Abstract
Tumor necrosis factor (TNF) signals through two membrane receptors, TNFR1 and TNFR2, and TNFR1 is known to be the major pathogenic mediator of chronic and acute inflammatory diseases. Present clinical intervention is based on neutralization of the ligand TNF. Selective inhibition of TNF receptor 1 (TNFR1) provides an alternative opportunity to neutralize the pro-inflammatory activity of TNF while maintaining the advantageous immunological responses mediated by TNFR2, including immune regulation, tissue homeostasis and neuroprotection. We recently humanized a mouse anti-human TNFR1 monoclonal antibody exhibiting TNFR1-neutralizing activity. This humanized antibody has been converted into an IgG1 molecule (ATROSAB) containing a modified Fc region previously demonstrated to have greatly reduced effector functions. Purified ATROSAB, produced in CHO cells, showed strong binding to human and rhesus TNFR1-Fc fusion protein and mouse embryonic fibroblasts transfected with a recombinant TNFR1 fusion protein with an affinity identical to the parental mouse antibody H398. Using chimeric human/mouse TNFR1 molecules, the epitope of ATROSAB was mapped to the N-terminal region (amino acid residues 1-70) comprising the first cysteine-rich domain (CRD1) and the A1 sub-domain of CRD2. In vitro, ATROSAB inhibited typical TNF-mediated responses like apoptosis induction and activation of NFκB-dependent gene expression such as IL-6 and IL-8 production. These findings open the way to further analyze the therapeutic activity of ATROSAB in relevant disease models in non-human primates.
Introduction
Tumor necrosis factor (TNF) is a pleiotropic cytokine and a central mediator of inflammation. Elevated levels of TNF are associated with various inflammatory diseases including rheumatoid arthritis, psoriasis and Crohn disease. Several TNF-neutralizing reagents have been approved for the treatment of these diseases, including a soluble TNF receptor, etanercept, as well as the anti-TNF antibodies infliximab, adalimumab, certolizumab pegol and golimumab and many more are under development.Citation1,Citation2 With over 1 million patients treated with TNF antagonists, therapeutic efficacy is well-documented.Citation3 However, global TNF inhibition over a prolonged period of time increases the risk of tuberculosis (TB) reactivation, serious infections and even malignancies.Citation4–Citation6 Consequently, medical information of all approved anti-TNF medicines includes extensive warnings and precautions.
Two TNF receptors (CD120a, TNFR1; CD120b, TNFR2) mediate signal transduction upon binding of TNF.Citation7 Pro-inflammatory responses are mainly mediated by the ubiquitously expressed TNFR1. TNFR1 is activated both by the membrane-bound form of TNF (mTNF) and soluble TNF (sTNF), which is produced from mTNF by proteolytic cleavage. In contrast, TNFR2, expressed in a more restricted manner, e.g., by immune cells, endothelial cells and neurons, can only be activated by mTNF. Activation of TNFR2 mainly induces anti-apoptotic signals and can lead to cell proliferation in vitro.Citation8 Furthermore, TNFR2 appears to play a role in tissue homeostasis and regeneration.Citation9
Selective inhibition of TNFR1 signaling has gained increasing attention as an alternative to global TNF neutralization, which affects both TNF receptors. The application of receptor-selective rather than global inhibition of TNF responses represents a paradigm shift in the present clinical practice of treating inflammatory diseases. Conceptually, the selective inhibition strategy targets the predominant pathogenic pathway and potentially leaves signals essential for tissue homeostasis and immunocompetence untouched. This will in the long run improve therapeutic efficacy by minimizing therapy-limiting unwanted side effects, such as recurrence of TB and risk of malignancies or increased relapse rates in multiple sclerosis (MS),Citation10 probably due to lack of TNFR2-mediated myelin regeneration.Citation11 Recently, a TNF mutein (R1antTNF) selectively neutralizing the activity of TNFR1 has been described.Citation12 This TNF mutein, administered either as unmodified or as PEGylated protein (PEG-R1antTNF), demonstrated therapeutic efficacy in acute murine hepatitis models and a murine collagen-induced arthritis model.Citation13,Citation14 The beneficial effect of selectively inhibiting TNFR1 was further supported by results from a dominant-negative TNF mutein (XPro1595), which is capable of forming inactive complexes with sTNF, thus selectively inhibiting the pro-inflammatory action mediated by TNFR1 while preserving the innate immunity to infections.Citation15,Citation16 TNFR1-selective inhibition can be also achieved with TNFR1-specific antibodies. For example, a monoclonal antibody, H398, with selectivity for human TNFR1, showed potent inhibition of TNF-mediated signal transduction and cytotoxicity.Citation17–Citation19
We recently generated a humanized version of H398.Citation20 This humanized antibody (IZI-06.1), produced as Fab fragment in E. coli, exhibited in vitro neutralizing activities comparable to that of the Fab fragment of the parental antibody. Hence, IZI-06.1 may open a new treatment option by selectively inhibiting TNFR1-mediated signal transduction. In the present study, we have converted IZI-06.1 into a full IgG1 antibody (ATROSAB). In order to avoid Fc-mediated effector functions, a heavy chain with greatly reduced antibody-dependent cell-mediated cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC) was used.Citation21 ATROSAB was produced in mammalian cells and showed a similar binding and neutralizing behavior as the parental mouse H398 IgG. Furthermore, using chimeric human/mouse receptor molecules, we mapped the epitope of ATROSAB to the first 70 amino acids of human TNFR1.
Results
Production and binding activity of IZI-06.1 IgG (ATROSAB).
The humanized anti-human TNFR1 antibody IZI-06.1,Citation20 was converted into a human IgG1 using a heavy chain with greatly reduced effector functions (IgG1e3).Citation21 This antibody (ATROSAB) was produced in CHO cells. A 25 L scale production of ATROSAB was performed in a wave system over a period of 15 days with a maximum cell density of more than 12 mio cells/mL. Purity and integrity was confirmed by SDS-PAGE analysis and size exclusion chromatography ( and B). ATROSAB showed strong binding to recombinant human TNFR1-Fc composed of the extracellular region of TNFR1 fused to the human IgG1 Fc region (). The parental antibody, H398, exhibited an identical binding in ELISA. The selectivity for TNFR1 was confirmed by flow cytometric analysis of ATROSAB using mouse embryonic fibroblasts (MEF) transfected with fusion proteins comprising the extracellular domain of TNFR1 or TNFR2, respectively, fused to the intracellular domain of human Fas (TNFR1-Fas; TNFR2-Fas).Citation22 In this assay, binding was only seen with MEF-TNFR1-Fas, but not with MEF-TNFR2-Fas ( and B). Binding of ATROSAB to MEF-TNFR1-Fas was comparable to that of H398 as shown by a titration of antibody concentration (). The EC50 values were approximately 0.1 nM for both ATROSAB and H398. Next, we investigated species specificity with recombinant mouse TNFR1-Fc and rhesus TNFR1-Fc fusion proteins. In ELISA, binding of the two antibodies was observed for human and rhesus TNFR1-Fc, but not mouse TNFR1-Fc ().
Affinity measurements.
The affinity of ATROSAB for TNFR1 was determined by quartz crystal microbalance measurements using immobilized TNFR1-Fc. ATROSAB bound with sub-nanomolar affinity to human and rhesus TNFR1-Fc, similar to the affinity of H398 for human TNFR1-Fc and rhesus TNFR1-Fc ( and ). An approximately 10-fold reduced affinity was measured for monovalent scFv IZI-06.1, due to a faster off-rate, indicating that binding of ATROSAB and H398 to the dimeric TNFR1-Fc fusion proteins is influenced by avidity effects.
Antagonistic activity of ATROSAB.
ATROSAB inhibited in a dose-dependent manner the TNF-induced apoptosis of Kym-1 cells (). In this assay, a TNF concentration that resulted in 90% cytotoxicity was used. About half-maximal cytotoxicity, i.e., 55% viable cells, was observed at 60 nM for ATROSAB and 8 nM for H398, respectively. We then investigated the effects of ATROSAB on TNF-induced secretion of IL-6 from HeLa cells and IL-8 from HT1080 cells, respectively. TNF induced strong secretion of IL-6 from HeLa cells in a dose-dependent manner, reaching approximately 700 pg/ml of IL-6 after incubation with 4 nM TNF (200 ng/ml) for 18 h. Similarly, TNF induced secretion of IL-8 from HT1080 cells reached approximately 7,000 pg/ml after incubation with 4 nM TNF for 18 h ( and D). ATROSAB and H398 inhibited release of IL-6 from HeLa cells and IL-8 from HT1080 cells induced by 20 pM TNF (1 ng/ml) in a dose dependent manner ( and B). In these assays, the IC50 values were 60 nM for ATROSAB and 6 nM for H398 for inhibition of IL-6 release () and for inhibition of IL-8 release (), respectively. Incubation of HeLa cells or HT1080 cells (in the absence of TNF) with ATROSAB and H398, respectively, resulted in only marginal induction of cytokine release at a very narrow dose range. At concentrations around 10 nM only, slightly elevated IL-6 levels were observed (40–60 pg/ml vs. 15 pg/ml of untreated cells), corresponding to 3–4.5% of the response at a comparable TNF concentration (4 nM). For IL-8, the level was increased from 80 pg/ml of untreated cells to approximately 200 pg/ml after incubation with the antibodies, corresponding to approximately 2% of the equivalent TNF response. Human IgG included as negative control had no effect on cytokine release.
Plasma half-life.
Half-life of ATRSOAB and H398 was determined after a single dose intravenous (i.v.) injection into CD1 mice. Concentrations of the antibodies over a period of 7 days were measured by ELISA, which detects functional antibody molecules. Both antibodies showed a similar elimination from the blood with terminal half-lives of 10.5 ± 2.8 d for ATROSAB (n = 2) and 8.1 ± 1.5 d for H398 (n = 3) ().
Epitope mapping.
Because H398 and ATROSAB do not bind to mouse TNFR1, we applied a domain swapping strategy for epitope mapping (). Binding to these chimeric TNFR1-Fc molecules was analyzed by ELISA. All constructs were capable of binding human TNF, although constructs 3 and 4 showed slightly reduced binding compared with the other constructs, nevertheless demonstrating that the chimeric molecules retained ligand-binding activity. No binding of ATROSAB and H398 was seen with chimeric molecules when the human CRD1 and 2 (construct 3) or only the CRD1 was substituted by the corresponding mouse domains (construct 4). Further, no binding was seen when only the A1 domain of CRD1 of human TNFR1 was exchanged with the corresponding mouse sequence (construct 5). Binding was strongly reduced also with mouse TNFR1 containing the human CRD1 (construct 7) or the human A1 domain of CRD1 (construct 6), indicating that further regions are required for full binding. Extension of the human portion to include sub-domain A1 of CRD2 resulted in a chimeric TNFR1 to which H398 and ATROSAB show strong binding (construct 8). Thus, the epitope resides in the N-terminal region of TNFR1 covering residues 1–70. Within this region, 15 residues are different between human TNFR1 and mouse TNFR1, while only one residue is different between human and rhesus TNFR1 (). This residue (Ile 21) is substituted by a valine in rhesus and mouse TNFR1. Several of the residues different between human and mouse TNFR1 are exposed in the interaction site of the receptor with TNF, including Pro23, Gln24, Tyr30, Asn31, Ser57, Ser 59, His66 and His69. In order to further narrow down the epitope, we exchanged P23 and Gln24, located in sub-domain A1 of CRD1, by the corresponding mouse residues in the chimeric TNFR1 h1-2A1/m2B2-4 (construct 10, and B). These mutations completely abolished binding of ATROSAB and H398 under the applied assay conditions.
Discussion
Here, we describe the generation of an IgG1 derivative (ATROSAB) of a humanized TNFR1-specific antagonistic monoclonal antibody.Citation20 The IgG format was chosen because of its long half-life, established production and increased binding due to bivalency. In order to avoid induction of antibody-mediated effector functions, ATROSAB possesses a mutated Fc-region previously described to exhibit greatly reduced ADCC and CDC.Citation21
Receptor-selective inhibition by ATROSAB and the parental mouse antibody resulted in blocking of distinct signaling pathways of TNFR1, as shown by inhibition of TNF-mediated cell death, as well as NFκB induced IL-6 and IL-8 release. Both cytokines are biomarkers of inflammation and are elevated, e.g., during episodes of active disease, in rheumatoid arthritis.Citation23 The antagonistic activity of the murine H398 and the humanized monovalent Fab was described to be based on interference with ligand binding.Citation17,Citation19,Citation20 Using deletion mutants, the epitope recognized on TNFR1 was previously described to include the membrane-distal CRD1. By using a domain swapping strategy for chimeric mouse/human TNFR1-Fc fusion proteins, we now show that the epitope recognized by ATROSAB and H398 also includes subdomain A1 of CRD2, i.e., the entire epitope is covered by amino acids 1–70 in the N-terminal region of TNFR1. The finding that subdomain A1 of CRD2 is also required for antibody binding hints toward steric blockage as cause for neutralization of TNF action. The structure of TNFR1 with bound TNF () shows that the identified epitope region at least partially overlaps with the TNF binding site, which is mainly located in CRD2 and CRD3.Citation24 Additionally, site directed mutagenesis revealed that residues Pro23 and Gln24 of subdomain A1 of CRD1 directly contribute to antigen and species specificity. This is of interest because CRD1 is not directly involved in ligand binding,Citation24 but is critically involved in TNFR1 signaling. CRD1 controls high affinity ligand binding by stabilizing the conformation of the subsequent CRD2,Citation25 and removal of CRD1 results in loss of ligand binding.Citation25–Citation27 In addition, CRD1 comprises a homophilic receptor/receptor interaction site, the pre-ligand-binding assembly domain (PLAD), which is essential for generation of functional TNFR signal complexes.Citation26,Citation28 Hence, binding of ATROSAB to CRD1 could not only displace TNF by steric hindrance or by inducing a conformational change, but could also interfere with homotypic PLAD interactions, thereby blocking the formation of functional TNFR signal complexes.
ATROSAB showed a slightly reduced antagonistic activity compared to H398. This is probably not due to altered affinity since affinities of both antibodies for recombinant human TNFR1 were similar as determined by quartz crystal microbalance measurements and in flow cytometry measurements using TNFR1-expressing cells. Currently, we cannot exclude that ATROSAB and H398 bind in a slightly different way or to a slightly different area within the identified region (aa 1–70) containing the epitope. Further epitope mapping by site directed mutagenesis of exposed residues will provide insights into the exact localization of the conformational epitope of ATROSAB and H398 and the mechanism of ligand blocking.
In the absence of TNF, for both antibodies (H398 and ATROSAB) a minor stimulatory activity was revealed at a very narrow dose range by sensitive in vitro assays with established cell lines. This marginal effect of the bivalent antibodies on the cytokine release might be caused by some cross-linking of receptors because, for monovalent Fab fragments of ATROSAB and H398 in the same assays, no stimulatory activity could be discerned over a 4-log dose range (data not shown). However, when compared with the cellular response to TNF treatment, this minor activity of bivalent antibodies appears negligible, amounting at peak levels to 2–5% of a genuine TNF response. Moreover, on freshly isolated human peripheral blood T cells and granulocytes, no agonistic activity of the TNFR1 specific antibodies could be discerned in the TNF-dependent cellular response models of T-cell activation and O2- production, respectively (unpublished data).
Importantly, we could demonstrate binding of ATROSAB to rhesus TNFR1 with a similar affinity as for human TNF-R1, thus allowing in vivo evaluation of ATROSAB in rhesus monkeys. The collagen-induced arthritis (CIA) model is the recognized standard for potential RA therapeutics and could be already reproducibly induced in rhesus macaques.Citation29 Because of the well-established proximity (physiological, anatomical, genetic, microbiological and immunological) with humans, CIA in rhesus monkeys represents a very useful preclinical model for evaluation of safety and efficacy of novel therapiesCitation30 and enables the analysis of ATROSAB's neutralizing activity and safety in non-human primates.
TNFR1-selective antagonists, such as ATROSAB, may become new therapeutic options for diseases against which currently marketed anti-TNF therapeutics have failed or even exacerbate disease progression, including multiple sclerosis, congestive heart failure, metabolic diseases (e.g., type II diabetes), cytokine release syndrome, septic shock, acute (e.g., stroke) and chronic (e.g., Alzheimer and Parkinson disease) neurodegenerative diseases. The potential application of TNFR1 antagonists for central nervous system (CNS) diseases requires a thorough consideration of bioavailability. This point needs to be specifically addressed in further preclinical studies in appropriate animal disease models where the antibodies are crossreactive to the species TNFR1, such as baboon and rhesus monkeys or huTNFR1 k/i mouse lines that are presently under development. Irrespective of this, we argue that clinical experience suggests that in several acute or chronic inflammatory CNS diseases, e.g., MS, the blood brain barrier is disturbed and can be leaky to various extents, thus enabling access of IgG molecules to the site of wanted action.Citation31,Citation32 ATROSAB could be an especially useful therapeutic alternative in diseases already known to respond to anti-TNF treatment and particularly in those diseases where specific blockage of TNFR1 and maintenance of TNFR2 function appears as a promising therapeutic approach.
Materials and Methods
Materials.
Horseradish peroxidase (HRP)-conjugated anti-mouse IgG (Fc specific) antibody, HRP-conjugated anti-human IgG (whole molecule, Fc specific, Fab specific) antibodies, respectively, were purchased from Sigma (Taufkirchen, Germany). PE-labeled anti-mouse (whole molecule) and anti-human IgG (γ-chain specific) antibodies, respectively, were purchased from Sigma (Taufkirchen, Germany). Mouse embryonic fibroblasts (MEF) transfected with TNFR1-Fas (MEF-TNFR1-Fas) and TNFR2-Fas (MEF-TNFR2-Fas), respectively, were grown in RPMI 1640 medium, 5% FCS, 2 mM L-glutamine, 2 µg/ml puromycin.Citation22 The human rhabdomyosarcoma cell line Kym-1 was grown in RPMI 1640 medium, 10% FCS, 2 mM L-glutamine and HT1080wt cells and HeLa cells were grown in RPMI 1640 medium, 5% FCS, 2 mM L-glutamine.
Production of IZI-06.1 IgG (ATROSAB).
DNA encoding the light and heavy chain of ATROSAB, including Igκ signal sequences and codon-optimized for production in CHO cells, was produced synthetically (Geneart, Regensburg, Germany). The light chain (LC) DNA was cloned as BamHI/NotI fragment into shuttle vector pCV072 (Celonic GmbH, Jülich, Germany) and the heavy chain (HC) DNA was cloned as KasI/NheI fragment into pFUSE (InVivoGen, Toulouse, France). pFUSE-HC was digested with SmiI (SwaI) and the resulting blunt end fragment containing the entire HC expression cassette was cloned into pCV072-LC digested with PsiI. In this bicistronic expression cassette, the light chain is under the control of the PhEF1-HTLV promotor and the heavy chain gene is controlled by the PCMV enhanced promotor. The stably transfected CHO cells were grown in CDM4PermAB (Thermo Fischer, Erembodegem, Belgium) and cultivated in fed-batch mode in a 25 L wave bioreactor system (Sartorius Stedim, Melsungen, Germany) with a soy hydrolisate feeding solution (Kerry Biosciences, Almere, The Netherlands). Antibody was purified from cell culture supernatant by using Protein A chromatography (GE Healthcare, Uppsala, Sweden), followed by a membrane intermediate step with Sartobind Q single Sep mini (Sartorius Stedim. Melsungen, Germany). Final product was obtained via a buffer exchange step.
Production of TNFR1-Fc fusion proteins.
DNA encoding the extracellular region of human TNFR1 (aa 29–211), rhesus TNFR1 (aa 27–209) and mouse TNFR1 (aa 30–212) was produced synthetically (Geneart, Regensburg, Germany), introducing appropriate restriction sites between the individual domains and cloned into pSecTagL1-Fc (modified from pSecTag-FcHis).Citation33 Chimeric human/mouse TNFR1-Fc fusion proteins were generated by exchanging the different regions between human and mouse TNFR1-Fc. HEK293 cells were transfected with plasmid DNA using lipofectamine (Invitrogen, Karlsruhe, Germany) and stably transfected clones were selected in the presence of zeocin as described.Citation34 Cells were expanded in RPMI, 5% FCS, 2 mM L-glutamine to 90% confluence. For protein production, the medium was substituted with Opti-MEM I (Invitrogen, Karlsruhe, Germany) and supernatant was collected every 3–4 days. Proteins were purified from cell culture supernatant by protein A chromatography. In brief, supernatants were adjusted to pH 8 by adding 1/10 volume of 1 M TrisHCl, pH 8.0 and loaded onto a protein A-sepharose CL-4B column (Sigma, Taufkirchen, Germany). Bound protein was eluted with 100 mM glycine pH 3.0, neutralized by adding 1/10 volume 1 M TrisHCl, pH 8.0 and protein containing fractions were dialyzed against PBS. Protein concentrations were determined photometrically and purity was analyzed by SDS-PAGE and immunoblotting using an HRP-conjugated anti IgG (Fc specific) antibody (Sigma, Taufkirchen, Germany).
Protein characterization.
Size exclusion chromatography (SEC) was performed by HPLC using a BioSuite™ 250, 5 µm HR SEC (Waters GmbH, Eschborn, Germany). The following standard proteins were used: apoferritin (443 kDa), β-amylase (200 kDa), bovine serum albumin (67 kDa), carbonic anhydrase (29 kDa), aprotinin (6.5 kDa).
Affinity measurements.
Affinities of the antibodies were determined by quartz crystal microbalance measurements (QCM; Attana A-100 C-Fast system, Stockholm, Sweden). Binding experiments were performed in PBS 0.005% Tween 20 at a flow rate of 25 to 35 µl/min and temperature was controlled at 20°C. The TNFR1-Fc fusion proteins were chemically immobilized on an Attana carboxyl sensor chip by amine coupling at a concentration of 50 µg/ml according to the manufacturer's protocol resulting in a signal increase (frequency shift) of approximately 200 Hz. Antibodies were analyzed at concentration between 62.5 and 3.9 nM (4 measurements per concentration). The chip was regenerated with 10 mM glycine-HCl, pH 3.0. Buffer injections were performed prior to each sample injection to use as a reference in Attester Evaluation. Data were collected by Attester 3.0 (Version 3.1.1.8, Attana, Stockholm, Sweden) and analyzed by ClampXP.Citation35 A mass transport model was fitted to the data.Citation36
ELISA.
Recombinant human TNFR1-Fc fusion protein was immobilized in 96-well plates (50 ng/well in PBS) overnight at 4°C. After 2 h blocking with 2% (w/v) dry milk/PBS, recombinant antibody fragments were titrated in duplicates and incubated for 1 h at RT. Detection was performed with HRP-conjugated anti-human IgG (Fab-specific) antibody and HRP-conjugated anti-mouse IgG (Fc-specific) using TMB substrate (1 mg/ml TMB, sodium acetate buffer pH 6.0, 0.006% H2O2). The reaction was stopped with 50 µl of 1 M H2SO4. Absorbance was measured at 450 nm in an ELISA-reader.
Flow cytometry.
Binding to TNFR1-Fas or TNFR2-Fas transfected MEF cells was analyzed by flow cytometry. Cells (2 × 105) were incubated with dilution series of antibodies for 4 h at 4°C.Citation37 Cells were then washed with PBS and bound antibodies were detected with PE-labeled goat anti-mouse or anti-human antibody. Cells were analyzed by flow cytometry (Cytomics FC 500, Beckmann-Coulter, Krefeld, Germany). Data were evaluated with the program WinMDI, version 2.9 and fitted with GraphPrism software (La Jolla, CA, USA) from three independent binding curves.
Cytotoxicity.
Kym-1 cells (1.5 × 104 cells/100 µl) were grown in 96-well plates overnight. A constant amount of human soluble TNF (1.25 ng/ml in medium) was applied after pre-incubation with antibodies in triplicates (concentrations as indicated in the figures) in medium for 1 h. After 7 h, cells were stained by crystal violet (20% methanol, 0.5% crystal violet) for 15 min. The wells were washed with H2O and air-dried. The dye was resolved with methanol for 15 min and optical density at 550 nm was determined (Tecan infinite M200, Crailsheim, Germany).
IL-6 and IL-8 assays.
HT1080 cells (2.0 × 105 cells/100 µl) were grown in 96-well plates overnight. The next day, the medium was exchanged to remove constitutively produced IL-8 and the cells were incubated in duplicates together with serial dilutions of human soluble TNF for additional 18 h. Induction of IL-8 production and secretion into the culture supernatant was determined by an IL-8-Sandwich ELISA (ImmunoTools, Friesoythe, Germany) according to the manufacturer's protocol. In addition, cells were incubated with serial dilutions of antibodies in the absence and presence of TNF (1 ng/ml) and analyzed for IL-8 secretion after 18 h of incubation. In the same way, we analyzed the inhibitory effects of the antibodies on TNF-mediated secretion of IL-6 from HeLa cells using an IL-6 sandwich ELISA (ImmunoTools, Friesoythe, Germany) according to the manufacturer's protocol.
Plasma half-life.
CD1 mice (18–22 weeks, weight between 34 and 43 g) received an intravenous injection of 25 µg purified antibody (ATROSAB, H398) in a total volume of 150 µl. Blood samples (50 µl) were taken at 3 min, 1, 2, 4 and 7 days from the tail and incubated on ice for 15 min. Clotted blood was centrifuged at 13,000 g for 10 min and serum samples stored at −20°C. Serum concentrations were then determined by ELISA as described above. Terminal half-lives were calculated using concentrations at day 1 to 7.
Abbreviations
| ATROSAB | = | anti-TNF receptor one-specific antibody |
| TNF | = | tumor necrosis factor |
| TNFR1 | = | TNF receptor 1 |
Figures and Tables
Figure 1 Characterization of ATROSAB. (A) SDS-PAGE analysis of purified ATROSAB (4 µg/lane, Coomassie staining) analyzed under non-reducing (1) or reducing (2) conditions. (B) Size exclusion chromato-graphy of ATROSAB (the position of standard proteins is indicated). (C) ELISA of ATROSAB and H398 for binding to human TNFR1-Fc.
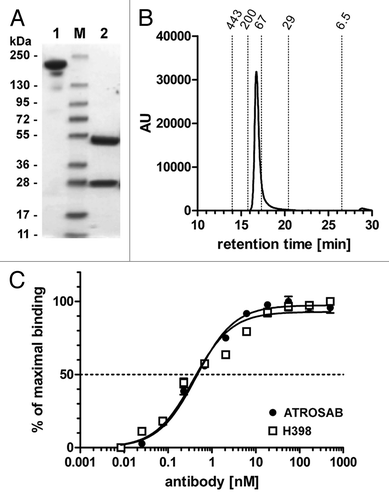
Figure 2 Flow cytometry analysis of binding of ATROSAB to mouse embryonic fibroblasts (MEF) transfected with human TNFR1-Fas (A) or human TNFR2-Fas (B). (C) Titration of binding of ATROSAB and H398 to MEF-TNFR1-Fas (n = 3).
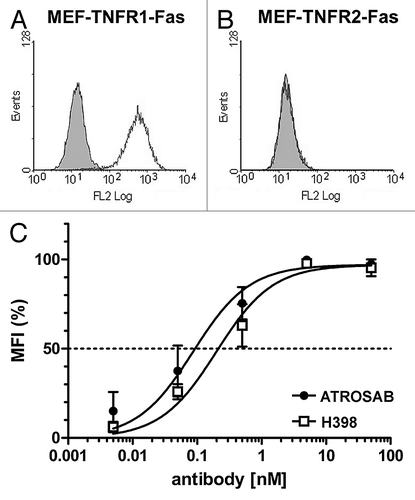
Figure 3 (A) SDS-PAGE analysis of purified human TNFR1-Fc (1 and 4), mouse TNFR1-Fc (2 and 5) and rhesus TNFR1-Fc (3 and 6) (4 µg/lane, Coomassie staining) analyzed under reducing (1–3) and non-reducing (4–6) conditions. (B) ELISA of binding of ATROSAB and H398 (5 µg/ml) to purified human TNFR1-Fc, rhesus TNFR1-Fc and mouse TNFR1-Fc (100 ng/well). Binding was detected by HRP-conjugated anti-moIgG (Fc-specific) antibody or anti-human Fab antibody, respectively. Binding of an anti-human Fc antibody (anti-Fc) was included as coating control.
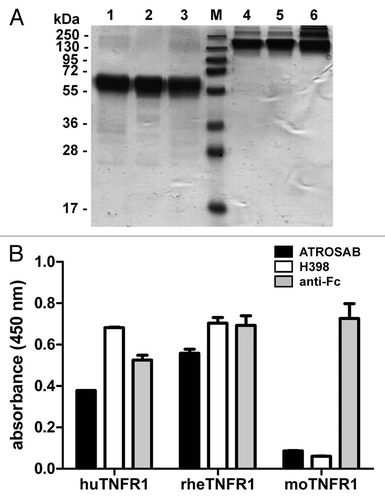
Figure 4 Determination of affinity of H398 and ATROSAB for binding to human and rhesus TNFR1-Fc by quartz crystal microbalance (QCM) measurements. (A) Binding of H398 to human TNFR1-Fc, (B) binding of ATROSAB to human TNFR1-Fc, (C) binding of H398 to rhesus TNFR1-Fc and (D) binding of ATROSAB to rhesus TNFR1-Fc.
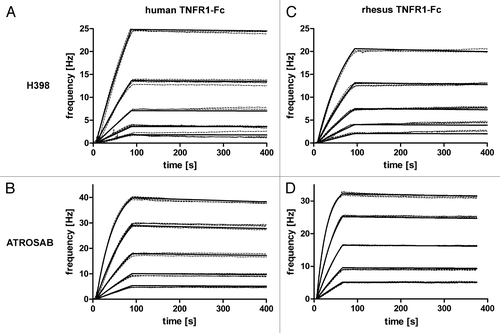
Figure 5 Inhibition of TNF-mediated cytotoxicity (1.25 ng/ml TNF) on Kym-1 cells by ATROSAB and H398. Cells were analyzed after 6 h by crystal violet staining (n = 3). Maximum (10% viability of control) and half maximum (55% viability of control) are displayed in dotted lines.
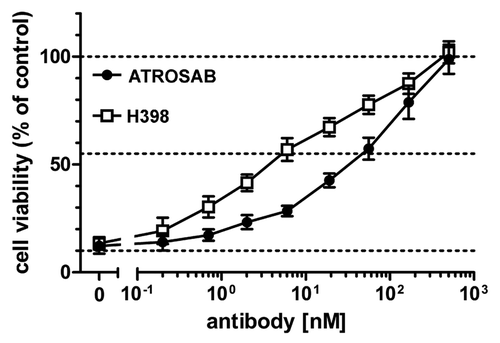
Figure 6 Inhibition of IL-6 and IL-8 secretion induced by TNF by ATROSAB and H398. HeLa cells (A) or HT1080 cells (B) were incubated with TNF (1 ng/ml) and increasing concentrations of ATROSAB or H398 and cytokine secretion were determined by ELISA (n = 3). Human IgG (huIgG) was incluced as negative control. In the same way, effects of antibodies on cytokine secretion in the absence of TNF were determined. Compared with TNF, both antibodies had only marginal effects on IL-6 (C) and IL-8 (D) secretion.
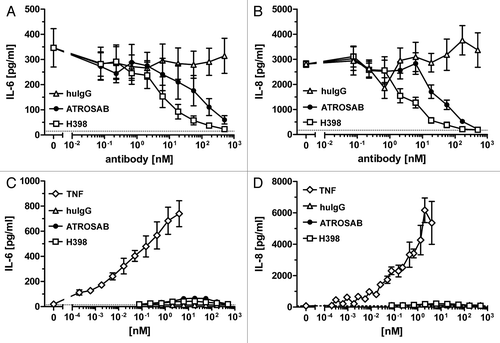
Figure 7 Plasma half-lives of ATROSAB and H398 after a single dose i.v. injection (25 µg) into CD1 mice. Serum concentrations of antibodies were determined by ELISA.
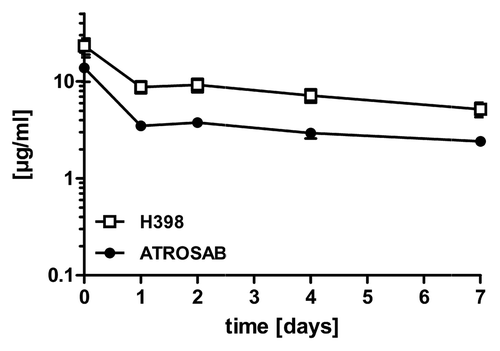
Figure 8 (A) Epitope mapping of ATROSAB and H398 using wild-type and chimeric human/mouse TNFR1-Fc fusion proteins. Antibodies (0.1 nM) were analyzed by ELISA for binding to the TNFR1-Fc fusion proteins. His-tagged human TNF (huTNF) was included as control. (B) Sequence comparison of the identified epitope region (aa 1–70) of human (huTNFR1), mouse (moTNFR1) and rhesus (rhTNFR1) TNFR1. Cysteine residues are marked with grey boxes and the two positions (P23, Q24) analyzed by site-directed mutagenesis are marked by asterisks.
Figure 9 (A) Structure of TNF (red) bound to TNFR1 (blue). The identified epitope region is marked in green. (B) A single TNFR1 chain. The two positions (P23, Q24) identified by mutagenesis to contribute to binding of ATROSAB and H398 are highlighted in dark green.
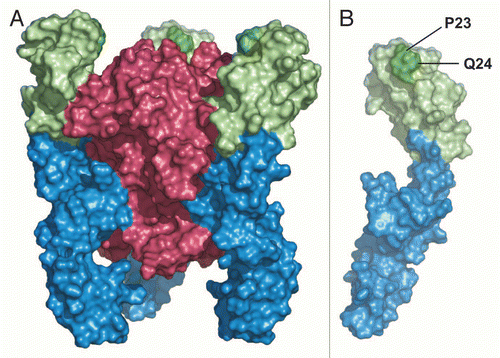
Table 1 Binding kinetics of H398 and ATROSAB
Acknowledgements
This work was supported by a grant from EC FP6, Project NeuroproMiSe, contract # LSHM-CT-2005-018637.
References
- Tracey D, Klareskog L, Sasso EH, Salfeld JG, Tak PP. Tumor necrosis factor antagonist mechanisms of action: a comprehensive review. Pharmacol Ther 2008; 117:244 - 279
- Kontermann RE, Scheurich P, Pfizenmaier K. Antagonists of TNF action—clinical experience and new developments. Expert Opin Drug Discov 2009; 4:279 - 292
- Schiff MH, Burmester GR, Kent JD, Pangan AL, Kupper H, Fitzpatrick SB, et al. Safety analyses of adalimumab (HUMIRA) in global clinical trials and US postmarketing surveillance of patients with rheumatoid arthritis. Ann Rheum Dis 2006; 65:889 - 894
- Bongartz T, Sutton AJ, Sweeting MJ, Buchan I, Matteson EL, Montori V. Anti-TNF antibody therapy in rheumatoid arthritis and the risk of serious infections and malignancies: systematic review and meta-analysis of rare harmful effects in randomized controlled trials. JAMA 2006; 295:2275 - 2285
- Desai SB, Furst DE. Problems encountered during anti-tumour necrosis factor therapy. Best Pract Res Clin Rheumatol 2006; 20:757 - 790
- Wallis RS. Tumour necrosis factor antagonists: structure, function and tuberculosis risks. Lancet Infect Dis 2008; 8:601 - 611
- Locksley RM, Killeen N, Lenardo MJ. The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell 2001; 104:487 - 501
- Wajant H, Pfizenmaier K, Scheurich P. Tumor necrosis factor signaling. Cell Death Differ 2003; 10:45 - 65
- Wallach D, Kovalenko A. 12th international TNF conference: the good, the bad and the scientists. Cytokine Growth Factor Rev 2009; 20:259 - 269
- TNF neutralization in MS: results of a randomized, placebo-controlled multicenter study. The Lenercept Multiple Sclerosis Study Group and The University of British Columbia MS/MRI Analysis Group. Neurology 1999; 53:457 - 465 Anonymous
- Arnett HA, Mason J, Marino M, Suzuki K, Matsushima GK, Ting JP. TNFalpha promotes proliferation of oligodendrocyte progenitors and remyelination. Nat Neurosci 2001; 4:1116 - 1122
- Shibata H, Yoshioka Y, Ohkawa A, Minowa K, Mukai Y, Abe Y, et al. Creation and X-ray structure analysis of the tumor necrosis factor receptor-1-selective mutant of a tumor necrosis factor-alpha antagonist. J Biol Chem 2008; 283:998 - 1007
- Shibata H, Yoshioka Y, Ohkawa A, Abe Y, Nomura T, Mukai Y, et al. The therapeutic effect of TNFR1-selective antagonistic mutant TNFalpha in murine hepatitis models. Cytokine 2008; 44:229 - 233
- Shibata H, Yoshioka Y, Abe Y, Ohkawa A, Nomura T, Minowa K, et al. The treatment of established murine collagen-induced arthritis with a TNFR1-selective antagonistic mutant TNF. Biomaterials 2009; 30:6638 - 6647
- Zalevsky J, Secher T, Ezhevsky SA, Janot L, Steed PM, O'Brien C, et al. Dominant-negative inhibitors of soluble TNF attenuate experimental arthritis without suppressing innate immunity to infection. J Immunol 2007; 179:1872 - 1883
- Olleros ML, Vesin D, Lambou AF, Janssens JP, Ryffel B, Rose S, et al. Dominant-negative tumor necrosis factor protects from Mycobacterium bovis Bacillus Calmette Guerin (BCG) and endotoxin-induced liver injury without compromising host immunity to BCG and Mycobacterium tuberculosis. J Infect Dis 2009; 199:1053 - 1063
- Thoma B, Grell M, Pfizenmaier K, Scheurich P. Identification of a 60 kD tumor necrosis factor (TNF) receptor as the major signal transducing component in TNF responses. J Exp Med 1990; 172:1019 - 1023
- Kruppa G, Thoma B, Machleidt T, Wiegmann K, Kronke M. Inhibition of tumor necrosis factor (TNF)-mediated NFkappaB activation by selective blockade of the human 55 kDa TNF receptor. J Immunol 1992; 148:3152 - 3157
- Moosmayer D, Dubel S, Brocks B, Watzka H, Hampp C, Scheurich P, et al. A single-chain TNF receptor antagonist is an effective inhibitor of TNF mediated cytotoxicity. Ther Immunol 1995; 2:31 - 40
- Kontermann RE, Munkel S, Neumeyer J, Muller D, Branschadel M, Scheurich P, et al. A humanized tumor necrosis factor receptor 1 (TNFR1)-specific antagonistic antibody for selective inhibition of tumor necrosis factor (TNF) action. J Immunother 2008; 31:225 - 234
- Armour KL, Clark MR, Hadley AG, Williamson LM. Recombinant human IgG molecules lacking Fcgamma receptor I binding and monocyte triggering activities. Eur J Immunol 1999; 29:2613 - 2624
- Krippner-Heidenreich A, Tubing F, Bryde S, Willi S, Zimmermann G, Scheurich P. Control of receptor-induced signaling complex formation by the kinetics of ligand/receptor interaction. J Biol Chem 2002; 277:44155 - 44163
- Terato K, Arai H, Shimozuru Y, Fukuda T, Tanaka H, Watanabe H, et al. Sex-linked differences in susceptibility of cynomolgus monkeys to type II collagen-induced arthritis. Evidence that epitope-specific immune suppression is involved in the regulation of type II collagen autoantibody formation. Arthritis Rheum 1989; 32:748 - 758
- Banner DW, D'Arcy A, Janes W, Gentz R, Schoenfeld HJ, Broger C, et al. Crystal structure of the soluble human 55 kd TNF receptor-human TNFbeta complex: implications for TNF receptor activation. Cell 1993; 73:431 - 445
- Branschadel M, Aird A, Zappe A, Tietz C, Krippner-Heidenreich A, Scheurich P. Dual function of cysteine rich domain (CRD) 1 of TNF receptor type 1: conformational stabilization of CRD2 and control of receptor responsiveness. Cell Signal 2009; 22:404 - 414
- Chan FK, Chun HJ, Zheng L, Siegel RM, Bui KL, Lenardo MJ. A domain in TNF receptors that mediates ligand-independent receptor assembly and signaling. Science 2000; 288:2351 - 2354
- Siegel RM, Frederiksen JK, Zacharias DA, Chan FK, Johnson M, Lynch D, et al. Fas preassociation required for apoptosis signaling and dominant inhibition by pathogenic mutations. Science 2000; 288:2354 - 2357
- Boschert V, Krippner-Heidenreich A, Branschadel M, Tepperink J, Aird A, Scheurich P. Single chain TNF derivatives with individually mutated receptor binding sites reveal differential stoichiometry of ligand receptor complex formation for TNFR1 and TNFR2. Cell Signal 2010; 22:1088 - 1096
- Bakker NP, van Erck MG, Zurcher C, Faaber P, Lemmens A, Hazenberg M, et al. Experimental immune mediated arthritis in rhesus monkeys. A model for human rheumatoid arthritis?. Rheumatol Int 1990; 10:21 - 29
- t'Hart BA, Amor S, Jonker M. Evaluating the validity of animal models for research into therapies for immune-based disorders. Drug Discov Today 2004; 9:517 - 524
- Minagar A, Alexander JS. Blood-brain barrier disruption in multiple sclerosis. Mult Scler 2003; 9:540 - 549
- Waubant E. Biomarkers indicative of blood-brain barrier disruption in multiple sclerosis. Dis Markers 2006; 22:235 - 244
- Muller D, Trunk G, Sichelstiel A, Zettlitz KA, Quintanilla M, Kontermann RE. Murine endoglin-specific single-chain Fv fragments for the analysis of vascular targeting strategies in mice. J Immunol Methods 2008; 339:90 - 98
- Muller D, Karle A, Meissburger B, Hofig I, Stork R, Kontermann RE. Improved pharmacokinetics of recombinant bispecific antibody molecules by fusion to human serum albumin. J Biol Chem 2007; 282:12650 - 12660
- Myszka DG, Morton TA. CLAMP: a biosensor kinetic data analysis program. Trends Biochem Sci 1998; 23:149 - 150
- Myszka DG. Kinetic analysis of macromolecular interactions using surface plasmon resonance biosensors. Curr Opin Biotechnol 1997; 8:50 - 57
- Benedict CA, MacKrell AJ, Anderson WF. Determination of the binding affinity of an anti-CD34 single-chain antibody using a novel, flow cytometry based assay. J Immunol Methods 1997; 201:223 - 231