Abstract
Recombinant antibodies may be engineered to obtain improved functional properties. Functional mapping of the residues in the binding surfaces is of importance for predicting alterations needed to yield the desired properties. In this investigation, 17 single mutation mutant single-chain variable fragments (scFvs) of the anti-idiotypic antibody anti-TS1 were generated in order to functionally map amino acid residues important for the interaction with its idiotype TS1. Residues in anti-TS1 determined to be very important for the interaction were identified, Y32L, K50L, K33H, and Y52H, and they were distributed adjacent to a centrally located hydrophobic area, and contributed extensively to the interaction energy (≥2.5 kcal/mol) in the interaction. Quantitative ELISA assays, BIAcore technologies and three-dimensional surface analysis by modeling were employed to visualize the consequences of the mutations. The expression levels varied between 2 - 1,800 nM as determined by ELISA. All the 17 scFvs displayed higher dissociation rates (60 - 1,300 times) and all but two of them also faster association rates (1.3 - 56 times). The decrease in affinity was determined to be 1.6 - 12,200 times. Two of the mutants displayed almost identical affinity with the wild type anti-TS1, but with a change in both association and dissociation rates. The present investigation demonstrates that it is possible to generate a large panorama of anti-idiotypic antibodies, and single out a few that might be of potential use for future clearing and pre-targeting purposes of idiotypic-anti-idiotypic interactions.
Introduction
Numerous possible applications for monoclonal antibodies (mAb) and antibody fragments in biotechnology, diagnostics and therapy exist. As treatments for malignant tumors, administered radiolabeled mAbs may form complexes with selected tumor antigens and subsequently inhibit growth of the tumor cells.Citation1 A major limitation with this approach is the remaining, non-targeting radiolabeled mAbs in the circulation and their uptake in normal tissues, which causes unintended exposure. Different methods to clear the primary, i.e., idiotypic (Id), mAb from the circulation have been presented and one is to use a secondary, i.e., anti-idiotypic (anti-Id) mAb.Citation2–Citation4 The mAbs included in this study are the Id TS1, which targets cytokeratin 8 (CK8) and the anti-Id, which targets TS1. The anti-Id anti-TS1 has been used successfully to clear the circulationCitation5,Citation6 of excess of the tumor localizing mAb TS1.Citation7,Citation8 It has also been shown that pre-formed complexes of the single-chain variable fragment (scFv) variants of both TS1 and anti-TS1 result in a higher tumor uptake of TS1 scFv both in vitro and in vivo.Citation9
Many antibodies produced for therapy must be genetically engineered to achieve high expression levels, improved folding, high solubility, reduced HAMA, increased stability in vivo and optimal binding. A detailed description of the interactions between an Id mAb paratope, its antigen and an anti-Id mAb may enable more effective engineering since the effect of mutations intended to improve the properties and attributes of mAbs may be more reliably predicted.Citation10–Citation14
Many research groups have analyzed the structural basis of antibody-antigen recognition.Citation11,Citation15–Citation17 It has been demonstrated that amino acid residues involved in establishing the complementary determining region (CDR) conformations are less frequently involved in antigenic interactions compared to residues in other positions in the CDR. Correlations between antigen type and the length and amino acid composition of the CDR and between type of antigen and residue position involved in the binding have been demonstrated.Citation18–Citation22 The length of the CDR influences its shape and binding to antigen through formation of pockets, grooves or flat surfaces. In the antigen combining site (ACS) of antibodies, there are usually between 12–20 amino acid residues that contribute to the interaction between the antibody and its antigen and, if any of these interacting residues are altered, the effect is most likely a reduced affinity.Citation16,Citation19 The interaction between an antibody and its antigen, however, is mainly established by three to six very important and crucial residues.
Interaction kinetics between two proteins can be described by their association and dissociation rates, but it is difficult to predict the effects of particular mutations, especially on the dissociation rate, because a precise knowledge of the shape and chemistry of the binding interface would be required. The association rate of two proteins can be enhanced by favorable long-range electrostatic attractions that guide proteins towards each other by affecting the orientation and the time the two proteins are in close proximity.Citation23–Citation25 The association rate is therefore easier to predict and, if the desired result of a mutation is to change the association rate, residues affecting the electrostatic interactions outside the paratope should most likely be altered. In vivo, antibody diversity is created by a number of methods, including amino acid substitutions that are more frequent at certain positions,Citation26 insertion and deletion of segmentsCitation27 and variation in the association between the heavy and the light chain variable region.Citation28,Citation29 In order to regulate the function of therapeutic proteins, such as antibodies, control of both the association and dissociation rates is important.
Site-directed mutagenesis is frequently used both to regulate affinity, as well as other properties, and analyze the binding surfaces of antibodies and antigens.Citation11,Citation13,Citation14,Citation22,Citation30 The effect of a certain mutation can be studied by methods such as enzyme-linked immunosorbent assay (ELISA)Citation30,Citation31 and optical biosensors based on surface plasmon resonance (SPR).Citation32
In this investigation, we functionally mapped amino acid residues important for the anti-TS1 interaction with TS1 by performing site-directed mutagenesis of a scFv construct of anti-TS1. Targets for mutagenesis were selected based on two previous studies that included amino acid residue interaction statistics, homology modeling and chemical modification of anti-TS1 and TS1.Citation21,Citation33 SPR-based BIAcore systems were used to study the effects of the mutations on interaction kinetics and affinity.
Results
Mutagenesis, purification, quantification and SDS-PAGE.
The sequences of the 17 anti-TS1scFv mutants cloned in pET 26 were confirmed to be correct by sequencing of the insert in both directions. The expression level of the anti-TS1 scFv mutants varied and the concentration of scFv obtained from the media ranged from 2–1,800 nM as determined by quantitative ELISA. SDS-PAGE analyses of two of the ion exchange purified anti-TS1 mutants and the purified wild-type anti-TS1 scFv used as a standard in the quantitative ELISA are shown in .
Biacore® studies.
BIAcore studies were performed on the wild type and the 17 mutant scFvs. The kd and ka constants were determined using the BIAevaluation 3.2 (BIAcore, Uppsala, Sweden); presents a typical plot for one of the anti-TS1 scFv mutants. A plot of rate constants for the association, ka, versus dissociation, kd, of the mutant anti-TS1 scFv, wild-type anti-TS1 scFv and the anti-TS1 IgG is presented in .
The negative control demonstrated that the remaining impurities in the samples after ion exchange chromatography purification did not affect the BIAcore measurements. All dilutions with measurable concentration displayed the same kinetic rates. The anti-TS1 scFv wild type demonstrated a ka value of 1.15 × 105 M−1s−1 and a kd value of 2.2 × 10−6 s−1. Compared to the anti-TS1 IgG,Citation6 with a ka value of 2.2 × 105 M−1s−1 and a kd value of 2.6 × 10−6 s−1, the affinity of the scFv was reduced 1.6 times, although, as the dissociation rate of the anti-TS1 scFv was above 10−5 s−1, which corresponds to a complex half-life of more than 13 h, the dissociation time used in the experimental set up (10 min) was too short to accurately determine the dissociation rate constant.
All 17 scFv mutants displayed higher dissociation rates (60–1,300 times) and all except D101 and Y96 also demonstrated faster association rates (1.3–56 times) compared to the wild type. Three mutated residues, D100aAH and D101AH and Y96AL had a more explicit negative effect on the binding and between 800–12,200 times lower affinities were calculated for these mutants (). Seven additional mutations, Y32AH, K33AH and Y52HA, as well as Y32AL, H34AL, K50AL and V94AL, also demonstrated a high affinity reduction (between 48–164 times) and the nine other mutants displayed affinity reductions in the range of 1.6–17 times. Four of these nine mutants (Y32AH, D56AH and H30aAL and R54AL) displayed increased association rates (36–56 times) and increased dissociation rates (78–1,790 times). As shown in , the mutations of D56AH and R54AL demonstrated similar affinity as the wild type (iso-affinity).
Three-dimensional structures.
The homology model of anti-TS1 ACS indicated that the molecule displays a surface that is mainly polar, with a centrally located region that is mainly hydrophobic. This hydrophobic region involves CDR3 of the heavy chain and also, to some extent, the CDR3 of the light chain. The model also showed that the anti-TS1 ACS displays several positively and a few negatively charged areas scattered over the surface. A front view of the three-dimensional model of anti-TS1 colored by polarity and charge is presented in . None of the 17 mutations resulted in any visible structural changes in the model. The surface of the model was also colored by accessibility (data not shown) and if approximately 20% or more of a residue was visible, then we considered it as accessible. The accessibility of each mutated residue in the homology model and the frequency of involvement in antigen binding for the selected residue positions,Citation22 as well as the affinity changes determined by BIAcore, are displayed in . No homologies between the epitope of CK8Citation34 and the distribution of functionally important residues in the three-dimensional structure of anti-TS1 ACS were seen.
Discussion
In antibody engineering, the ability to functionally map the interaction site on antibodies is very important. Detailed descriptions of interactions are necessary to facilitate understanding of the manner in which antibodies bind to antigenic surfaces and predict the effects of mutations.Citation19–Citation22,Citation30,Citation33,Citation35 A system involving three molecular partners, e.g., an antibody that interacts with an antigen and an anti-Id antibody that binds competitively, is complex to study. Some anti-Id antibodies mimic the structure of the antigen,Citation36,Citation37 some demonstrate functional mimicryCitation38,Citation39 and some show no resemblance to the antigen.Citation40 Anti-Id anti-TS1 antibodies that bind the Id anti-CK8 TS1 have shown potential in preclinical experiments as clearing agents. When administered in a scFv format simultaneously with TS1 scFv, anti-TS1 contributes to an increased accumulation of TS1 in the tumor.Citation9
We have previously studied interactions between the therapeutic mAbs TS1, its anti-Id antibody, anti-TS1 and its target antigen CK8. We identified the interacting surfaces of TS1 and CK8 and we showed mutual inhibition of CK8 and anti-TS1 to binding with TS1, although they do not show any obvious structural similarities.Citation4,Citation10,Citation33,Citation34 In this study, we used site-directed mutagenesis of anti-TS1 scFv to identify amino acid residues that were functionally important for the anti-TS1 interaction with TS1 to further improve understanding of this complex system. Amino acid residues in anti-TS1 were selected for mutagenesis based on a computer model that displayed accessibility of the residues and the effects of chemical modification of anti-TS1 alone or complexed with TS1.Citation33 We also based the selection of residues to be mutated on statistical calculationsCitation21 of the probability of certain amino acid residues in the CDRs of anti-TS1 to be involved in the interaction with TS1.
The properties of the IgG and scFv versions of the wild-type antibody as measured on the BIAcore were very similar. The reason for the lack of increased functional affinity of the IgG form based on its bivalent binding is most likely due to the stiff hinge region of this anti-TS1 IgG antibody, which makes it difficult for anti-TS1 to bind with both arms to immobilized TS1 in the BIAcore. We previously studied the formation of complexes between TS1 and anti-TS1 by electron microscopy and our results revealed that the complexes formed between TS1 and anti-TS1 were mainly ring shaped structures comprising four or more antibodies.Citation5
All 17 mutants demonstrated an increase in their dissociation rate constant (kd) of between 60–1,300 times, which probably occurred because we selected residues that were expected to have a negative effect on the binding. Fifteen mutants demonstrated an increase in their association rate constant (ka) of between 1.3–56 times, indicating that a truncation of these residues in the ACS to the less polar amino acid alanine had some advantage on the association. Due to this effect on binding, all analyzed residues should be considered important for the interaction with TS1 or stabilization of the structure of anti-TS1 or both.
Based on the observations in this and other studies, three residues, D101H, R24L and H34L, were considered structurally important. D101H and H34L had low accessibility in the homology model () and, according to Almagro,Citation22 these positions have low antigen binding frequency (). D101H is structurally important because of its interaction with the arginine at position 94 in the heavy chain, which establishes the structure required for antigen binding.Citation41 The arginine at position 24 probably forms an intramolecular salt bridge with D70, both of which are found in the light chain.Citation42 Vargas-Madrasso and Paz-Garcia demonstrated that position 34 in the light chain frequently participates in the interface by contacting residues in the heavy chain.Citation29 We therefore consider the role of H34L, which is not visible from the surface of anti-TS1, as structural ().
For the mutants D100aAH and Y96AL, the association rate constants were increased only slightly while the dissociation rate constant increased more than 1,000 times (). D100aH is accessible () and positioned close to the center of CDR3 with its side chain oriented towards the center of the combining site (antigen binding frequency 33%Citation22). The side chain of Y96L is also oriented towards the antigen binding site, but is less accessible and has an antigen binding frequency of 42%.Citation22 It has been demonstrated that both position 100aH and position 96L are involved in stabilization of the VH/VL interface.Citation29 The roles for D100aH and Y96L were therefore considered as probably both structurally important and important for the interaction with TS1.
The additional 12 mutated residues were considered important for the interaction since all but two residues (D56AH and R54AL, which displayed nearly iso-affinity with the wild type) contributed >1 kcal/mol to the interaction ( and ). Four of these 12 residues, Y32L, K50L, Y52H and K33H, were considered very important for the interaction because they contributed extensively to the interaction energy (≥2.5 kcal/mol) reducing the affinity between 48–164 times ( and ). With the exception of K33H, all of these residues are accessible in the homology model of anti-TS1 and the residue positions have an antigen binding frequency between 33–73% ( and ).
It has been demonstrated that 3–6 residues in the ACS contribute to most of the interaction energy and these important residues tend to cluster together in the center of the ACS.Citation35,Citation43 When a residue contributes to the binding, it must be protected from the solvent and therefore ACS usually contains hydrophobic regions that desolvate the interacting interface.Citation35 The homology model of anti-TS1 indicates that the molecule is mainly polar, but displays a centrally located region that is hydrophobic. This hydrophobic region involves CDR3 in the heavy chain, but also to some extent the CDR3 in the light chain. Residues Y32L, K50L, V94L and Y96L, as well as K33H, Y52H and D100aH, are positioned adjacent to this hydrophobic region ().
Collis et al. compared the residue distribution in CDR loops and loops of other proteins, and tyrosine (Y) and tryptophan (W) were demonstrated to be over-represented in CDR loops in antibodies that bind proteins, while cysteine (C), aspartic acid (D), glutamic acid (E), glycine (G), lysine (K), proline (P) and valine (V) were demonstrated to be significantly under-represented.Citation18 We have demonstrated that, besides Y and D, K and V in anti-TS1 are also important for the binding to TS1. Interestingly, lysine (K) and valine (V) are residues that are under-represented as specificity determining residues in antibodies,Citation22 but it has been demonstrated that approximately 50% of the binding energy derived from the antigen is attributed to the residues arginine (R), lysine (K), asparagine (N) and aspartic acid (D).Citation44 In our previous study, chemical modification indicated that K was not important for the interaction with TS1.Citation33 The reason for these different results could be explained by the fact that chemical modification adds a chemical group to the side chains while the alanine substitution reduces the side chain to a methyl group.Citation45
Many studies have demonstrated that the shape of the ACS varies with the antigen. Antibodies that bind protein antigen are usually flatter, while often a groove can be seen in antibodies that bind peptides.Citation21 The ACS of TS1 and anti-TS1 are mainly flat as expected; however, the light chain CDR1 is protruding in both antibodies and TS1 contains a groove that forms a C-shape around the CDR3 in the heavy chain.Citation33 In addition to this geometric fit, both the groove in TS1 and the protruding parts of anti-TS1 are partly hydrophobic, which make hydrophobic interactions possible between these sites.Citation10 Using chemical modification, it was demonstrated that histidine was important for TS1 interaction with anti-TS1 and not CK8 and the only histidine found in the ACS is located in this groove.Citation33 Interestingly, despite the fact that anti-TS1 and CK8 compete for the TS1 binding, no resemblance could be seen between the ACS of anti-TS1 and the epitope of CK8.
The model also displays several positively charged areas, and some negative, that are scattered over the surface. The surface of anti-TS1 was colored by electrostatic potential (data not shown) using the Swiss-Pdb, Deep View version 3.7 program, which illustrated the entire Fv region as positively charged, while TS1 is more negatively charged. If the desired result of a mutation is to increase the association rate, then it is most likely that the long range electrostatic interactions should be altered. Relatively fast association rates have been achieved for several protein complexes by optimizing the electrostatic complementarity of their binding sites.Citation46,Citation47 Replacing four residues with alanine as single-mutants, Y32H, D56H, H30aL and R54L, increased the association between 36–56 times; these residues are positioned in the periphery of the ACS of anti-TS1 (). For the Y32AH mutation (), the increased association could occur because the tyrosine side chain hides a large part of the positively charged residue R94H (and also to some extent D101H). Mutant anti-Id antibodies with similar affinity as the wild type, but with higher association and dissociation rate constants as mutants D56AH and R54AL, are expected to bind faster to TS1 in the circulation and compete differently with the antigen at the tumor site. This could possibly cause a further increase on the tumor uptake of TS1 scFv in pre-formed scFv complexes studied earlier.Citation9 The present work has functionally mapped the ACS of anti-TS1 and generated a large panorama of anti-Id antibodies with different interacting properties, which may help in the design of a system with improved clearing and tumor delivery.
Material and Methods
Hybridoma cell lines and monoclonal antibodies.
Hybridoma cell lines producing monoclonal antibody TS1,Citation48 which targets cytokeratin 8 (CK8) and its anti-Id, anti-TS1,Citation6 were cultured as described.Citation48 The mAbs were purified from cell culture media using a Protein G column (Pharmacia Biotech) and eluted with 0.1 M glycine/HCl buffer pH 2.3. Fractions were neutralized and stored at −20°C.
Site-directed mutagenesis.
Site-directed mutagenesis of 17 amino acid residues was performed on purified pET 26 plasmid with the anti-TS1scFv gene,Citation49 gene bank accession numbers AJ884574 (anti-TS1 heavy chain variable region) and AJ884575 (anti-TS1 light chain variable region), using Quick-Change Site-Directed Mutagenesis kit as described by the manufacturer (Stratagen). Four tyrosine (Y), three histidine (H), two lysine (K), three aspartic acid (D), two arginine (R), one tryptophan (W), one phenylalanine (F) and one valine (V) residues were selected for mutagenesis based mainly on the chemical modification results (Y, R and D), surface exposureCitation33 and statistical importance of actual amino acid position in the Fv part (K, H, W, V and F).Citation21 Each of the 17 amino acids selected for mutagenesis was replaced with the small and hydrophobic amino acid alanine, using mutagenic primers, designed according to the manufacturer (Stratagen, La Jolla, CA, USA). Mutated plasmids were systematically verified by sequencing of the antibody fragments on both strands using T7 promotor and T7 terminator primers (Novagen). For the sequencing, the Big Dye Terminator cycle sequencing kit (PE Biosystems) and the ABI Prism 310 Genetic Analyzer (PE Biosystems) were used. Each mutated plasmid was transformed to the E. coli strain Rosetta DE3 (Novagen).
Expression and purification.
Expression of the scFv was performed by culturing the transformed E. coli Rosetta DE3 strains in 400 ml LB with kanamycin 30 µg/ml and chloramphenicol 75 µg/ml for approximately 16 h at 30°C to an OD600 value between 3–3.5. The expression vector with the scFv gene contains a pelB leader to enable transportation of the scFv to the periplasmic space. Isopropyl β-D-1-thiogalactopyranoside (IPTG) was added to a final concentration of 1 mM to induce expression and glycine and Triton X-100 were added to a final concentration of 2 and 1% respectively, to release the scFv into the culture media.Citation50 The expression of scFv into the media was performed at 20°C overnight. Cell cultures were centrifuged and culture supernatant were obtained, concentrated approximately 200 times and dialysed against 20 mM Na-phosphate buffer pH 6.5. The dialysed samples (corresponding to approximately 200 ml culture supernatant) were filtered through a 0.45 µm filter (Acrodisc syringe filter, PALL, Gelman laboratory), applied to a cation exchange chromatography column (Hi Trap Sp HP, Amersham Biosciences) in 20 mM Na-phosphate buffer pH 6.5 and eluted with a continuous NaCl gradient.
SDS-PAGE.
The cation exchange chromatography purified wild type and scFv mutants, as well as Ni-NTA affinity and cation exchanged purified wild-type anti-TS1 scFv, used in quantitative ELISA were concentrated five times with trichloroacetic acid and analysed on SDS-PAGE (4% stacking and 12% separating gel) performed according to Laemmli.Citation51 The SDS-PAGE gels were stained with Coomassie brilliant blue.
Quantitative ELISA.
Microtiter plates (Nunc) were coated overnight at 4°C with 100 µl/well of polyclonal goat anti mouse Fab (SIGMA) in 50 mM Tris pH 7.4, 0.5 M NaCl (TBS) at a concentration of 2.5 µg/ml. The plates were washed three times for five minutes with TBS, pH 7.4 with 0.05% Tween 20 (TBST), before adding the cation exchange chromatography purified scFv mutants in duplicate, undiluted and serially diluted 1:3 in ten steps. Sample incubation was performed overnight at 4°C. The plates were washed with TBST as before and polyclonal goat anti-mouse Fab conjugated with alkaline phosphatase (SIGMA) at a concentration of 2.5 µg/ml was added and incubated over night at 4°C. After washing as before, the plates were developed with 3 mM p-nitrophenyl phosphate in 50 mM 2-amino-2-metyl-1-propanol, 1 mM MgCl2 and pH 10.0. The absorbance was read at 405 nm and the samples were quantified using a standard curve of the wild type anti-TS1 scFv with a measuring range from 0.4–40 nM.
Kinetic studies using BIAcore®.
A BIAcore® 2000 with the BIAcore 2000 control software version 3.2 (BIAcore, Uppsala, Sweden) was used for the kinetic studies. For the evaluation of the sensograms at concentrations higher than 25 nM, the Langmuir model was used. For concentrations below 25 nM, binding with the mass transfer model was used. Local Rmax was used to correct for the bulk effect seen in some samples. For all kinetic measurements, the CM5 sensor chip and the amine coupling kit with 1-ethyl-3-(3-dimetylaminopropyl) carbodiimide (EDC) and N-hydroxysuccinimide (NHS) was used. Immobilization of TS1 to approximately 1,500 resonance units (RU) on the sensor chip CM5 was performed with 30 µg TS1 IgG/ml Na-acetate buffer pH 4.5 and standard procedures using the amine coupling kit. The samples (wild type and the 17 mutant scFvs) were dialyzed against running buffer and, because of large variation in concentrations, all samples were analyzed undiluted and diluted 1:3, 1:9, 1:27 and 1:81 with a continuous flow rate of 30 µl/min in HBS-EP pH 7.4 (Hepes 10 mM, NaCl 150 mM, EDTA 3.4 mM, P20 surfactant 0.05%) as running buffer. A negative control with ion exchange purified culture supernatant from E. coli with pET 26b without a scFv insert was included in the experimental setup to study the effect of impurities in the BIAcore system. The association and dissociation time between immobilized TS1, the wild type and the 17 mutant scFvs (cation exchange chromatography purified) were measured for 180 seconds and 600 seconds, respectively. The changes in free energy (ΔΔG = kcal/mol) for each anti-TS1 mutant interacting with TS1 were calculated using the equilibrium dissociation constant KD [kd/ka (M)], the ideal gas constant R (1.989 cal/Kmol), the temperature T (298 K) and the following equation ΔΔG= (-RTlnKD)mutant − (-RTlnKD)wild type.
Surface analysis.
A three-dimensional model of the variable region (Fv) of anti-TS1 wild type was generated as described previously.Citation33 Using the Deep View version 3.7,Citation52 the 17 anti-TS1 mutations were made in the model, energy minimized and the modeled mutant under investigation was compared to the wild type model. The same software was also used to color the surface of the anti-TS1 wild type homology model by polarity and charge and by accessibility.
Abbreviations
| ACS | = | antigen combining site |
| CDR | = | complementary determining region |
| CK | = | cytokeratin |
| Fr | = | frame |
| ELISA | = | enzyme-linked immunosorbent assay |
| ka | = | association rate constant |
| kd | = | dissociation rate constant |
| mAb | = | monoclonal antibody |
| scFv | = | single chain fragment variable |
Figures and Tables
Figure 1 SDS PAGE analysis of 2 of the ion exchange purified anti-TS1 mutants and the purified wild type anti-TS1 scFv used as standard in the quantitative ELISA. Lane 1: anti-TS1 scFv wild type 28 kDa (purified for the quantitative ELISA). Lane 2: anti-TS1 scFv mutant (low concentration approximately 10 nM). Lane 3: anti-TS1 scFv mutant (high concentration approximately 100 nM).

Figure 2 A representative kinetic plot, Ru signal versus sec, for one anti-TS1 scFv mutant (T97) at different concentrations (1,068 nM, 356 nM, 119 nM, 40 nM and 13 nM) by BIAcore as described (Langmuir model with local Rmax was used).
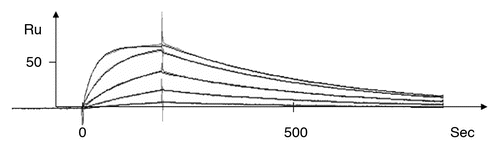
Figure 3 A plot of the association rate and dissociation rate constants of the 17 mutants. The wild-type anti-TS1 scFv and the anti-TS1 IgG demonstrating that all mutants display a higher dissociation rate constant (60–1,300) and 15 of the mutants a higher association rate constant (1.3–56) than the wild type scFv. Above and to the right in the figure, the affinity constant (KA) is presented by numbers and displayed as diagonal lines in the plot. The affinity for the mutants was reduced between 1.6–12,200 times.
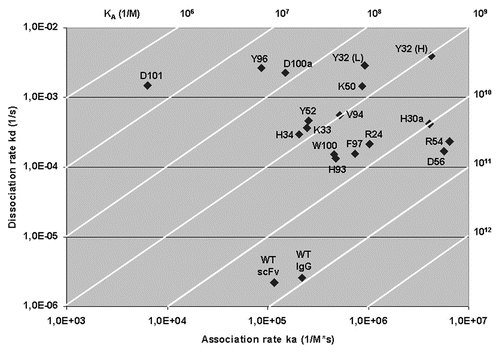
Figure 4 A three-dimensional model of anti-TS1 viewing the complementary determining regions (CDR) and antigen binding surface from the front. The upper part of the model represents the CDR of the heavy chain and the lower part the light chain. Grey color represents hydrophobic areas, yellow color represents polar parts, blue represent positive charges and red represent negative charges. Accessible residues are depicted with circles. Amino acids within thick black circles resulted in a substantial reduction (>2.5 kcal/mol) in binding energy (2.6–3.9 kcal/mol). Amino acids within thin black circles resulted in a moderate reduction in binding energy (1.6–2.3 kcal/mol). Amino acids with dashed circles resulted in a low reduction in binding energy (0.2–0.3 kcal/mol). One letter abbreviation of amino acids is used.
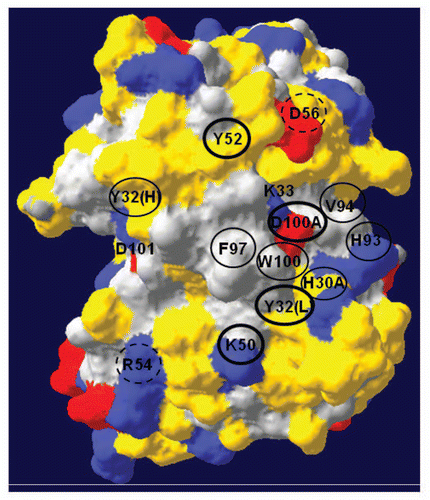
Table 1 For each amino acid residue position selected for mutagenesis, the table presents the approximate relative accessibility, the reduction in binding energy, the affinity reduction and the antigen binding frequency for antibodies that bind protein antigensCitation22
Acknowledgements
This research was supported by Swedish Cancer Research Council (grants number: 4445) and Karlstad University, Sweden.
References
- Milenic DE, Brady ED, Brechbiel MW. Antibody-targeted radiation cancer therapy. Nat Rev Drug Discov 2004; 3:488 - 499
- Sandstrom P, Johansson A, Ullen A, Rathsman S, Riklund-Ahlstrom K, Stigbrand T. Idiotypic-anti-idiotypic antibody interactions in experimental radioimmunotargeting. Clin Cancer Res 1999; 5:3073 - 3078
- Sharkey RM, Boerman OC, Natale A, Pawlyk D, Monestier M, Losman MJ, et al. Enhanced clearance of radiolabeled murine monoclonal antibody by a syngeneic anti-idiotype antibody in tumor-bearing nude mice. Int J Cancer 1992; 51:266 - 273
- Ullen A, Ahlstrom KR, Heitala S, Nilsson B, Arlestig L, Stigbrand T. Secondary antibodies as tools to improve tumor to non tumor ratio at radioimmunolocalisation and radioimmunotherapy. Acta Oncol 1996; 35:281 - 285
- Johansson A, Erlandsson A, Eriksson D, Ullen A, Holm P, Sundstrom BE, et al. Idiotypic-anti-idiotypic complexes and their in vivo metabolism. Cancer 2002; 94:1306 - 1313
- Ullen A, Sandstrom P, Ahlstrom KR, Sundstrom B, Nilsson B, Arlestig L, et al. Use of anticytokeratin monoclonal anti-idiotypic antibodies to improve tumor:nontumor ratio in experimental radioimmunolocalization. Cancer Res 1995; 55:5868 - 5873
- Eriksson D, Joniani HM, Sheikholvaezin A, Lofroth PO, Johansson L, Riklund Ahlstrom K, et al. Combined low dose radio- and radioimmunotherapy of experimental HeLa Hep 2 tumours. Eur J Nucl Med Mol Imaging 2003; 30:895 - 906
- Riklund KE, Makiya R, Sundstrom B, Back O, Henriksson R, Hietala SO, et al. Inhibition of growth of HeLa cell tumours in nude mice by 125I-labeled anticytokeratin and antiPLAP monoclonal antibodies. Anticancer Res 1991; 11:555 - 560
- Jafari R, Holm P, Sandegren J, Stigbrand T, Sundstrom BE. Localization of complexed anti-cytokeratin 8 scFv TS1-218 to HeLa HEp-2 multicellular tumor spheroids and experimental tumors. Cancer Biotherapy and Radiopharmaceuticals 2010; 25:455 - 463
- Holm P, Jafari R, Sundstrom BE. Functional mapping and single chain construction of the anti-cytokeratin 8 monoclonal antibody TS1. Mol Immunol 2007; 44:1075 - 1084
- Iacob RE, Keck Z, Olson O, Foung SK, Tomer KB. Structural elucidation of critical residues involved in binding of human monoclonal antibodies to hepatitis C virus E2 envelope glycoprotein. Biochim Biophys Acta 2008; 1784:530 - 542
- Jermutus L, Honegger A, Schwesinger F, Hanes J, Pluckthun A. Tailoring in vitro evolution for protein affinity or stability. Proc Natl Acad Sci USA 2001; 98:75 - 80
- Reichmann D, Rahat O, Albeck S, Meged R, Dym O, Schreiber G. The modular architecture of protein-protein binding interfaces. Proc Natl Acad Sci USA 2005; 102:57 - 62
- Worn A, Pluckthun A. Stability engineering of antibody single-chain Fv fragments. J Mol Biol 2001; 305:989 - 1010
- Braden BC, Poljak RJ. Structural features of the reactions between antibodies and protein antigens. FASEB J 1995; 9:9 - 16
- Davies DR, Cohen GH. Interactions of protein antigens with antibodies. Proc Natl Acad Sci USA 1996; 93:7 - 12
- Shiroishi M, Tsumoto K, Tanaka Y, Yokota A, Nakanishi T, Kondo H, et al. Structural consequences of mutations in interfacial Tyr residues of a protein antigen-antibody complex. The case of HyHEL-10-HEL. J Biol Chem 2007; 282:6783 - 6791
- Collis AV, Brouwer AP, Martin AC. Analysis of the antigen combining site: correlations between length and sequence composition of the hypervariable loops and the nature of the antigen. J Mol Biol 2003; 325:337 - 354
- Dougan DA, Malby RL, Gruen LC, Kortt AA, Hudson PJ. Effects of substitutions in the binding surface of an antibody on antigen affinity. Protein Eng 1998; 11:65 - 74
- Janin J, Chothia C. The structure of protein-protein recognition sites. J Biol Chem 1990; 265:16027 - 16030
- MacCallum RM, Martin AC, Thornton JM. Antibody-antigen interactions: contact analysis and binding site topography. J Mol Biol 1996; 262:732 - 745
- Almagro JC. Identification of differences in the specificity-determining residues of antibodies that recognize antigens of different size: implications for the rational design of antibody repertoires. J Mol Recognit 2004; 17:132 - 143
- Gabdoulline RR, Wade RC. On the protein-protein diffusional encounter complex. J Mol Recognit 1999; 12:226 - 234
- Sheinerman FB, Honig B. On the role of electrostatic interactions in the design of protein-protein interfaces. J Mol Biol 2002; 318:161 - 177
- Albeck S, Unger R, Schreiber G. Evaluation of direct and cooperative contributions towards the strength of buried hydrogen bonds and salt bridges. J Mol Biol 2000; 298:503 - 520
- Ramirez-Benitez MC, Almagro JC. Analysis of antibodies of known structure suggests a lack of correspondence between the residues in contact with the antigen and those modified by somatic hypermutation. Proteins 2001; 45:199 - 206
- Ohlin M, Borrebaeck CA. Insertions and deletions in hypervariable loops of antibody heavy chains contribute to molecular diversity. Mol Immunol 1998; 35:233 - 238
- Khalifa MB, Weidenhaupt M, Choulier L, Chatellier J, Rauffer-Bruyere N, Altschuh D, et al. Effects on interaction kinetics of mutations at the VH-VL interface of Fabs depend on the structural context. J Mol Recognit 2000; 13:127 - 139
- Vargas-Madrazo E, Paz-Garcia E. An improved model of association for VH-VL immunoglobulin domains: asymmetries between VH and VL in the packing of some interface residues. J Mol Recognit 2003; 16:113 - 120
- Weidenhaupt M, Khalifa MB, Hugo N, Choulier L, Altschuh D, Vernet T. Functional mapping of conserved, surface-exposed charges of antibody variable domains. J Mol Recognit 2002; 15:94 - 103
- Friguet B, Chaffotte AF, Djavadi-Ohaniance L, Goldberg ME. Measurements of the true affinity constant in solution of antigen-antibody complexes by enzyme-linked immunosorbent assay. J Immunol Methods 1985; 77:305 - 319
- Malmborg AC, Borrebaeck CA. BIAcore as a tool in antibody engineering. J Immunol Methods 1995; 183:7 - 13
- Erlandsson A, Holm P, Ullen A, Stigbrand T, Sundstrom BE. Studies of the interactions between the anticytokeratin 8 monoclonal antibody TS1, its antigen and its anti-idiotypic antibody alphaTS1. J Mol Recognit 2003; 16:157 - 163
- Johansson A, Sandstrom P, Ullen A, Behravan G, Erlandsson A, Levi M, et al. Epitope specificity of the monoclonal anticytokeratin antibody TS1. Cancer Res 1999; 59:48 - 51
- Bogan AA, Thorn KS. Anatomy of hot spots in protein interfaces. J Mol Biol 1998; 280:1 - 9
- Fields BA, Goldbaum FA, Ysern X, Poljak RJ, Mariuzza RA. Molecular basis of antigen mimicry by an anti-idiotope. Nature 1995; 374:739 - 742
- Gaida FJ, Pieper D, Roder UW, Shively JE, Wagener C, Neumaier M. Molecular characterization of a cloned idiotypic cascade containing a network antigenic determinant specific for the human carcinoembryonic antigen. J Biol Chem 1993; 268:14138 - 14145
- Johnson G, Moore SW. Functional idiotypic mimicry of an adhesion- and differentiation-promoting site on acetylcholinesterase. J Cell Biochem 2004; 91:999 - 1009
- Kolesnikov AV, Kozyr AV, Alexandrova ES, Koralewski F, Demin AV, Titov MI, et al. Enzyme mimicry by the antiidiotypic antibody approach. Proc Natl Acad Sci USA 2000; 97:13526 - 13531
- Weissenhorn W, Chen YH, Riethmuller G, Rieber EP, Weiss EH. VH-related idiotopes detected by site-directed mutagenesis. A study induced by the failure to find CD4 anti-idiotypic antibodies mimicking the cellular receptor of HIV. J Immunol 1992; 149:1237 - 1241
- Kipriyanov SM, Moldenhauer G, Martin AC, Kupriyanova OA, Little M. Two amino acid mutations in an anti-human CD3 single chain Fv antibody fragment that affect the yield on bacterial secretion but not the affinity. Protein Eng 1997; 10:445 - 453
- Morea V, Lesk AM, Tramontano A. Antibody modeling: implications for engineering and design. Methods 2000; 20:267 - 279
- Liang S, Liu Z, Li W, Ni L, Lai L. Construction of protein binding sites in scaffold structures. Biopolymers 2000; 54:515 - 523
- Jackson RM. Comparison of protein-protein interactions in serine protease-inhibitor and antibody-antigen complexes: implications for the protein docking problem. Protein Sci 1999; 8:603 - 613
- Kaplan H, Long BG, Young NM. Chemical properties of functional groups of mouse immunoglobulins of the IgA, IgG2a and IgM classes. Biochemistry 1980; 19:2821 - 2827
- Schreiber G, Fersht AR. Rapid, electrostatically assisted association of proteins. Nat Struct Biol 1996; 3:427 - 431
- Selzer T, Albeck S, Schreiber G. Rational design of faster associating and tighter binding protein complexes. Nat Struct Biol 2000; 7:537 - 541
- Sundstrom BE, Nathrath WB, Stigbrand TI. Diversity in immunoreactivity of tumor-derived cytokeratin monoclonal antibodies. J Histochem Cytochem 1989; 37:1845 - 1854
- Erlandsson A, Eriksson D, Johansson L, Riklund K, Stigbrand T, Sundstrom BE. In vivo clearing of idiotypic antibodies with antiidiotypic antibodies and their derivatives. Mol Immunol 2006; 43:599 - 606
- Yang J, Moyana T, MacKenzie S, Xia Q, Xiang J. One hundred seventy-fold increase in excretion of an FV fragment-tumor necrosis factor alpha fusion protein (sFV/TNF-alpha) from Escherichia coli caused by the synergistic effects of glycine and triton X-100. Appl Environ Microbiol 1998; 64:2869 - 2874
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970; 227:680 - 685
- Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis 1997; 18:2714 - 2723