Abstract
Antibody charge variants have gained considerable attention in the biotechnology industry due to their potential influence on stability and biological activity. Subtle differences in the relative proportions of charge variants are often observed during routine biomanufacture or process changes and pose a challenge to demonstrating product comparability. To gain further insights into the impact on biological activity and pharmacokinetics (PK) of monoclonal antibody (mAb) charge heterogeneity, we isolated the major charge forms of a recombinant humanized IgG1 and compared their in vitro properties and in vivo PK. The mAb starting material had a pI range of 8.7-9.1 and was composed of about 20% acidic variants, 12% basic variants, and 68% main peak. Cation exchange displacement chromatography was used to isolate the acidic, basic, and main peak fractions for animal studies. Detailed analyses were performed on the isolated fractions to identify specific chemical modification contributing to the charge differences, and were also characterized for purity and in vitro potency prior to being administered either subcutaneously (SC) or intravenously (IV) in rats. All isolated materials had similar potency and rat FcRn binding relative to the starting material. Following IV or SC administration (10 mg/kg) in rats, no difference in serum PK was observed, indicating that physiochemical modifications and pI differences among charge variants were not sufficient to result in PK changes. Thus, these results provided meaningful information for the comparative evaluation of charge-related heterogeneity of mAbs, and suggested that charge variants of IgGs do not affect the in vitro potency, FcRn binding affinity, or the PK properties in rats.
Introduction
Monoclonal antibodies (mAbs) are now well-established pharmacological therapeutic modalities based on the understanding and identification of key targets involved in inflammatory, oncologic and autoimmune diseases. Heterogeneity of purified antibodies (immunoglobulins, Ig) based on the simple chemical modifications of selected amino acids sitesCitation1–Citation3 is of considerable importance in the biotechnology field. The IgG class, whose molecular weight is approximately 150 kDa, comprises approximately 85% of the Ig in normal human serum, as well as most mAb therapeutics currently marketed or in development. A typical IgG is composed of two identical Fabs (fragment, antigen binding, 55 kDa) and an Fc (fragment, crystallizable, 35 kDa) domain assembled in the Y shape motif. In IgG, the unique complementarity-determining regions (CDRs) usually define the antigen specificity and reside in the variable fragment (Fv) portion of the Fab. The hinge region provides flexibility for the two Fabs relative to the Fc and affects bivalent antigen binding and activation of Fc effector functions. The Fc portion of IgG participates in pH-dependent electrostatic interactions with the neonatal Fc receptor (FcRn), which protects the mAb from degradation and partially accounts for their relatively long serum half-lives.Citation4,Citation5 In general, mAbs, like many proteins, have charge heterogeneity that optimizes the balance of gaining favorable electrostatic interactions and determines their structure, stability, binding affinity, chemical properties and hence their biological reactivity.Citation6–Citation10
mAbs have gained significant attention as potential therapeutics due to the high degree of specificity in binding to target antigens, ability to initiate immune response to the target antigen and long serum persistence,Citation11 thereby reducing the need for frequent dosing. As advances in antibody engineering, including hybridoma and phage-display technologies, have enabled the development of murine, chimeric, humanized and fully human mAbs, numerous novel therapeutic mAbs currently approved for the treatment of diseases, including infliximab and rituximab, have been mass produced. For biopharmaceutical development, product consistency and long shelf life are important factors that provide flexibility in manufacturing and supply management. It has also become apparent that mAbs are more heterogeneous than previously thought or reported.Citation12 During manufacture, various forms of microheterogeneity in size, charge and other parameters occur due to enzymatic processes or spontaneous degradation and modifications. mAbs undergo chemical degradation via several different mechanisms, including oxidation, deamidation, isomerization and fragmentation, that result in the formation of various charge variants and heterogeneity, thus modifying their isoelectric pH (pI) values.Citation13–Citation16
The different post-translational modifications seen in mAbs are presented in a recent comprehensive review in ref. Citation16. The major chemical degradation pathways for mAbs are shown in . Chemical and enzymatic modifications such as deamidation and sialylation, respectively, result in an increase in the net negative charge on the mAbs and cause a decrease in pI values, thereby leading to formation of acidic variants.Citation17–Citation20 C-terminal lysine cleavage results in the loss of net positive charge and leads to acidic variant formation.Citation13,Citation21 Another mechanism for generating acidic variants is the formation of various types of covalent adducts, e.g., glycation, where glucose or lactose can react with the primary amine of a lysine residue during manufacturing in glucose-rich culture media or during storage if a reducing sugar is present in the formulation.Citation18,Citation19,Citation22,Citation23 Formation of the basic variants can result from the presence of C-terminal lysine or glycine amidation, succinimide formation, amino acid oxidation or removal of sialic acid, which introduce additional positive charges or removal of negative charges; both types of modifications cause an increase in pI values.Citation10,Citation13,Citation20,Citation24–Citation28
Although substantial knowledge and experience with the degradation pathways that are active during production in cell 6culture, purification, formulation and storage of therapeutic mAbs has accumulated, the biopharmaceutical industry continues to characterize microheterogeneity thoroughly in order to demonstrate batch-to-batch consistency and predict shelf-life of these complex protein molecules. The current challenge is to understand the effects that mAb microheterogeneity may have on efficacy, potency, immunogenicity and clearance. Evidence has been presented that deliberately modifying the pI of an antibody by approximately one pI unit or more can give noticeable differences in the pharmacokinetics (PK) of an intact mAb.Citation29,Citation30 Most studies of antibody charge modifications have involved intravenous (IV) administration; in contrast, there is little information regarding the effects of charge on the PK of subcutaneously (SC) administered mAbs. Passage through the interstitium to the vascular or lymphatic capillaries can present a barrier to efficient drug absorption after SC administration.Citation31,Citation32 Interstitial diffusion of mAbs is likely to be influenced by their charge and their electrostatic interactions with negatively charged plasma-derived proteins present within the interstitial area underlying the dermis of the skin.Citation31 Therefore, additional studies are required to determine the impact on FcRn binding and PK of such charge differences following IV and SC injections. Elucidating the impact of mAb charge heterogeneity on biological activity requires isolation or enrichment of different charged variant forms in significant quantities to perform testing. The goals of this work were to separate the major charge variants (acidic, basic and main peak fractions) of a humanized IgG1 mAb, characterize some of their biophysical characteristics, FcRn and antigen binding properties and compare their PK in IV vs. SC administration in rats.
Results
Separation and characterization of charge variants.
The cation exchange chromatography (IEC) elution profile of the starting mAb material used in this study is shown in . Three distinct areas were noted. The early and late-eluting peaks were termed the acidic and basic variants, respectively. The most abundant peak was termed the main peak. The starting mAb had 20% acidic, 68% main and 12% basic variants. We developed a general method to separate the charge variants using displacement chromatography that was successfully scaled-up to generate gram quantities of material from a single chromatography run in high resolution (data not shown). The scaled-up displacement method allowed sufficient quantities of material to be generated for the material-intensive studies described here. The cation exchange chromatograms in show the profiles of the three charge variant fractions collected after separation by displacement chromatography. The individual charge variant pools were formulated at 30 mg/mL in 20 mM histidine acetate, 120 mM sucrose, 0.02% polysorbate 20 at pH 6.0.
Concentration, osmolality and pH were measured for all the charge variant formulations; results are shown in . The protein concentration in the final formulations of all samples was 30 mg/mL at pH 6.0 and their osmolality was between 161 and 171 mOsm/kg. The endotoxin level, as measured by limulus amebocyte lysate (LAL) testing, ranged between 0.01–0.2 EU/mg for all three components, whereas the endotoxin for the starting mAb was <0.01 EU/mg. This level was maintained well within the limit of <5 EU/kg for human dosing studies (USP 85). The low level of endotoxin showed that the aseptic techniques utilized during displacement chromatography and formulation are adequate for maintaining sterility and preventing significant increase in pyrogen levels throughout the process. The isolated charge variants were further characterized using a panel of analytical techniques (). IEC analysis () of the acidic, basic and main peak fractions showed that the isolated materials were 95, 94 and 94% pure, respectively. The individual charge variant fractions were analyzed by size exclusion chromatography (SEC) and typical elution profiles are shown in . The size distribution for the acidic and basic variants, as compared to the main peak and starting mAb, appeared to be comparable. The higher aggregate level in the basic variant (10%) as compared to all other mAb samples (≤0.3%) was most likely due to the selective enrichment of aggregates in the basic variant region. This enrichment may be due to the fact that aggregates tend to have higher affinity for cation-exchangers and would elute in the basic region, i.e., rear of the displacement train, during displacement on the cation exchange column. Previous studies have shown no significant changes in the total mass balance of the aggregates in the collected material upon further SEC analysis (data not shown). The protein conformation of the isolated acidic and basic variants and main peak were further characterized using circular dichroism (CD). The CD spectra for all the variants were identical in both the near and the far-UV region (data not shown) suggesting that no secondary and tertiary differences were detected for any of the charge variants. This finding also indicated that no changes occurred during purification using displacement chromatography.
Detailed analyses were performed on the basic and acidic charge variant fractions to identify specific modifications and will be the described in detail in a future publication (manuscript in preparation). Briefly, the specific modifications found for the charge variants are given in . Basic variants were in the basic variant (10%) as compared to all other mAb samples (0.3%) was most likely due to the selective enrichment of aggregates in the basic variant region. This enrichment may be due to the fact that aggregates tend to have higher affinity for cation-exchangers and would elute in the basic region, i.e., rear of the displacement train, during displacement on the cation exchange column. Previous studies have shown no significant changes in the total mass balance of the aggregates in the collected material upon further SEC analysis (data not shown). The protein conformation of the isolated acidic and basic variants and main peak were further characterized using circular dichroism (CD). The CD spectra for all the variants were identical in both the near and the far-UV region (data not shown) suggesting that no secondary and tertiary differences were detected for any of the charge variants. This finding also indicated that no changes occurred during purification using displacement chromatography. Detailed analyses were performed on the basic and acidic charge variant fractions to identify specific modifications and will be the described in detail in a future publication (manuscript in preparation). Briefly, the specific modifications found for the charge variants are given in . Basic variants were identified by peptide mapping and comprised a mixture of heavy chain C-terminal and light chain N-terminal forms. Heavy chain forms had Lys removed from one or both of the C-termini. Light chain forms retained the Val-His-Ser leader sequence at the N-terminus. Acidic variants were comprised of a mixture of sialylated, reduced disulfide, cross-linked (non-reducible), glycated and deamidated forms. Deamidation occurred at heavy chain residues Asn391 and Asn436, which correspond to residues 389 and 434, respectively, in the Kabat numbering system.Citation33 Glycation was identified by MS of the reduced acidic variant fraction (not shown) and the levels of were 3.5% based on boronate chromatography analysis. However, no specific lysine containing peptides were identified by LC-MS, indicating that the glycation was distributed over multiple lysine residues and below the detection limit for peptide mapping.
The isoelectric points of the charge variants were determined using imaged capillary isoelectric focusing (icIEF). shows the mean pI values for all mAb charged isoforms. The pI of the mAb starting material was 8.7–9.1. The pI values for the acidic and basic variants and the main peak were 8.7–8.9, 9.1 and 9.0, respectively, which are within the range of the starting material. The pI values indicate that, in general, the pI of the isoforms correlate with elution position on the IEC assay, as well as with the position within the displacement train, which is expected.
To examine whether displacement chromatography would create new peaks or change the charge behavior of the mAb, a sample of the starting material was bound and displaced from the column. The displaced material was collected in a single pool (as opposed to fractionated) and analyzed via SEC and IEC. Results from this experiment showed no differences in the mAb before and after displacement chromatography (data not shown). This indicates that the displacement chromatography method used herein can be used to isolate charge variants that are representative of the species found in the starting mAb.
The displacer, which is a potential impurity from the displacement process, was quantified during the entire process by following its absorbance at λmax of 262 nm. No absorbance at this wavelength was observed along with the main peak and acidic charge variant peaks, indicating that the displacer was resolved from these protein peaks eluting off the column. However, a trace amount of the displacer was detected in the last few basic variant fractions via an IEC assay. The displacer in these aliquots was easily removed by buffer exchange and its absence confirmed by IEC (data not shown). The results obtained from the various analyses listed above indicated that the charge variants were sufficiently enriched, representative of the variants in the starting material, free of adventitious agents and contaminants and therefore suitable for conducting further in vitro and in vivo studies.
FcRn binding.
To assess whether IgG charge variations exhibit altered FcRn binding activities, the binding of each isolated charge variant fraction to immobilized rat FcRn was evaluated using the surface plasmon resonance (BIAcore T100). The results indicated a small decrease in the overall binding response at pH 6 of the acidic fraction compared with the main fraction, whereas the binding response of the basic and main fractions is comparable based on statistical analysis (). Based on our previous observations with another IgG, the rat FcRn expression system does not affect binding affinity. Binding of the IgG to rat FcRn expressed in HEK293 or Chinese hamster ovary (CHO) cells showed similar binding (data now shown).
For kinetic binding analysis, rat FcRn was also immobilized onto the sensor chip. While there was no measurable binding for any of the variant fractions to rat FcRn at pH 7.4 (data not shown), the binding activities could be clearly detected at pH 6. In addition, a simple 1:1 binding model was inadequate to fit the data (data not shown), consistent with the reported 2:1 stoichiometric ratio for the FcRn:IgG complex.Citation34 Therefore, a bivalent analyte binding model was applied; representative sensorgrams along with the residual plot for all charge variant fractions are shown in . The association and dissociation rate constants (ka and kd, respectively) and the equilibrium dissociation constants (KD) calculated from the kinetic analyses are summarized in . The results demonstrate that rat FcRn binds to the acidic, main and basic fractions with an average KD1 of 6.96 ± 2.09, 5.38 ± 0.76 and 5.40 ± 1.07 × 10−7 M, respectively. These values were comparable to the measured affinity of the starting material, which had an average KD1 of 5.67 ± 0.82 × 10−7 M. No major discrepancy was observed between the experimental data and the fitted curves, as shown in the residual plots as well as the low chi-squared values (ranging from 0.173–0.351), suggesting that the bivalent analyte model adequately describes the interaction of the rat FcRn and the IgG charge variants.
Numerous studies have indicated a correlation between the affinity for FcRn binding and the serum half-life of mAbs in rodents and primates.Citation4,Citation35 The acidic fraction of humanized IgG1 exhibited a decrease in overall binding response to FcRn at pH 6 compared to the basic and main fractions. In contrast, no appreciable binding was observed for any of the fractions at pH 7.4. Since the limits for serum half-life differences from subtle decrease or increase in FcRn-binding affinity have not been determined, we evaluated the PK of all the charge variants in rats to determine whether their charge distribution alters the serum half-life (see below).
Anti-proliferation potency assay.
To assess the in vitro potency, we measured the ability of each mAb fraction to inhibit the proliferation of a cultured target cell line and calculated the mean activity expressed in percent activity compared with reference material. The percent specific activity showed 83 and 95% for the acidic and basic variants, respectively, whereas the main peak and the starting material showed 98 and 100% activity (). The acidic fractions showed slightly lower potency, but are within assay variability. Based on these results, the potencies of individual mAb variant fractions were equivalent.
Pharmacokinetics in rats.
The response of each charge variants in the PK assay was studied to show the assay's sensitivity to different charge variants. Our results indicated that all charge variants gave equivalent responses in the PK assay (data not shown). In the IV study, the primary endpoint for PK analysis compared parameter area under the curve for 1–14 days (AUC0–14) across groups. These studies have demonstrated that the majority of AUC is in the first 14 days; in addition, anti-therapeutic antibody (ATA) formation that confounds PK analysis occurs during the first 10 days and beyond. The corresponding results are given in . The IgG1 acidic variant, main peak variant and starting material were 907, 891 and 942 µg*day/mL, respectively. The ratio of geometric means of acidic to starting material was 0.963 with a 90% CI of 0.905–1.03 and the ratio of geometric means of main peak to starting material was 0.946 with a 90% CI of 0.884–1.01; both met the criterion for bioequivalence. Furthermore, other PK parameters such as Cmax and half-life also exhibited similarity across cohorts. Therefore, the antibody from the three groups exhibited similar exposure at this dose level.
For rats dosed subcutaneously, group mean concentration-time profiles following subcutaneous administration are shown in . The IgG1 acidic and basic variants, main peak and starting material were 408, 395 and 383 and 411 µg*day/mL, respectively (). The ratio of geometric means of the acidic to starting material was 0.992 with a 90% CI of 0.925–1.063. The ratio of geometric means of the basic to starting material was 0.932 with a 90% CI of 0.811–1.072. The ratio of geometric means of main peak to starting material was 0.961 with a 90% CI of 0.872–1.058. All comparisons to starting material met the exhibited similarity across cohorts. Therefore, the antibody from the three groups exhibited similar exposure at this dose level. For rats dosed subcutaneously, group mean concentration-time profiles following subcutaneous administration are shown in . The IgG1 acidic and basic variants, main peak and starting material were 408, 395 and 383 and 411 µg*day/mL, respectively (). The ratio of geometric means of the acidic to starting material was 0.992 with a 90% CI of 0.925–1.063. The ratio of geometric means of the basic to starting material was 0.932 with a 90% CI of 0.811–1.072. The ratio of geometric means of main peak to starting material was 0.961 with a 90% CI of 0.872–1.058. All comparisons to starting material met the criterion for bioequivalence. A comprehensive list of parameters showing that Tmax and Cmax values were also very similar across groups is summarized in .
Overall, following a single 10 mg/kg IV or SC dose in rats of all charge variants, the serum concentration-time profiles were all similar (). Thus, there were no differences in the PK parameters between all charge variants (), including a relatively long terminal half-life of approximately 8.09–10.1 days. In fact, the SC administration did not amplify the impact of charge variants on PK parameters compared to IV administration. Similar PK exposure based on AUC0-14 was observed ranging from 891–942 µg*day/mL and 383–411 µg*day/mL following all mAb fractions administered via the IV and SC route of administration, respectively. Furthermore, there was no statistical difference in the primary endpoint, AUC0–14, when comparing acidic, basic and main peak to the starting material. The similarity in Tmax is further evidence suggesting that there is no significant change in the absorption phase pharmacokinetics of charge variants.
Discussion
The PK and distribution of mAbs are attributed to many factors, including: equilibration with the protein space of the extracellular fluid,Citation36 interaction with the Fc portion of the mAb with Fc receptors,Citation4,Citation5 and possibly electrostatic interaction due to cell membrane surface charge and antibody charge.Citation37,Citation38 Previous reports have shown that deliberate modification of the pI of an antibody by approximately one pI unit or more can give noticeable differences in the PK of an intact antibody.Citation30,Citation39 These deliberately engineeredCitation30 and chemically modified mAbs with acidic,Citation40–Citation44 basicCitation45–Citation48 and neutralCitation49,Citation50 molecules are important approaches that can be used to modulate the magnitude of the overall ionic nature of the antibody population, thereby manipulating their PK behavior while preserving antigen binding specificity and potency.Citation30,Citation51–Citation53 Moreover, apart from the chemical nature of the appended group, the site of modification, e.g., tyrosines, lysines, cysteines, oligosaccharides, has also been shown to affect the in vitro and in vivo properties of mAbs.Citation54
Routine manufacturing of mAbs produces charge variants whose biological impact is not well understood. Displacement chromatography, a highly efficient purification process for isolating charge variants, was used to isolate acidic, main and basic peak mAb charge variants from a therapeutic mAb drug substance manufactured at large scale using a routine manufacturing process. The isolated charge variant fractions had similar pIs (8.7–9.1), fragment and aggregate levels and responses in the PK (data not shown) and potency assays as the starting material. These results indicate that the modifications giving rise to the charge differences and the chromatographic isolation did not impact the potency or ability of the PK assay to detect the mAb. Therefore, the isolated materials have properties similar to and are representative of, the variants in the starting material. These results also show that displacement chromatography is a mild technique that has the resolution and capacity to enable preparation of mAb charge variants in sufficient quality and quantity for in vivo and in vitro studies.
PK differences can result from non-specific electrostatic interactions or altered FcRn binding. As FcRn is responsible for the protection of IgG from systemic elimination through pH dependence,Citation35 changing the binding of IgG to FcRn generally translates to deliberate control of serum IgG half-life.Citation55 Therefore, the contribution of charge differences to rat PK and IgG interaction with rat FcRn was tested with the isolated charge variants. Although slight changes in binding of acidic variants to rat FcRn were detected, these changes did not translate to PK difference compared to the starting material. Of the specific acidic variants identified, deamidation of Asn436 and glycation at some Fc Lys residues could affect FcRn binding. Alanine scanning of the human IgG1 for the human FcRn receptorCitation56 showed that the Asn434Ala variant (KabatCitation33 numbering, which corresponds to Asn436 in the mAb charge variant) resulted in a 3.5-fold increase in binding to FcRn. This increased binding of the Ala mutant suggests that deamidation at Asn436 could affect FcRn binding. Lys288Ala, Lys317Ala and Lys360Ala variants also altered FcRn binding and therefore glycation of these residues could potentially impact FcRn binding. However, no specific lysines were identified in the LC-MS analysis indicating that the low level of glycation observed was distributed over the entire mAb and not localized to the Fc region. Similarly, sialylation is not expect to alter binding to FcRn.Citation57 The basic variants identified in this mAb are not expected to impact FcRn binding as the modifications to the N- and C-termini () are away from the receptor binding sites on the antibody.Citation58
In a recent paper, Yeung et al.Citation59 studied the direct relationship between FcRn-binding affinity in vitro and PK properties in vivo. In particular, their data gave insight into the affinity improvement limit for an engineered FcRn variant to achieve improved PK. Importantly, variants with less than 3- to 4-fold difference in FcRn-binding affinity are not expected to show measurable difference in PK compared with wild type. All other charge variants had indistinguishable rat FcRn-binding affinity at pH 6, which is considered physiologically relevant for the FcRn-IgG interaction.Citation59,Citation60 The PK in rats of acidic, main and basic charge variant fractions administered SC and acidic and main peak charge variant fractions administered IV, were similar to the starting material and each other. As the major mechanisms involved in mAb disposition and dynamics are largely conserved across species,Citation61 our pre-clinical rat model is expected to represent non-specific, charge-related effects on kinetics in humans due to the presence of similar negatively charged structures that decorate the surface of most mammalian cells.Citation62 In this context, the rat model used in the present study is expected to be relevant for predicting charge related effects on PK in humans.
The results presented here demonstrate that mAb charge heterogeneity generated during routine manufacturing had minimal, if any, effect on the FcRn binding, potency or PK properties of an IgG1 in healthy rats. These findings may be readily applied to similar IgG1 mAb therapeutics. Evidence based on this work and othersCitation30,Citation49,Citation59,Citation63,Citation64 indicates that minor changes in the nature of ionic charge resulting in pI differences less than approximately one unit are not expected to affect the biological function of mAbs and may not require extensive PK comparability assessments. This study improves our understanding of the effects of charge variants on activity and PK and enhances our ability to generate safe and efficacious antibody drugs.
Materials and Methods
Antibodies.
All sample handling steps for isolation of charge variant fractions were aseptically performed under a laminar flow hood. All materials and reagents used for this study were ensured to be pyrogen-free, either by LAL testing or material certification. The mAb analyzed was expressed in Chinese hamster ovary (CHO) cells and was a humanized IgG1 that does not react with the rat counterpart of its human target antigen, and shows activity in vivo. This purified mAb was characterized using ion exchange chromatography and the profile showed the presence of three distinct components: the acidic (20%) and basic variants (12%) and the main peak (68%).
Displacement chromatography and characterization of charge variant fractions.
Separation of charge variants. mAb charge variant fractions were isolated using displacement chromatography. Briefly, a GE AKTA Explorer chromatography system was used for all runs. A 10 × 600 mm GE Tricorn column was packed with GE Source 15S cation resin to a bed height of 585 mm and all processes were performed at ambient temperature. In general the antibody starting material (at pH 6.1) was loaded on to the column to a load density of ∼30 mg/mL. The loaded column was then washed with equilibration buffer (30 mM MES, 10 mM NaCl, pH 5.85) to remove any non-binding molecules and adjust the pH of the bound antibody and column. Next, 10 column volumes (CV) of the displacer (5 mM Sachem SP1, 27 mM MES, 9 mM NaCl, pH 5.75) was loaded at 30 cm/hr to displace the bound mAb. The displacement train was fractionated at the column outlet. Every fifth fraction was then analyzed using an ion exchange column on an HPLC (IEC method described below) to determine the fraction purity.
Fractions of charge variants with >90% purity were pooled and transferred into an Amicon Ultra-15 (30,000 MWCO from Millipore, Bedford, MA), concentrated and buffer exchanged with 20 mM histidine acetate buffer, pH 6.0. The appropriate amounts of a concentrated sucrose and polysorbate 20 solutions were added to the individual pools to obtain a final concentration of 30 mg/ml charge variant, 120 mM sucrose and 0.02% polysorbate 20. The samples were sterile-filtered with 0.22 µm PVDF membrane filters (50 mL Durapore Vacuum-Driven Filter Device, Millipore, Bedford, MA, USA). The individual charge variants in the above formulation were tested using size-exclusion and ion-exchange chromatography and checked for protein content, pH, osmolality and endotoxin levels using the methods described below.
Protein concentration measurements. The protein concentration of isolated charge variant fractions was determined by measurement of the UV-absorbance on an Agilent 8453 spectrophotometer (Sunnyvale, CA, USA) via volumetric sample preparation. The samples were blanked against the formulation buffer and the absorbance was measured at Amax at 278–279 nm.
Osmolality. The osmolality was measured by freezing point method using an Advanced Instrument Model 3320 and calibrated on the same day with 100 and 500 mOsm/kg Clinitrol Controline osmolality standards (Advanced Instrument, Norwood, MA, USA).
Size exclusion chromatography. Size variant distribution was determined by size exclusion chromatography (SEC) using a TosoHaas column G3000 SWXL (Tosoh Biosep, LLC, Montgomeryville, PA, USA) at ambient temperature on an Agilent 1200 HPLC (Santa Clara, CA, USA). Samples were eluted over 30 minutes with a 0.2 M potassium phosphate, 0.25 M potassium chloride, pH 6.2 mobile phase monitored by a UV detector at 280 nm. Chromatograms were integrated using Dionex Chromeleon Software (Sunnyvale, CA, USA) and relative percent peak areas obtained.
Circular dichroism. Far and near-UV circular dichroism (CD) spectroscopy was used to ensure that no major structural change resulted in the tertiary structure of mAbs upon modification of different residues. Experiments were conducted using a Jasco 815 spectropolarimeter (Easton, MD, USA). Solution ellipticities for the near UV and the far UV were measured from 340-240 nm and from 190–250 nm in potassium 10 mM phosphate buffer (pH 6.0) respectively. Studies were conducted at 0.8 mg/ml mAb concentration using 10 mm path length cell for measuring the CD spectra in the near UV region and 0.5 mg/ml mAb concentration in a 1 mm path length cell for measuring the far UV region. Multiple scans, at a resolution of 0.1 nm and a scan rate of 10 nm/min, were accumulated and averaged in order to improve signal to noise ratio. Buffer spectra obtained with the same acquisition parameters were subtracted from the averaged scans for IgG1 solutions.
Limulus amoebocyte lysate assay for endotoxin determination. Endotoxin levels were measured to confirm comparability and remained within predetermined study criteria post-formulation of the charge variants from the starting material. All samples were endotoxin tested by the chromogenic LAL method using a Kinetic-QCL assay kit with a BioWhittaker Kinetic-QCL reader (BioWhittaker, Wokingham, UK), according to manufacturer's instructions. This assay is a quantitative, kinetic assay for the detection of Gram-negative bacterial endotoxin. Gram-negative bacteria catalyze the activation of a pro-enzyme in the LAL. The activated enzyme catalyzes the splitting of p-nitroaniline (pNA) from the colorless substrate Ac-Ile-Glu-Ala-Arg-pNA. The concentration of the endotoxin in a sample is calculated from its reaction time by comparison to the reaction time of the standard curve.
Capillary isoelectric focusing for pI determination. Imaged capillary isoelectric focusing (icIEF) is known to be an effective tool with high separation resolution in biomolecular analysis. This technique has been confirmed to be suitable for fast determination of pI of proteins and is able to distinguish subtle differences in charge.Citation65,Citation66 Briefly, the pI of each charge variant fraction was determined by icIEF using an iCE280 analyzer (Convergent Bioscience) with a fluorocarbon-coated capillary cartridge (100 µm × 5 cm). Capillary isoelectric focusing was achieved using a mixture of 0.35% methyl cellulose, 5% total carrier ampholytes (3–10 Pharmalyte) and 0.1% pI markers varying from 5.8 to 9.8 (5.8, 6.6, 7.0, 7.5, 8.2, 9.5 and 9.8) in purified water. The anolyte was 80 mM phosphoric acid and the catholyte was 100 mM sodium hydroxide, both in 0.1% methyl cellulose. All samples were diluted, mixed with the ampholyte solution and then focused by introducing a potential of 1,500 volts for 1 min, followed by a potential of 3,000 volts for 5 min. The final protein concentration in the ampholyte and sample mixture was 0.25 mg/mL. The separation was monitored at 280 nm.
Ion exchange chromatography. Charge variants were separated on a 4.0 × 250 mm Dionex ProPac cation exchange (IEC) column WCX-10. The mobile phase was a MES buffer at pH 6.0 at a flow rate of 1.0 mL/min. Bound forms were eluted with a sodium chloride gradient. The column temperature was 42°C and detection was at 280 nm.
To quantify the amount of acidic forms due to sialic acid, the acidic variant fraction was analyzed with and without treatment with neuraminidase, which removes sialic acid. The difference in the percent peak area between treated and untreated samples enabled quantitation of acidic forms containing sialic acid.
HPLC-MS of peptide digests. Acidic variants were sulfitolyzed, digested with endoproteinase Asp-N and analyzed by reverse phase HPLC on a Zorbax 300SB-C8 column. Mobile phase A was 0.1% TFA in deionized water and mobile phase B was 0.08% TFA in isopropyl alcohol. Absorbance was monitored at 214 nm and masses were determined by electrospray mass spectrometery using a Thermo Fisher LTQ.
Basic variants were reduced, carboxymethylated, digested with trypsin and analyzed by reverse phase HPLC on a VYDAC C-18 column. Solvent A was water, solvent B was acetonitrile and both had 0.1% trifluoroacetic acid (TFA). A 0–60% gradient was used to separate the peptides. Absorbance was monitored at 214 nm and masses determined by electrospray mass spectrometry using an API3000.
Capillary electrophoresis-sodium dodecyl sulfate (CE-SDS). Acidic variant fractions were analyzed by both reduced and non-reduced CE-SDS. Samples were labeled with 5-carboxytetramethylrhodamine succinimidyl ester. After removal of excess dye, the samples were either treated with iodoacetamide (non-reduced sample) at 70°C for 5 min or DTT (reduced sample) at 70°C for 20 min. The samples were analyzed on a Beckman PA 800 capillary electrophoresis system equipped with a 50 um diameter uncoated fused-silica capillary. Samples were injected electrokinetically and separation was performed at a constant voltage of 15 kV over a 40 min period. The sieving matrix was SDS-MW gel buffer (Beckman Coulter) and the capillary maintained at a temperature of 20°C. The migration of labeled components was monitored by laser induced fluorescence detection using excitation at 488 nm and the emission at 560 nm.
Boronate affinity chromatography. A TSK-Gel boronate-5PW column (Tosoh), 7.5 × 75 mm, 10 µm particle size was used to determine glycation levels. The affinity-based separation occurred such that the non-glycated molecules flow through the column equilibrated in 0.1 M HEPES, 0.2 M NaCl, 25 mM Tris, pH 8.6, then the glycated molecules are single-step eluted from the resin with the same buffer that also contained 0.5 M sorbitol. Column temperature was 40°C and the mobile phase flow-rate was 1.0 mL/minute. Each sample was run in triplicate with 100 µL injections of a 1:1 dilution in equilibration buffer (50 µg loads). Eluent absorbance was monitored at 280 nm to determine the percentage of glycated mAb in each charge variant fraction.
Affinity measurements binding to rat FcRn.
The coding regions of the rat FcRn α-chain ectodomain (Met1-Ser309) and the full-length β2-microglobulin light chain (rβ2m) were generated by gene synthesis (Blue Heron). The coding regions of rat FcRn and r β2m were subcloned into a previously described pRK mammalian cell expression vector.Citation67 For expression and purification of rat FcRn, human embryonic kidney (HEK293) cells were transfected using FUGENE (Roche Applied Science) according to the manufacturer's protocol. After 24 h of incubation with transfection complexes, cells were switched to serum-free PSO4 medium [Genentech; 1 g/L Pluronic F-68, 5.5 g/L combination nonselect medium (Life Technologies), 4.3 g/L glucose, 1.22 g/L sodium bicarbonate, 0.1 g/L gentamicin sulfate (pH 7.1); 350 milliosmolar] supplemented with 5 mg/L recombinant bovine insulin and trace elements and grown for 7 days. Cells were pelleted by centrifugation and rat FcRn was purified from the culture supernatants by pH-dependent binding to a human IgG-Sepharose (Amersham) column. Briefly, supernatants were acidified to pH 5.8 with 50 mM MES and flowed over a 4 mL hIgG-Sepharose column at ∼1.5 mL/min. After washing with >10 column volumes of wash buffer (20 mM MES, 150 mM NaCl, pH 5.8), bound rat FcRn was eluted with 20 mM HEPES, 150 mM NaCl, pH 8.0. Eluted proteins were then concentrated and further purified by size exclusion chromatography on a Superdex 200 column (Amersham) with PBS pH 6.0 as the running buffer. Fractions containing monomeric FcRn were pooled, and the concentration was determined on a Nanodrop 8000 spectrometer (Thermo Scientific) using a mass extinction coefficient of 1.95 Lcm−1g−1 at 280 nm.
All binding and kinetics studies were performed on recombinant rat FcRn, by surface plasmon resonance technology using a Biacore T-100™ instrument (GE Healthcare, Piscataway, NJ, USA). All experiments were carried out at 37°C. Rat FcRn (5 µg/mL) was immobilized onto two of the four flow cells (FCs) of a Series S CM5 sensor chip (GE Healthcare), FC2 and FC4, using a standard amine coupling procedure according to the manufacturer's protocol. The immobilization levels were approximately 40 response units (RU) per flow cell. The other two FCs, FC1 and FC3, were subjected to the same immobilization protocol in the absence of coupling ligand (rat FcRn) to serve as in-line background reference flow cells for FC2 and FC4, respectively, in subsequent experiments. All mAb samples were diluted to 25 nM and 250 nM in running buffer (PBS, 0.05% polysorbate, pH 6.0) and were injected for 2 min at a flow rate of 50 µL/minute. Surfaces were regenerated between cycles by two sequential injections of PBS, pH 7.4 (35 seconds per injection at 50 µL/min). To ensure that any differences seen between different charge variants were beyond assay variation, a statistical experimental design was used in which eight independent runs for each sample were carried out in random order using two separate sensor chips. The reference-corrected binding responses were reported 5 seconds before the end of each injection.
Experiments measuring the kinetics of the interaction between rat FcRn and mAb charge variants were performed essentially as described above, except that each injection consisted of a 5 min association phase and a 6 min dissociation phase. The first dissociation rate constant (kd1), first association rate constant (ka1) and the first dissociation equilibrium constant (KD1) from kinetic analyses were calculated with a bivalent analyte model using the BIAevaluation Software (version 3.2; GE Healthcare). Four independent experimental runs were carried out using two separate sensor chips and the results were averaged.
Anti-proliferation assay.
To assess the in vitro potency of mAb charge variants, a cell culture assay was used to measure their ability to inhibit the proliferation of cells expressing the receptor. Briefly, cells were seeded in 96-well tissue culture microtiter plates and incubated overnight at 37°C under 5% CO2 to allow cell attachment. The following day, the culture medium was removed and serial dilutions of each mAb fraction were added to the plates. The plates were then incubated for four days at 37°C under 5% CO2 and the relative number of viable cells was quantified indirectly using a redox dye, alamarBlue® according to the manufacturer's protocol. The control materials were also tested using the same assay parameters and conditions. Each mAb concentration was assayed in triplicate and the changes in color as measured by fluorescence were directly proportional to the number living cells in the culture. The absorbance of each well was then measured on a fluorescence 96-well plate reader. The results, expressed in relative fluorescence units (RFU), were plotted against the antibody concentration and a parallel line analysis program was used to estimate the anti-proliferative activity of the antibody relative to the reference material.
Intravenous and subcutaneous pharmacokinetic studies in normal rats.
The PK studies were conducted in two parts using Sprague-Dawley male rats. The first was an IV study that evaluated statistically the acidic charge variant, main peak and starting material. In this study, a total of 36 male rats, weighing 279–317 g and 8 weeks in age, were assigned to three different groups, each consisting of 12 rats per group. All animals received a single IV bolus dose of 10 mg/kg via jugular cannula. Blood samples (approximately 0.2 mL) from the jugular vein of each animal were collected pre-dose and post-dose at 5 min; 1, 4, 8, 24, 32 h and 2, 4, 7, 10, 14, 21, 28 and 35 days. Blood samples were collected in serum separators. Diluted serum sample were assayed for antibody concentrations using a receptor-binding direct Enzyme Linked ImmunoSorbent Assay (ELISA). In this particular assay, a recombinant antigen receptor specific for this mAb molecule was used as the capture reagent and mouse anti-human IgG Fc conjugated to horseradish peroxidase (Jackson ImmunoResearch Laboratories, Inc., Catalog # 209-035-098) was used as the detection reagent. The minimum dilution of sample for the ELISA was 1/100 and the minimum quantifiable concentration was 400 ng/mL. In brief, 96-well polystyrene microtiter plates (Nunc, Thermo Fisher Scientific, catalog # 439454) were coated with 2 mg/mL of the antigen with 0.05 M sodium carbonate buffer and incubated for 2–6 h at 2°C–8°C. The coat solution was then removed from the wells and the wells were blocked with approximately 200 µL of assay buffer (phosphate buffer saline/0.5% bovine serum albumin/0.05% polysorbate 20/0.033% proclin 300) for 1–2 h at room temperature with agitation. The wells were then washed with 400 µL × 6 of PBS Buffer (phosphate buffer saline/0.05% Polysorbate 20). Standards (in duplicate), controls (in duplicate) and serially diluted samples were added to the plate at 100 µL and incubated at room temperature for 2 h ± 5 min with agitation. The wells were then washed with 400 µL of PBS buffer six times. Mouse anti-human IgG Fc conjugated to horseradish peroxidase was diluted 1:1,500 in assay buffer and added to each well at 100 µL. The plates were incubated at room temperature for 1 h ± 5 min with agitation and wells were washed six times with 400 µL of PBS Buffer, before 100 µL of a 1:1 mixture of tetramethyl benzidine (TMB) peroxidase substrate (0.4 g/L TMB) [Kirkegaard & Perry Labs (KPL) 50-76-0] and peroxidase solution B, 0.02% hydrogen peroxide (KPL 50-65-00) was added to each well. After approximately 15 min of incubation, the reaction was stopped with the addition of 100 µL of 1 M phosphoric acid to each well. The yellow color developed by conversion of the substrate was measured on a Biotek ELX800 plate reader using two filters, 450 nm for detection absorbance and 630 nm for reference absorbance.
The second was a subcutaneously administered study conducted with 48 male rats that evaluated statistically the acidic and basic charge variant, main peak and starting material. The animals (12 per group), weighing 279–317 grams and 8 weeks of age were assigned to four different groups. Each rat was administered a 0.1 mL subcutaneous dose in the right lateral flank at 10 mg/kg. Approximately 0.2 mL of blood was collected from the tail vein for each animal at pre-dose and post-dose at 1, 3, 6 and 24 h and days 2, 3, 4, 7, 10 and 14. The concentrations of the antibody in serum were determined using a receptor-binding ELISA as described above.
Pharmacokinetic data analysis.
The serum mAb time concentration data for all groups in both studies were analyzed using non-compartmental analysis (Model 200, WinNonlin Pro, Version 5.0.1; Pharsight Corporation; Mountain View, CA, USA). For plotting purposes, all LTR values were converted to ½*LTR, with a value of 0.2 µg/mL. Based on prior experience with this mAb, AUC0–14 was chosen as the primary endpoint for this study.
Financial Disclosure
All authors are employees of Genentech, Inc., a member of the Roche Group and hold financial interest in Roche.
Figures and Tables
Figure 1 Chromatographic profiles obtained from a (A) IEC shown in full scale and (B) SEC shown in expanded scale for all charge variant fractions, starting material and buffer blanks. See for numerical values obtained from these analyses.
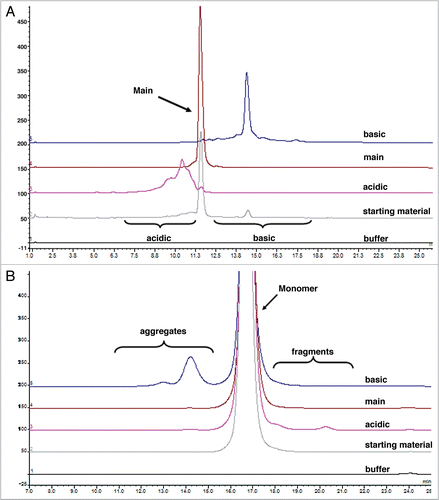
Figure 2 Representative sensorgrams of fitted data from the kinetic analysis of all charge variant fractions (A) acidic, (B) main, (C) basic and (D) starting material to immobilized rat FcRn. Black lines are fitted curves using a bivalent binding model. All sample concentrations (from bottom to top) are 62.5, 125, 250, 500, 1,000, 2,000 and 4,000 nM. Residual plot shows the difference between experimental and fitted data for every point in the sensorgram. Also see for clarity.
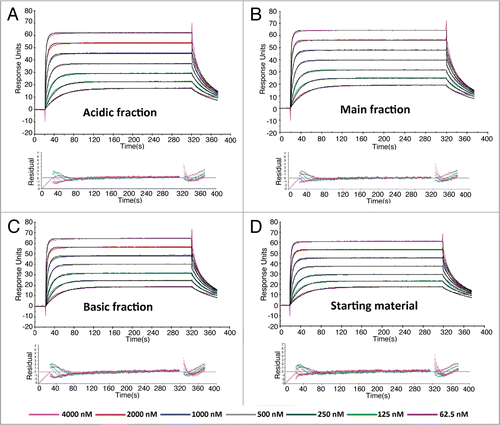
Figure 3 Linear changes in serum concentrations versus time for all charge variant fractions following single (A) intraveneous or (B) subcutaneous administration of 10 mg/kg in normal rats. The assay minimum quantifiable concentration was 0.4 µg/mL in rat serum. Results are mean ± SD (n = 12 rats/time point).
Table 1 Major chemical degradation pathways which are a common source of charge-related heterogeneity of therapeutic IgG1 mAbs
Table 2 Analytical results to assess all mAb charge variant fractions
Table 3 Analytical characterization of isolated charge variant fractions
Table 4 Comparison of all charge variant fractions to evaluate their kinetics of binding to rat FcRn, binding responses (in response unit) and their in vitro binding specific activity
Table 5 Pharmacokinetic parameters of all mAb fractions in normal rats
Acknowledgements
We would like to thank Michelle G. Schweiger and the In-Vivo Studies Group for carrying out the pharmacokinetic studies, Jennifer Visich for her support and for her helpful discussions of the pharmacokinetic data, Bert Gunter and Cherry Lei for statistical analysis of the FcRn binding and PK data, Will McElroy for skillfully carrying out the icIEF experiments, C. Andrew Boswell and Daniela Bumbaca for editorial efforts. We also would like to express appreciation to the many colleagues and peers who have contributed to expanding our knowledge of charge-related heterogeneity in antibodies and regret the absence herein of many valuable reference citations due to space limitations.
References
- Seiler FR, Gronski P, Kurrle R, Lüben G, Harthus HP, Ax W, et al. Monoclonal antibodies: their chemistry, functions and possible uses. Angew Chem Int Ed Engl 1985; 24:139 - 160
- Cohen S. General structure and heterogeneity of immunoglobulins. Proc R Soc Lond B Biol Sci 1966; 166:114 - 123
- Boswell CA, Deng R, Lin K, Putnam WS, Lei C, Theil FP, et al. Mrsny R, Daugherty A. In vitro-in vivo correlation of pharmacokinetics, pharmacodynamics and metabolism for antibody therapeutics. Proteins and Peptides: Pharmacokinetic, Pharmacodynamic and Metabolic Outcomes 2010; New York NY: Informa Healthcare 1 - 14
- Ober RJ, Radu CG, Ghetie V, Ward ES. Differences in promiscuity for antibody-FcRn interactions across species: implications for therapeutic antibodies. Int Immunol 2001; 13:1551 - 1559
- Carter P. Improving the efficacy of antibody-based cancer therapies. Nature Rev Cancer 2001; 1:118 - 129
- Boonyaratanakornkit BB, Park CB, Clark DS. Pressure effects on intra- and intermolecular interactions within proteins. Biochem Biophys Acta 2002; 1595:235 - 249
- Zhang W, Czupryn MJ. Free sulfhydryl in recombinant monoclonal antibodies. Biotechnol Prog 2002; 18:509 - 513
- Collins K. Charge density-dependent strength of hydration and biological structure. Biophys J 1997; 72:65 - 76
- Perkins M, Theiler R, Lunte S, Jeschke M. Determination of the origin of charge heterogeneity in a murine monoclonal antibody. Pharm Res 2000; 17:1110 - 1117
- Harris RJ, Kabakoff B, Macchi FD, Shen FJ, Kwong MJ, Andya DS, et al. Identification of multiple sources of charge heterogeneity in a recombinant antibody. J Chromatogr B 2001; 752:233 - 245
- Sidhu SS. Full-length antibodies on display. Nat Biotech 2007; 25:537 - 538
- Carson KL. Flexibility—the guiding principle for antibody manufacturing. Nat Biotech 2005; 23:1054 - 1058
- Harris RJ, Shire SJ, Winter C. Commercial manufacturing scale formulation and analytical characterization of therapeutic recombinant antibodies. Drug Dev Res 2004; 61:137 - 154
- Wang W, Singh S, Zeng DL, King K, Nema S. Antibody structure, instability and formulation. J Pharm Sci 2007; 96:1 - 26
- He XZ, Que AH, Mo JJ. Analysis of charge heterogeneities in mAbs using imaged CE. Electropheresis 2009; 30:714 - 722
- Vlasak J, Ionescu R. Heterogeneity of monoclonal antibodies revealed by charge-sensitive methods. Curr Pharm Biotechnol 2008; 9:468 - 481
- Chirino AJ, Mire-Sluis A. Characterizing biological products and assessing comparability following manufacturing changes. Nat Biotech 2004; 22:1383 - 1391
- Lyubarskay Y, Houde D, Woodard J, Murphy D, Mhatre R. Analysis of recombinant monoclonal antibody isoforms by electrospray ionization mass spectrometry as a strategy for streamlining characterization of recombinant monoclonal antibody charge heterogeneity. Anal Biochem 2006; 348:24 - 39
- Zheng JY, Janis LJ. Influence of pH, buffer species and storage temperature on physicochemical stability of a humanized monoclonal antibody LA298. Int J Pharm 2006; 308:46 - 51
- Chumsae C, Gaza-Bulseco G, Sun J, Liu H. Comparison of methionine oxidation in thermal stability and chemically stressed samples of a fully human monoclonal antibody. J Chromatogr B 2007; 850:285 - 294
- Antes B, Amon S, Rizzi A, Wiederkum S, Kainer M, Szolar O, et al. Analysis of lysine clipping of a humanized lewis-Y specific IgG antibody and its relation to Fc-mediated effector function. J Chromatogr B 2007; 852:250 - 256
- Huang L, Lu J, Wroblewski VJ, Beals JM, Riggin RM. In vivo deamidation characterization of monoclonal antibody by LC/MS/MS. Anal Chem 2005; 77:1432 - 1439
- Andya JD, Maa YF, Costantino HR, Nguyen PA, Dasovich N, Sweeney TD, et al. The effect of formulation excipients on protein stability and aerosol performance of spray-dried powders of a recombinant humanized anti-IgE monoclonal antibody. Pharm Res 1999; 16:350 - 358
- Wypych J, Li M, Guo A, Zhang Z, Martinez T, Allen MJ, et al. Human IgG2 antibodies display disulfide-mediated structural isoforms. J Biol Chem 2008; 283:16194 - 16205
- Dillon TM, Ricci MS, Vezina C, Flynn GC, Liu YD, Rehder DS, et al. Structural and functional characterization of disulfide isoforms of the human IgG2 subclass. J Biol Chem 2008; 283:16206 - 16215
- Wright A, Morrison SL. Effect of C2-associated carbohydrate structure on Ig effector function: studies with chimeric mouse-human IgG1 antibodies in glycosylation mutants of Chinese hamster ovary cells. J Immunol 1998; 160:3393 - 3402
- Johnson KA, Paisley-Flango K, Tangarone BS, Porter TJ, Rouse JC. Cation exchange-HPLC and mass spectrometry reveal C-terminal amidation of an IgG1 heavy chain. Anal Biochem 2007; 360:75 - 83
- Pan H, Chen K, Chu L, Kinderman F, Apostol I, Huang G. Methionine oxidation in human IgG2 Fc decreases binding affinities to protein A and FcRn. Protein Sci 2009; 18:424 - 433
- Sharifi J, Khawli LA, Hornick JL, Epstein AL. Improving monoclonal antibody pharmacokinetics via chemical modification. Q J Nucl Med 1998; 42:242 - 249
- Igawa T, Tsunoda H, Tachibana T, Maeda A, Mimoto F, Moriyama C, et al. Reduced elimination of IgG antibodies by engineering the variable region. Protein Eng Des Sel 2010; 23:385 - 392
- McLennan DN, Porter CJH, Charman SA. Subcutaneous drug delivery and the role of the lymphatics. Drug Discovery Today: Technologies 2005; 2:89 - 96
- Schmid-Schonbein GW. Microlymphatics and lymph flow. Physiol Rev 1990; 70:987 - 1028
- Kabat EA, Wu TT, Perry HM, Gottesman KS, Foeller C. Sequences of Proteins of Immunological Interest 9:Fifth Edition Bethesda United States Public Health Service, National Institutes of Health 3243 Publication Number 1991
- Martin WL, Bjorkman PJ. Characterization of the 2:1 complex between the class I MHC-related Fc receptor and its Fc ligand in solution. Biochem 1999; 38:12639 - 12647
- Roopenian DC, Akilesh S. FcRn: the neonatal Fc receptor comes of age. Nat Rev Immunol 2007; 7:715 - 725
- O'Connor SW, Bale WF. Accessibility of circulating immunoglobulin G to the extravascular compartment of solid rat tumors. Cancer Res 1984; 44:3719 - 3723
- Goldenberg NM, Steinberg BE. Surface charge: A key determinant of protein localization and function. Cancer Res 2010; 70:1277 - 1280
- Khawli LA, Epstein AL. Exploration of novel strategies to enhance monoclonal antibodies targeting. Q J Nucl Med 1997; 41:25 - 35
- Boswell CA, Sun X, Niu W, Weisman GR, Wong EH, Rheingold AL, et al. Comparative in vivo stability of copper-64-labeled cross-bridged and conventional tetraazamacrocyclic complexes. J Med Chem 2004; 47:1465 - 1474
- Boswell CA, Brechbiel MW. Development of radioimmunotherapeutic and diagnostic antibodies: an inside-out view. Nucl Med Biol 2007; 34:757 - 778
- Gangopadhyay A, Petrick AT, Thomas P. Modification of antibody isoelectric point affects biodistribution of 111-indium-labeled antibody. Nucl Med Biol 1996; 23:257 - 261
- ten Kate CI, Fischman AJ, Rubin RH, Fucello AJ, Riexinger D, Wilkinson RA, et al. Effect of isoelectric point on biodistribution and inflammation: imaging with indium-111-labelled IgG. Eur J Nucl Med 1990; 17:305 - 309
- Perera RM, Zoncu R, Johns TG, Pypaert M, Lee FT, Mellman I, Lloyd JO, et al. Internalization, intracellular trafficking and biodistribution of monoclonal antibody 806: a novel anti-epidermal growth factor receptor antibody. Neoplasia 2007; 9:1099 - 1110
- Hong G, Bazin-Redureau MI, Scherrmann JM. Pharmacokinetics and organ distribution of cationized colchicine-specific IgG and Fab fragments in rat. J Pharm Sci 1999; 88:147 - 153
- Lee HJ, Pardridge WM. Monoclonal antibody radiopharmaceuticals: cationization, pegylation, radiometal chelation, pharmacokinetics and tumor imaging. Bioconjug Chem 2003; 14:546 - 553
- Pardridge WM, Kang YS, Diagne A, Zack JA. Cationized hyperimmune immunoglobulins: pharmacokinetics, toxicity evaluation and treatment of human immunodeficiency virus-infected human-peripheral blood lymphocytes-severe combined immune deficiency mice. J Pharmacol Exp Ther 1996; 276:246 - 252
- Bickel U, Lee VMY, Pardridge WM. Pharmacokinetic differences between 111In- and 125I-labeled cationized monoclonal antibody against b-amyloid in mouse and dog. Drug Delivery 1995; 2:128 - 135
- Khawli LA, Glasky MS, Alauddin MM, Epstein AL. Improved tumor localization and radioimaging with chemically modified monoclonal antibodies. Cancer Biother Radiopharm 1996; 11:203 - 215
- Khawli LA, Mizokami MM, Sharifi J, Hu P, Epstein AL. Pharmacokinetic characteristics and biodistribution of radioiodinated chimeric TNT-1, -2 and -3 antibodies modified with biotin. Cancer Biother Radiopharm 2002; 17:359 - 370
- Iznaga-Escobar N, Mishra AK, Perez-Rodriguez R. Factors affecting pharmacokinetics of monoclonal antibodies: a review article. Meth Find Exp Clin Pharmacol 2004; 26:123 - 127
- Epstein AL, Khawli LA, Hu P. Torchilin V, Muzykantov A. Tumor necrosis treatment and imaging of solid tumors. Biomedical Aspects of Drug Targeting 2003; New Rochelle, NY Mary Ann Liebert 249 - 274
- Winkelhake JL. Effects of chemical modification of antibodies on their clearance from the circulation. J Bio Chem 1977; 252:1865 - 1868
- Torchilin VP, Klabanov AL, Nossiff ND, Slinkin MA, Strauss HW, Haber E, et al. Monoclonal antibody modification with chelate-linked high molecular weight polymers: major increases in polyvalent cation binding without loss of antigen binding. Hybridoma 1987; 6:229 - 240
- Rodwell JD, Alvarez VL, Lee C, Lopes AD, Goers JW, King HD, et al. Site-specific covalent modification of monoclonal antibodies: in vitro and in vivo evaluations. Proc Natl Acad Sci USA 1986; 83:2632 - 2636
- Petkova SB, Akilesh S, Sproule TJ, Christianson GJ, Khabbaz HA, Brown AC, et al. Enhanced half-life of genetically engineered human IgG1 antibodies in a humanized FcRn mouse model: potential application in humorally mediated autoimmune disease. Int Immunol 2006; 18:1759 - 1769
- Shields RL, Namenuk AK, Hong K, Meng YG, Rae J, Briggs J, et al. High resolution mapping of the binding site on Human IgG1 for FcγRI, FcγRII, FcγRIII and FcRn and design of IgG1 variants with improved binding to the FcγR. J Biol Chem 2001; 276:6591 - 6604
- Jones AJS, Papac DI, Chin EH, Keck R, Baughman SA, Lin YS, et al. Selective clearance of glycoforms of a complex glycoprotein pharmaceutical caused by terminal N-acetylglucosamine is similar in humans and cynomolgus monkeys. Glycobiology 2007; 17:529 - 540
- Sondermann P, Oosthuizen V. Mediation and Modulation of Antibody Function. Biochem Soc Transacti 2002; 30:481 - 486
- Yeung YA, Leabman MK, Marvin JS, Qiu J, Adams CW, Lien S, et al. Engineering human IgG1 affinity to human nonatal Fc receptor: impact of affinity improvement on pharmacokinetics in primates. J Immunol 2009; 182:7663 - 7671
- Wang E, Brown PS, Aroeti B, Chapin SJ, Mostov KE, Dunn KW. Apical and basolateral endocytic pathways of MDCK cells meet in acidic common endosomes distinct from a nearly-neutral apical recycling endosome. Traffic 2000; 1:480 - 493
- Tang L, Persky AM, Hochhaus G, Meibohm B. Pharmacokinetic aspects of biotechnology products. J Pharm Sci 2004; 93:2184 - 2204
- Muchmore EA, Varki NM, Fukuda M, Varki A. Developmental regulation of sialic acid modifications in rat and human colon. FASEB J 1987; 1:229 - 235
- Onda M, Kreitman RJ, Vasmatzis G, Lee B, Pastan I. Reduction of the non-specific animal toxicity of anti-Tac(Fv)-PE38 by mutations in the framework regions of the Fv which lower the isolelectric point. J Immunol 1999; 163:6072 - 6077
- Putnam WS, Prabhu S, Zheng Y, Subramanyam M, Wang YMC. Pharmacokinetic, pharmacodynamic and immunogenicity comparability assessment strategies for monoclonal antibodies. Trends Biotechnol 2010; 28:509 - 516
- Meert C, Guo A, Novick S, Han M, Pettit D, Balland A. Evaluation of pI marker Sources for cIEF characterization of a therapeutic antibody. Chromatographia 2007; 66:963 - 968
- Wu J, Huang T. Peak identification in capillary isoelectric focusing using the concept of relative peak position as determined by two isoelectric point markers. Electrophoresis 2006; 27:3584 - 3590
- Eaton DL, Wood WI, Eaton D, Hass PE, Hollingshead P, Wion K, et al. Construction and characterization of an active factor VIII variant lacking the central one-third of the molecule. Biochem 1986; 25:8343 - 8347