µl/min and then injecting 5 mM NaOH for 60 s at 5 µl/min to remove any non-covalently bound protein after each binding cycle. The samples to be analyzed were diluted in HBS-P (10 mM HEPES, pH 7.4, 150 mM NaCl, 0.005% Surfactant-44-P20) and injected at a flow rate of 5 µl/min. The contact time (association phase) was 3 min for the antibodies at a concentration between 1–5 nM. The contact time (association phase) was 3 min, the dissociation time (washing with running buffer) 5 min for each molecule at a flow rate of 30 µl/min. All interactions were performed at 25°C (standard temperature). Signals were detected at a rate of one signal per second.
pH dependent fluorescence.
To measure the pH dependent fluorescence of the antibody-Citrine fusion proteins, we used a microtiter plate fluorescence reader Safire2 (Tecan, Austria). pH steps of 2.0, 4.0, 5.0, 5.5, 6.0, 7.4 and 9.0 were generated by adjusting PBS using either HCl or NaOH. A pH row of increasing values was generated in a microtiter plate and the different antibody-Citrine fusion proteins were added to a final concentration of 50 nM in total volume of 150 µl. Subsequently, the microtiter plate was shaken for 30 s at 750 rpm. Citrine fluorescence was excited at 516 nm and emission was measured at 529 nm. Both emission and excitation bandwidth were 5 nm. Each read was performed 10 times with an integration time of 40 µs.
Microscopy.
For microscopy, I24 cells were grown on glass coverslips to a density of about 50–70%. They were then treated with the antibody-Citrine fusion proteins at a concentration of 5 nM for 2 h on ice. Afterwards, the cells were washed in cold PBS and one set was fixed immediately. Another set of cells was incubated at 37°C for the times indicated. These cells were fixed at the time points indicated with paraformaldehyde. For staining, the fixed cells were washed, incubated with the blocking reagent GSDB and incubated with a rabbit anti-human kappa-light chains antibody (DAKO) at a concentration of 6.5 µg/ml for 1.5–2 h in a humidity chamber. After another wash, the cells were incubated with a Cy3-labeled goat anti-rabbit antibody (Molecular Probes) in a concentration of 28.6 µg/ml for 1.5 h in a humidity chamber. The cells were then washed. Next, the DNA was labeled with DAPI (Roche) at a concentration of 10 µg/ml for 2–3 min, washed again and covered with mounting medium. The cells were analyzed with a Leica SP20 confocal microscope.
FACS analysis.
For FACS analysis, MCF7 cells were detached by 15 min incubation in Accutase. After washing in FACS buffer (PBS containing 5% FCS) the cells were seeded in a 96-well rounded bottom microtiterplate (Corning Inc., Cat# 3799) to a final density of 3 × 105 and used immediately. The cells were incubated in the presence of 3.43 nM of antibody-Citrine fusion protein or isotype control antibody (Jackson Immunoresearch) or for 30 min on ice to allow binding, but prevent internalization. For detection of bound antibodies, a secondary Cy5 labeled antibody [Jackson Immunoresearch 709–15 176–1,490; Cy5 F(ab')2 Donkey anti-human IgG (H + L)] was added to a final concentration of 3.43 nM. The cells were subsequently washed in FACS buffer to remove unbound antibody. After washing, the cells with the small scFv module were incubated with DIG-Cy5 in a equimolar concentration. After another washing step 10,000 cells were analyzed with the FACS canto II (BD Biosciences). Cy5 was analyzed in the Cy5 channel, Citrine fluorescence was analyzed using the FITC channel.
FACS pH analysis.
To determine the pH dependency using FACS, one group of MCF7 cells were incubated in the presence of 50 mM NH4Cl for 1 h before the start of the experiment. All subsequent steps for this group were carried out in the presence of 50 mM NH4Cl. The cells were detached by 15 min incubation in Accutase. After washing in FACS buffer the cells were seeded in a 96-well rounded bottom microtiter plate (Corning Inc., Cat# 3799) to a final density of 3 × 105 and used immediately. The cells were incubated in the presence of 3.43 nM of antibody-Citrine fusion protein for 30 min on ice to allow binding, but prevent internalization. The cells were subsequently washed in FACS buffer to remove unbound antibody. The cells were measured immediately and again after 2 h with a FACS canto II (BD Biosciences). Citrine fluorescence was analyzed using the FITC channel.
Abbreviations
| AB | = | antibody |
| GFP | = | green fluorescent protein |
| YFP | = | yellow fluorescent protein |
| eGFP | = | enhanced green fluorescent protein |
| IGF-1R | = | type 1 insulin-like growth factor receptor |
| DIG | = | digoxigenin |
| (G4S)2 | = | Glycine-Glycine-Glycine-Glycine-Serine |
| IgG | = | immunoglobulin class G |
| scFv | = | single chain variable regions |
| N-terminus | = | amino terminus |
| C-terminus | = | carboxy terminus |
| 6xHis | = | Histidine-Histidine-Histidine-Histidine-Histidine-Histidine |
| HC | = | antibody heavy chain |
| LC | = | antibody light chain |
| LeY | = | Lewis-Y carbohydrate |
| SEC | = | size exclusion chromatography |
Figures and Tables
Figure 1 Composition of antibody-Citrine fusion protein formats. Three general formats were generated: either Citrine was added to the C-termini of the light chains (LC) (top row) or to the C-termini of the heavy chains (HC) (middle row) or Citrine was inserted between two single chain Fvs (scFvs) (bottom row). The two different heavy chain formats both use the “knobs-into-holes” technology. One format uses protein complementation (PCA) to generate a full Citrine molecule from two halves, the other format has full length (FL) Citrine at the C-terminus of only one HC. The format with two scFvs has a Hexa-Histidine tag (6x-His) for purification. The variable antibody regions used targeted either the DIG hapten (anti-DIG) or the IGF-1 receptor (anti-IGF-1R).
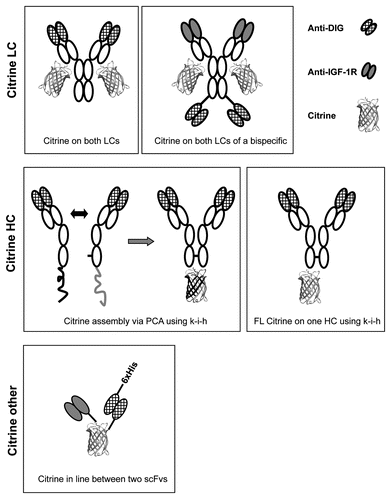
Figure 2 Expression and purification of antibody-Citrine fusion proteins. An example for Protein A purification (A) of a DIG-specific antibody with Citrine attached to the C-termini of the light chains from culture supernatants. Grey line pH, brown line conductivity, blue line 280 nm, red line 527 nm. The same protein was subjected to size exclusion chromatography (SEC) purification (B) using a Superdex200 column. Fractions collected between both red lines were pooled and used for further analysis. Blue line 280 nm, brown line conductivity, grey line pH. Analysis of the purified product (blue line) by analytical SEC (C) using a Superdex200 column at 280 nm shows the purity (98.7%) of the antibody-Citrine fusion protein. Reference mix (black line) with (from left) 670 kDa, 158 kDa, 44 kDa, 17 kDa and 1.3 kDa.
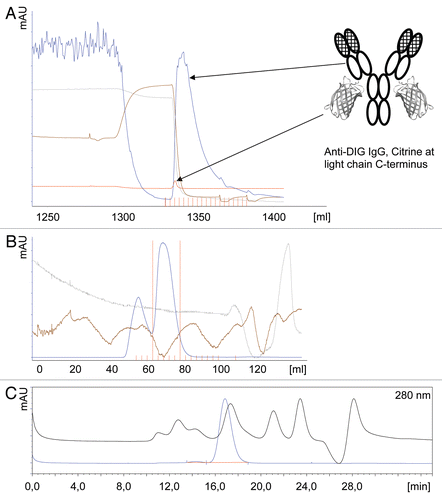
Figure 3 Antibody-Citrine fusion proteins retain the full binding specificity and affinity of their parent antibodies. Surface plasmon resonance (SPR) analysis was performed with the antibody formats indicated. All proteins were captured by a anti-human IgG antibody to the chip. To determine the binding affinities of the different formats, mono-digoxygenated myoglobin was injected in increasing concentrations. KD values were obtained using non-linear curve fitting (Langmuir). Affinities for each format are indicated in nM. Further information can be found in . Depictions of the molecules are described in .
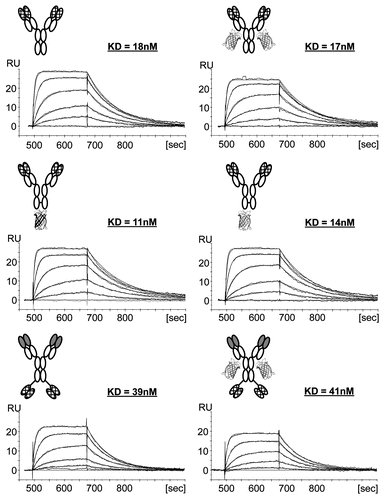
Figure 4 All antibody-Citrine fusion protein variants show pH-dependent Citrine fluorescence. 50 nM of the different antibody-Citrine fusion protein formats were incubated in buffer adjusted to the pH indicated. The sample was excited at 516 nm and the fluorescence was determined at 529 nm. Fluorescence was measured and normalized for all fusion proteins to generate a signal independent of the number of Citrine molecules. An anti-DIG IgG without Citrine was used as a negative control. Depictions of the molecules are described in .
Figure 5 Antibody-Citrine fusion proteins to detect cell binding and internalization. Cells were incubated on ice in the presence of an anti-IGF-1R antibody-Citrine fusion protein for 2 h. The cells were either fixed immediately after this period (upper part) or after overnight incubation at 37°C (lower part). A Cy3-labeled human kappa light chain (κLC) specific antibody was used to detect the antibody-Citrine fusion protein. Citrine fluorescence was measured in the FITC channel. Some sub-cellular structures show predominantly Citrine signal (Arrow).
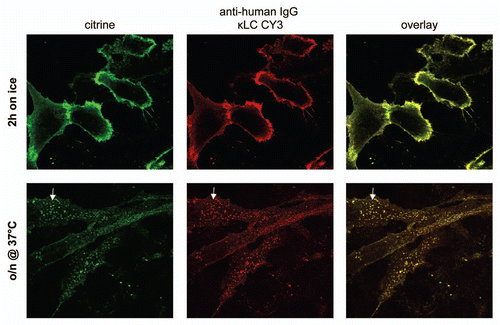
Figure 6 Antibody-Citrine fusion proteins to detect binding to live cells. Cells were incubated in the presence of antibody-Citrine fusion proteins and analyzed by FACS. The left hand parts (A–C) show detection of bound antibodies by APC-labeled secondary antibodies (Sec. AB), the left hand parts show detection by means of the Citrine fluorescence (A–C). The antibody fusion proteins analyzed were (A) full-size anti-IGF-1R antibodies (IGF-1R WT Citrine) and the (B) LeY carbohydrate (LeY WT Citrine), both with Citrine attached to the C-termini of their light chains. A small antibody format (scFv Citrine) with Citrine in line between a scFv specific for the IGF-1 receptor and the DIG hapten was used (C) as an example for a monovalent binding protein. Untreated cells (Cells) were used as the control in all experiments.
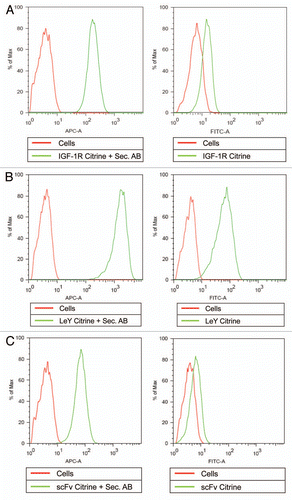
Figure 7 Antibody-Citrine fusions to track acidification in living cells. Cells incubated with or without NH4Cl (NH4) were treated on with an IGF-1 receptor specific antibody carrying Citrine on the C-termini of the light chains (IGF-1R Citrine) for 1 hour on ice. One fraction of both cell populations was analyzed in the FITC channel immediately (A) to monitor Citrine fluorescence. The other fraction was analyzed after 2 h at 37°C (B) to ensure antibody internalization. Untreated cells (Cells) were used as control.
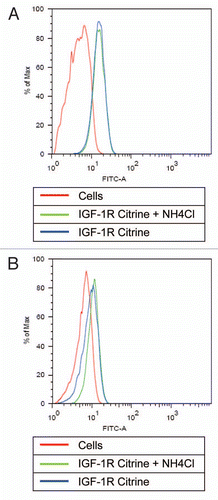
Table 1 Overview of produced recombinant antibodies
Table 2 Overview of binding abilities of DIG-specific Citrine formats
Additional material
Download Zip (740.6 KB)References
- Chalfie M, Tu Y, Euskirchen G, Ward WW, Prasher DC. Green fluorescent protein as a marker for gene expression. Science 1994; 263:802 - 805
- White J, Stelzer E. Photobleaching GFP reveals protein dynamics inside live cells. Trends Cell Biol 1999; 9:61 - 65
- Patterson GH, Knobel SM, Sharif WD, Kain SR, Piston DW. Use of the green fluorescent protein and its mutants in quantitative fluorescence microscopy. Biophys J 1997; 73:2782 - 2790
- Niswender KD, Blackman SM, Rohde L, Magnuson MA, Piston DW. Quantitative imaging of green fluorescent protein in cultured cells: comparison of microscopic techniques, use in fusion proteins and detection limits. J Microsc 1995; 180:109 - 116
- Song L, Hennink EJ, Young IT, Tanke HJ. Photobleaching kinetics of fluorescein in quantitative fluorescence microscopy. Biophys J 1995; 68:2588 - 2600
- Tsien RY. The green fluorescent protein. Annu Rev Biochem 1998; 67:509 - 544
- Casey JL, Coley AM, Tilley LM, Foley M. Green fluorescent antibodies: novel in vitro tools. Protein Eng 2000; 13:445 - 452
- Griep RA, van Twisk C, van der Wolf JM, Schots A. Fluobodies: green fluorescent single-chain Fv fusion proteins. J Immunol Methods 1999; 230:121 - 130
- Morino K, Katsumi H, Akahori Y, Iba Y, Shinohara M, Ukai Y, et al. Antibody fusions with fluorescent proteins: a versatile reagent for profiling protein expression. J Immunol Methods 2001; 257:175 - 184
- Olichon A, Surrey T. Selection of genetically encoded fluorescent single domain antibodies engineered for efficient expression in Escherichia coli. J Biol Chem 2007; 282:36314 - 36320
- Veettil S, Budisa N, Jung G. Photostability of green and yellow fluorescent proteins with fluorinated chromophores, investigated by fluorescence correlation spectroscopy. Biophys Chem 2008; 136:38 - 43
- Cubitt AB, Woollenweber LA, Heim R. Understanding structure-function relationships in the Aequorea victoria green fluorescent protein. Methods Cell Biol 1999; 58:19 - 30
- Ormo M, Cubitt AB, Kallio K, Gross LA, Tsien RY, Remington SJ. Crystal structure of the Aequorea victoria green fluorescent protein. Science 1996; 273:1392 - 1395
- Griesbeck O, Baird GS, Campbell RE, Zacharias DA, Tsien RY. Reducing the environmental sensitivity of yellow fluorescent protein. Mechanism and applications. J Biol Chem 2001; 276:29188 - 29194
- Heikal AA, Hess ST, Baird GS, Tsien RY, Webb WW. Molecular spectroscopy and dynamics of intrinsically fluorescent proteins: coral red (dsRed) and yellow (Citrine). P Natl Acad Sci USA 2000; 97:11996 - 12001
- Sbalzarini IF, Mezzacasa A, Helenius A, Koumoutsakos P. Effects of organelle shape on fluorescence recovery after photobleaching. Biophys J 2005; 89:1482 - 1492
- Li E, Placone J, Merzlyakov M, Hristova K. Quantitative measurements of protein interactions in a crowded cellular environment. Anal Chem 2008; 80:5976 - 5985
- Dundr M, McNally JG, Cohen J, Misteli T. Quantitation of GFP-fusion proteins in single living cells. J Struct Biol 2002; 140:92 - 99
- Hirschberg K, Miller CM, Ellenberg J, Presley JF, Siggia ED, Phair RD, et al. Kinetic analysis of secretory protein traffic and characterization of golgi to plasma membrane transport intermediates in living cells. J Cell Biol 1998; 143:1485 - 1503
- Holtke HJ, Seibl R, Burg J, Muhlegger K, Kessler C. Non-radioactive labeling and detection of nucleic acids. II. Optimization of the digoxigenin system. Biol Chem Hoppe Seyler 1990; 371:929 - 938
- Kessler C, Holtke HJ, Seibl R, Burg J, Muhlegger K. Non-radioactive labeling and detection of nucleic acids. I. A novel DNA labeling and detection system based on digoxigenin: anti-digoxigenin ELISA principle (digoxigenin system). Biol Chem Hoppe Seyler 1990; 371:917 - 927
- Ridgway JB, Presta LG, Carter P. ‘Knobs-into-holes’ engineering of antibody CH3 domains for heavy chain heterodimerization. Protein Eng 1996; 9:617 - 621
- Merchant AM, Zhu Z, Yuan JQ, Goddard A, Adams CW, Presta LG, et al. An efficient route to human bispecific IgG. Nat Biotechnol 1998; 16:677 - 681
- Nyfeler B, Michnick SW, Hauri HP. Capturing protein interactions in the secretory pathway of living cells. P Natl Acad Sci USA 2005; 102:6350 - 6355
- Nyfeler B, Hauri HP. Visualization of protein interactions inside the secretory pathway. Biochem Soc Trans 2007; 35:970 - 973
- Riedemann J, Macaulay VM. IGF1R signalling and its inhibition. Endocr Relat Cancer 2006; 13:33 - 43
- Chitnis MM, Yuen JS, Protheroe AS, Pollak M, Macaulay VM. The type 1 insulin-like growth factor receptor pathway. Clin Cancer Res 2008; 14:6364 - 6370
- Beuger V, Kunkele KP, Koll H, Gartner A, Bahner M, Burtscher H, et al. Short-hairpin-RNA-mediated silencing of fucosyltransferase 8 in Chinese-hamster ovary cells for the production of antibodies with enhanced antibody immune effector function. Biotechnol Appl Biochem 2009; 53:31 - 37
- Brinkmann U, Pai LH, FitzGerald DJ, Willingham M, Pastan I. B3(Fv)-PE38KDEL, a single-chain immunotoxin that causes complete regression of a human carcinoma in mice. P Natl Acad Sci USA 1991; 88:8616 - 8620
- Pastan I, Lovelace ET, Gallo MG, Rutherford AV, Magnani JL, Willingham MC. Characterization of monoclonal antibodies B1 and B3 that react with mucinous adenocarcinomas. Cancer Res 1991; 51:3781 - 3787
- Brinkmann U, Reiter Y, Jung SH, Lee B, Pastan I. A recombinant immunotoxin containing a disulfide-stabilized Fv fragment. P Natl Acad Sci USA 1993; 90:7538 - 7542
- Reiter Y, Brinkmann U, Jung SH, Pastan I, Lee B. Disulfide stabilization of antibody Fv: computer predictions and experimental evaluation. Protein Eng 1995; 8:1323 - 1331
- Burtrum D, Zhu Z, Lu D, Anderson DM, Prewett M, Pereira DS, et al. A fully human monoclonal antibody to the insulin-like growth factor I receptor blocks ligand-dependent signaling and inhibits human tumor growth in vivo. Cancer Res 2003; 63:8912 - 8921
- Gong Y, Yao E, Shen R, Goel A, Arcila M, Teruya-Feldstein J, et al. High expression levels of total IGF-1R and sensitivity of NSCLC cells in vitro to an anti-IGF-1R antibody (R1507). PLoS One 2009; 4:7273
- Maxfield FR, McGraw TE. Endocytic recycling. Nat Rev Mol Cell Biol 2004; 5:121 - 132
- Ohkuma S, Poole B. Fluorescence probe measurement of the intralysosomal pH in living cells and the perturbation of pH by various agents. P Natl Acad Sci USA 1978; 75:3327 - 3331
- Michnick SW, Remy I, Campbell-Valois FX, Vallee-Belisle A, Pelletier JN. Detection of protein-protein interactions by protein fragment complementation strategies. Methods Enzymol 2000; 328:208 - 230