Abstract
Therapeutic proteins circulating in blood are in a highly crowded, redox environment at high temperatures of ~37°C. These molecules circulate in the presence of enzymes and other serum proteins making it difficult to predict from in vitro studies the stability, aggregation or pharmacokinetics of a therapeutic protein in vivo. Here, we describe use of a high throughput capillary electrophoresis based microfluidic device (LabChip GXII) to obtain pharmacokinetics (PK) of a fluorescently labeled human mAb directly from serum. The non-labeled and labeled mAbs were evaluated in single dose rat PK studies using a traditional ELISA method or LabChip GXII, respectively. The fluorescent dye did not significantly alter clearance of this particular mAb, and PK parameters were comparable for labeled and unlabeled molecules. Further, from the CE profile we concluded that the mAb was resistant to fragmentation or aggregation during circulation. In a follow-up experiment, dimers were generated from the mAb using photo-induced cross-linking of unmodified proteins (PICUP) and labeled with the same fluorophore. The extent of dimerization was incomplete and some monomer and higher molecular weight species were found in the preparation. In rat PK studies, the serum concentration-time profile of the three entities present in the dimer preparation could be followed simultaneously with the GXII technology. While further studies are warranted, we believe this method could be adapted to obtain PK of different forms of antibodies (oxidized, deamidated or various glycosylated species) and other proteins.
Introduction
The science of analyzing drugs in biological fluids (bioanalysis) can be traced back to the early 1930s. An excellent review by Howard Hill describes the evolution of various technologies utilized for analyzing small molecules, and in recent years for large therapeutic proteins.Citation1 These technologies include GC chromatography in the 1950s, high-performance liquid chromatography (HPLC) methods in the 1960s and early 1970s, electrophoretic techniques in the 1970s, followed by various forms of ligand-binding assays, e.g., radio-immunoassays, enzyme-linked immunoassays and more recently the introduction of miniaturized, high throughput methods such as the automated Gyros system (Gyros AB, Uppsala, Sweden). Miniaturization of analytical methods (‘lab-on-a-chip’ approach) combined with higher throughput has resulted in a surge of publications in the application of microfluidics in the field of bioanalysis.Citation1,Citation2 The major advantages of miniaturization are that they allow investigators to analyze small volumes of material with very good sensitivity, provide better utilization of reagents, frequently use disposable technology, provide faster sampling times, remove the need for manual pipetting, and, importantly, provide versatility in detector capabilities.Citation2,Citation3 Detectors that are added to these devices range from optical to electrochemical detection and mass spectrometry-based methods.Citation4 Within the optical-based methods, fluorescence excitation and emission detector systems are popular due to their relative simplicity of use; however, more advanced methods include the use of fluorescence lifetime imaging (FLIM) and the many variations of surface enhanced Raman scattering (SERS).Citation5
The use of capillary electrophoresis (CE) to separate, by size, either nucleic acids, proteins or peptides, carbohydrates, lipids or single cells, as well as the use of CE to perform functional assays and monitor biomolecular interactions, is very well documented.Citation6 Microfluidic chip capillary electrophoresis sodium dodecyl sulfate (CE-SDS) is a robust platform that has found a wide range of applications in early discovery and process development. Xiaoyu Chen and colleagues described the use of microfluidic CE-SDS in bioproduction for analysis of glycans, fragments and aggregates.Citation7-Citation9 The technology was also utilized for titer evaluation and for the evaluation of the purity profiles of cell culture and fermentation samples. The authors argue that this is no longer a “nice-to-have” but a “must-have” enabling technology to meet the challenge of quickly bringing well-characterized drugs to the market.
Therapeutic proteins circulating in blood are immersed in a highly crowded, redox environment at temperatures of ~37°C.Citation10 Enzymes and other serum proteins could influence their behavior in vivo. Therefore, identification of high throughput technologies that help select, early in discovery, molecules that are stable in serum and show good pharmacokinetic (PK) properties is desirable because it could facilitate rapid screening of a larger pool of candidates. In addition to evaluating the PK of the molecules, more detailed understanding of their fate in the circulation such as aggregation, fragmentation, non-specific binding to serum proteins or rapid clearance is especially desirable. Further, technologies that facilitate analysis directly in serum are more advantageous in early discovery settings because they eliminate the need to run laborious methods that require extensive method development and internal standards to account for extraction inefficiencies.
LabChip GXII separation is based on size, with the resolution ranging from 14 to ~300 kDa. Analysis of each sample is fast, occurring in 40 sec, and the samples are analyzed directly from a 96-well plate. There was a 4 log linear range of detection (lower limit of detection was about 10 pg/μL) for pre-labeled proteins. Fluorophore conjugation to protein was enabled with the Pico Protein dye provided by the manufacturer. In this report, we used the LabChip GXII to analyze the serum concentration of a pre-labeled monoclonal antibody (mAb) that was administered intravenously to rats and also to evaluate the clearance of mAb dimers and other high molecular weight species that were created using the PICUP technology.Citation11 The cross-linking method resulted in formation of stable covalently linked aggregates. The types of aggregates created by the PICUP method are potentially different from those found in drug product after long-term storage. However, the intent of the present study was to evaluate clearance of stable covalent aggregates and not reversible aggregates, which may be of less concern.
The monoclonal antibody selected for the study was a well-characterized human IgG1 molecule with greater than 99% monomer content that targets a soluble antigen. The first question addressed in this study was whether the fluorophore had an effect on the PK of the antibody. To this end, unlabeled and labeled mAb were evaluated in single dose rat PK studies using either the traditional ELISA method or GXII respectively. In a follow-up experiment, dimers were generated from the same mAb using photo induced cross-linking of unmodified proteins (PICUP). The covalent dimer was also conjugated with a fluorophore and evaluated in rat PK. Dimerization was not complete, approximately 10% monomer was still present in the reaction mixture and, in addition, ~17% high molecular weight species was also formed during the cross-linking process. These results were confirmed by analytical ultracentrifugation. In a rat IV pharmacokinetic study, the serum concentration-time profile of the three entities present in the dimer solution could be followed simultaneously with the help of the GXII technology. This technology is especially promising because the method is applicable in early discovery settings but could be adapted to obtain PK of different antibody forms (oxidized, deamidated, various glycosylated species) and other therapeutic proteins such as enzymes. Future work will include a larger panel of mAbs to correlate PK obtained using both the GXII and traditional ELISA based methods.
Results
Incorporation of a fluorescent tag
Rapid evaluation of therapeutic proteins in vivo is an important part of candidate selection in discovery. To avoid time consuming development of quantitation methods for each candidate, a generic high throughput method was evaluated that is based on pre-labeling the protein with a small, high intensity, stable fluorophore, followed by sample analysis, including quantitation, on a capillary electrophoresis system under non-reducing conditions. We labeled the molecule with the Pico Protein dye supplied by the manufacturer. The fluorescent dye reacts with primary amines, thus labeling lysine residues and N-terminal amines. After quenching of the labeling reaction, the extent and position of the incorporated label was monitored by mass spectrometry after the molecule was reduced to its heavy and light chain subunits. The heavy chain has 31 lysine residues and the light chain has 11 lysine residues. As seen in , we observed a Gaussian distribution of label incorporated into the heavy chain (HC) where about 78% of the HC had incorporated 3–6 molecules and about 85% of the light chain (LC) had incorporated 1–3 molecules. We used surface plasmon resonance (Biacore) to evaluate antigen binding to labeled mAb. Fluorescently tagged mAb, captured on a CM-5 chip using goat anti-Fc antibody, was able to bind antigen, which suggested that the labeled mAb was correctly folded and the fluorescent tag did not interfere with antigen binding (results not shown).
Table 1. Fluorescent label incorporated (%) into heavy and light chain as determined by mass spectrometry
Sensitivity evaluation of labeled mAb in rat serum
The labeled mAb was spiked into Sprague Dawley (SD) rat serum at a concentration of 100 μg/mL and serially diluted to a final concentration of 0.2 μg/mL. Shown in is an overlay of the different electropherograms obtained at each concentration where in addition to the monomer, we also observed the presence of low amounts of aggregates and fragmented molecules that were easily resolved. The data viewed in digital gel format confirmed the presence of the labeled antibody, a small amount of aggregate, and fragments (). Plotting concentration of the labeled mAb against fluorescence intensity of the monomer () revealed good correlation (R2 = 0.99) in the concentration range evaluated.
Figure 1. Analysis of fluorescence on GXII: (A) Electropherogram of labeled mAb serially diluted in serum (B) Standard curve obtained after plotting fluorescence intensity vs. concentration (μg/mL) of the labeled monoclonal antibody spiked into rat serum.
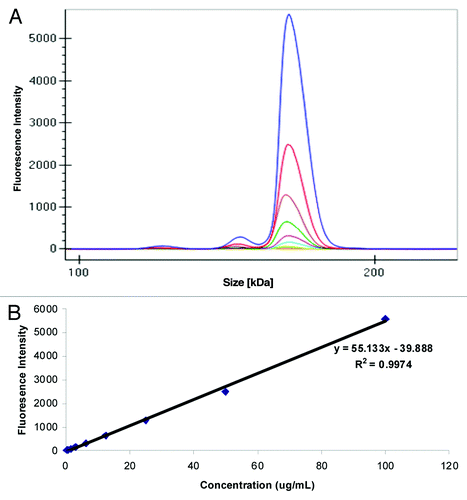
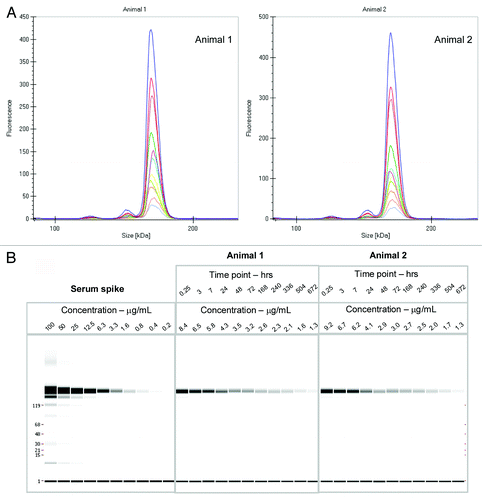
Figure 2. (A) Electropherogram of rat serum samples (two animals) dosed with labeled mAb and analyzed at various time points (0.25 h to day 28) (B) Digital gel image after serial dilution of labeled mAb spiked into rat serum in vitro, and analysis of rat serum samples from PK study with labeled antibody after a 1:10 dilution in denaturation buffer.
Pharmacokinetics of labeled mAb after intravenous administration in rats
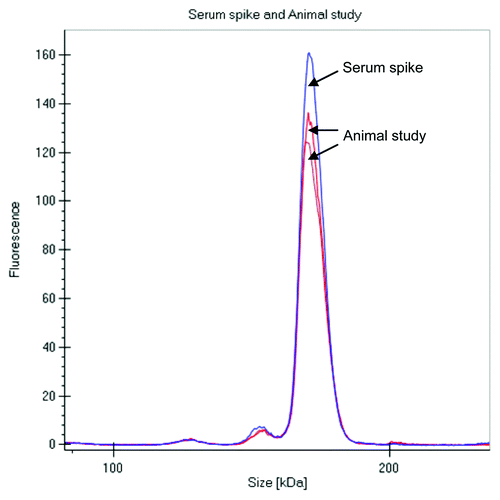
The labeled mAb was intravenously administered to two Sprague Dawley (SD) rats at 4 mg/kg dose, and serum samples were collected at multiple time points over a month, stored frozen and protected from light. When ready for analysis, the samples were diluted in the denaturation buffer provided by the manufacturer and directly analyzed on the GXII. There was no need for any extra purification or a clean up step prior to analyzing the samples. Data was either visualized as an electropherogram () or in digital gel format (); both indicated that the overall profiles of the antibody were similar out to 28 d. The electropherogram of labeled mAb analyzed from the animals at later time points were comparable to the labeled mAb directly spiked into serum (shown in ), with no additional fragments or novel high molecular weight species observed over time, indicating that the antibody was stable in the circulation over an extended period of time.
Figure 3. Comparison of electropherograms of labeled mAb spiked into serum and analysis after 3 d in a rat PK study (2 rats).
Comparison of the pharmacokinetic profile of labeled and non-labeled mAb
To evaluate if the fluorescent label incorporation had an effect on the PK properties of the antibody, the PK profile of the fluorescently tagged mAb was compared with historical PK data obtained with the native mAb in rats. The concentration-time profile of the labeled mAb analyzed on the GXII and compared with the PK profile of non-labeled mAb is shown in . The concentration of labeled mAb in rat serum was directly calculated from a calibration curve that plotted known spikes of labeled mAb against fluorescence intensity (shown in ). The concentration of non-labeled mAb was obtained using a conventional ELISA based assay.
Figure 4. Comparison of the pharmacokinetic profile of labeled mAb, analyzed on the GXII, vs. the non-labeled mAb analyzed by conventional ELISA methods revealed similar profiles.
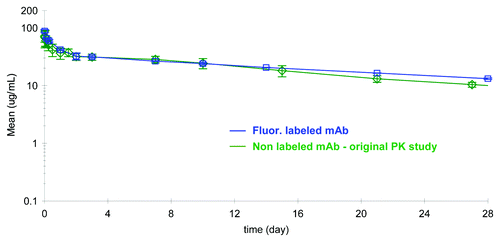
The PK profiles obtained using two different methods were remarkably similar and the PK parameters calculated from the two studies were comparable (). Unlabeled mAb and fluorescently tagged mAb exhibited low serum clearance of 0.20 or 0.16 mL/h/kg and small volumes of distribution of 104 or 113 mL/kg, respectively. The fluorescently tagged mAb appeared to have a somewhat longer average terminal half-life of 21 d in the two rats tested compared with the native protein half-life of 14 d (n = 4). This data revealed that the fluorophore attached to the mAb via lysine residues did not negatively influence the PK of the molecule, and additionally suggested that there was no indication of rat anti-human antibody response against either the native or the labeled protein.
Table 2. Pharmacokinetic parameters of labeled and non-labeled mAb
Preparation of covalent dimer using the PICUP method
Photo-induced coupling of unmodified proteins (PICUP) is a rapid and highly efficient method to cross-link proteins through the formation of a covalent bond between adjacent amino acids. Ruthenium(II) was photolyzed in the presence of persulfate to form Ru(III), which is a strong one electron oxidant. The Ru(III) is speculated to then attack residues such as tyrosine on the protein, thus creating a highly reactive radical that would either couple with a neighboring tyrosine residue (homo-atom linkage) or a nearby lysine or cysteine group (hetero-atom linkage) to cross-link proteins.
Using the PICUP method, we were able to create a large amount of covalently cross-linked dimer that was further purified using a preparative SEC column (data not shown). The purity of the covalent dimer was confirmed using analytical ultracentrifugation () and GXII ( and ). Interestingly, the presence of a small amount of residual monomer (10%) and higher molecular weight (HMW) aggregate (17%) was observed even after the purification step. The HMW aggregate was found to be primarily tetramers and low levels of hexamer ().
Figure 5. Purity and molecular mass of the cross-linked dimer verified by sedimentation velocity experiments.
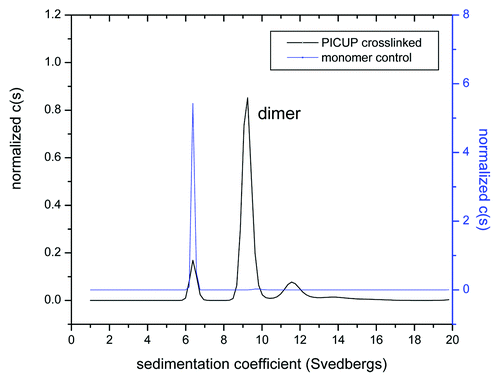
Figure 6. (A) Electropherogram of labeled dimer spiked into serum (B) Standard curve obtained after plotting fluorescence intensity vs. concentration (μg/mL) of labeled dimer.
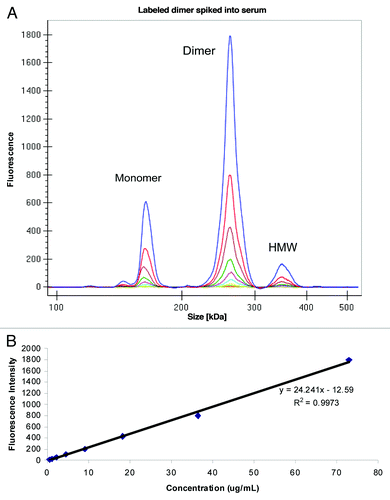
Figure 7. (A) Electropherogram of labeled dimer analyzed from serum of two animals after various time points following a 1:10 dilution in denaturation buffer (0.25 h to day 28) (B) Digital gel image after serial dilution of labeled dimer in serum in vitro and analysis of PK serum samples from two animals following a 1:10 dilution in denaturation buffer.
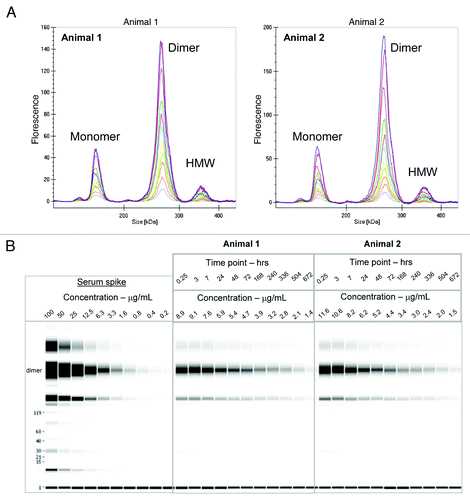
Table 3. Composition of the purified dimer preparation: identification of different species obtained in sedimentation velocity experiments
Evaluation of labeled dimer spiked into serum
To enable analysis of the dimer directly from serum, we labeled the purified dimer preparation with the Pico Protein dye supplied by the manufacturer. The extent of incorporation of dye was not evaluated in the dimer; however, the overall intensity of the dimer was found to be lower than the labeled monomer in the previous study. The labeled dimer preparation was then spiked into serum at a total protein concentration of 100 μg/mL and serially diluted to a final concentration of 0.2 μg/mL and analyzed on GXII. Because the GXII is a size-based separation method, we were able to obtain the intensity of the dimer peak without interference from either the monomer or the HMW species. An overlay of the different electropherograms obtained at each concentration of labeled dimer is shown in . The small amount of monomer and HMW species that co-purified with the dimer was easily resolved. Shown in is the data viewed in digital gel format that also showed the monomer and HMW species. A calibration curve obtained by plotting concentration of labeled dimer (73% of total protein spiked) against fluorescence intensity () revealed good correlation (R2 = 0.99) in the concentration range evaluated. Similar calibration curves (data not shown) could be obtained for the monomer (10%) and HMW species (17%).
Analysis of labeled dimer after administration to rats
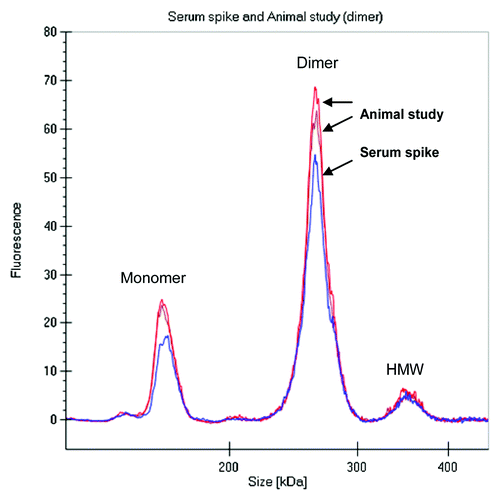
During the drug discovery process, early antibody purifications often result in heterogeneous mixtures containing monomer and some higher molecular weight species, e.g., dimers or oligomers. To evaluate if the PK of a mAb dimer might be different from that of a monomer, we compared the rat PK of the covalently linked fluorescently labeled dimer preparation described above to the PK properties of the fluorescent labeled monomer obtained earlier in Sprague Dawley (SD) rats. The labeled dimer was intravenously administered to two SD rats at 4 mg/kg dose, serum samples were collected and directly analyzed on the GXII. Data was either visualized as an electropherogram () or in digital gel format (). The electropherogram of labeled dimer analyzed from the animals was comparable to labeled dimer directly spiked into serum (shown in ).
Figure 8. Electropherogram of labeled dimer analyzed after spiking into serum and after recovery from animal (3 d) revealed similar profile.
Comparison of the pharmacokinetic profile of labeled mAb and labeled dimer
The comparison of the PK profiles of the labeled mAb and labeled dimer as analyzed on the GXII is shown in . The concentration of labeled dimer was calculated from a calibration curve that plotted known spikes of labeled dimer against fluorescence intensity of the dimer (). The PK profiles of the labeled mAb and labeled dimer were very similar (Fig. Nine and ) with the dimer showing a somewhat shorter terminal half-life (17 vs. 21 d) and the volume of distribution of the labeled dimer (Vss of 76 ml/kg) somewhat less than that of the labeled mAb (Vss of 113 ml/kg). While this data may be within the experimental error of the method, it is also possible that the dimer, because of its larger size, was not able to extravasate into the tissues as effectively as the mAb.
Figure 9. Comparison of the pharmacokinetic profile of labeled mAb and labeled dimer (n = 2 each) analyzed on the GXII revealed similar profiles.
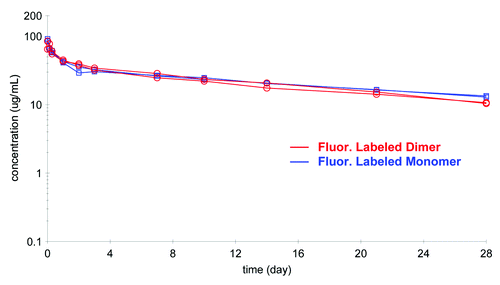
Table 4. Pharmacokinetic parameters of labeled monomer and dimer
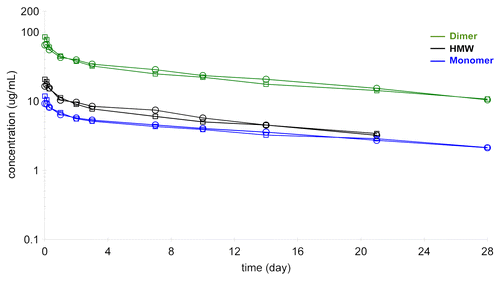
In the same experiment, it was possible to follow the serum profile of the other fractions in the dimer preparations, i.e., the leftover monomer (10% of total protein) and the high molecular weight species (17% of total protein). These molecules also showed the biphasic decline with extended elimination phase that is typical of antibodies (). The HMW species appeared to have comparable half-life to the dimer (16 d), which might be explained by the observation that the material was not actually high molecular weight aggregate, but a mixture of relatively small oligomers, tetramers and hexamer
Figure 10. Pharmacokinetic profile of purified dimer preparation, labeled monomer (10%) and labeled HMW fraction (17%) analyzed on the GXII.
Discussion
mAb therapeutic protein candidates in a variety of forms are filling the pipelines of major pharmaceutical and biotechnology companies. At least 30 antibody-based therapeutic proteins have been approved by the United States Food and Drug Administration.Citation12,Citation13 As of 2010, the number of mAb therapeutics in clinical development was estimated at ~240 with at least 40 novel molecules entering the clinic every year.Citation14 In such a competitive environment, the need for reliable high throughput methods to eliminate poor candidates early in the discovery cycle is desirable. Evaluation of the PK properties of therapeutic molecules and evaluation of physicochemical properties of these molecules are generally conducted by different groups and at different stages of development. Physicochemical characteristics such as aggregation, fragmentation and stability at 40°C are analyzed later in development in an optimized formulation buffer. However, PK parameters are generally obtained on a subset of candidates much earlier in the discovery phase. Correlating physicochemical properties to PK parameters is difficult and the use of in vitro stability assays to predict PK has not been established yet. Therefore, rodent PK is typically used directly to select the best candidates for development.
Therapeutics circulating in blood can be degraded by enzymes and serum components, and it is difficult to predict which molecules will tolerate the environment best.Citation10 Using the LabChip GXII we provide a means to capture in a single study both physicochemical characteristics and PK parameters of candidate molecules in a relatively higher throughput manner, directly from serum, with minimal sample manipulation. The GXII can also facilitate analysis of aggregates, fragments and novel adducts. The extended half-life of labeled mAb (21 d) vs. unlabeled mAb (14 d) is interesting. Since the number of animals used in the study was small, further studies are warranted to determine if these differences were significant or within experimental error. However, it is possible that modification of positively charged residues (lysines) with the fluorescent tag has the effect of lowering the isoelectric point (pI) of the labeled mAb-1, thereby extending its half-life. The other interesting observation in the present study was the comparable half-life of the labeled dimer (17 d) vs. labeled monomer (21 d). This is especially noteworthy as previous studies reveal that IgG molecules contain two FcRn-binding sites; further, reports of increased binding of aggregates to the FcRn receptor would predict an extended half-life for the labeled dimer.Citation15,Citation16 It is possible however, that the dimer was recognized more as a small immune complex and alternative immune complex clearance pathways contributed to its elimination. Further work is necessary with multiple antibodies and dimers to confirm the half-life trends observed.
Addition of the high intensity fluorescent label did not facilitate extensive clearance of the antibody or induce excessive aggregate formation (as often feared when introducing hydrophobic labels). Therefore, this methodology could be recommended for the early evaluation of different antibody preparations (e.g., glycoforms) or different proteins in vivo. Small animal body optical imaging using fluorescent probes for either ex vivo studies of isolated organs or whole body in vivo studies to evaluate distribution is a popular tool in pre-clinical work.Citation5,Citation17 Many of the fluorophores used in these studies emit in the near infrared spectrum as tissue absorption and auto fluorescence are reduced at these wavelengths.Citation5 Using the GXII we now present the possibility of also collecting physicochemical characteristics and concentration in serum of the molecules being studied; thereby, expanding the amount of information collected from a single study. The research and development of new fluorochromes that provide exquisite spatial resolution of molecules in vivo, as well as the burgeoning of microfluidic devices enabling high throughput analysis of various attributes of the molecule, herald promise in meeting the challenges of quickly bringing well-characterized drugs into the market.
Materials and Methods
Materials
Ethanolamine, N-ethyl maleimide, and dimethyl sulfoxide were purchased from Sigma-Aldrich (St. Louis, MO, USA). Sodium acetate, sodium phosphate (Na2HPO4), sodium chloride, and trifluoroacetic acid (TFA) were all purchased from JT Baker (Phillipsburg, NJ, USA). Ethylenediamine tetraacetic acid (EDTA), dithiothreitol (DTT), 5,5′-dithio-bis(2-nitrobenzoic acid) (DTNB), Iodoacetamide (IAA), N-ethylmaleimide (NEM) and acetonitrile were purchased from Sigma-Aldrich (St. Louis, MO, USA). Formic acid was purchased from EMD Chemicals (San Diego, CA, USA), and guanidine hydrochloride from Thermo Fisher Scientific (Rockford, IL, USA). Modified trypsin, (sequence Grade) was purchased from Promega (Madison, WI, USA), HT Pico Protein express kits, including chips and reagents, were purchased from Caliper Life Sciences (Hopkinton, MA, USA), N-glycanase (PNGase F), (Prozyme CA). The mAbs used in this study were produced at Abbott Bioresearch Center (Worcester, MA, USA). Amicon Ultra-15 centrifugal filter devices were purchased from Millipore (Billerica, MA, USA). Tris(2,2′-bipyridyl)ruthenium(II) chloride hexahydrate, Ru(bpy) and ammonium persulfate (APS) were purchased from Sigma-Aldrich (St. Louis, MO, USA).
Labeling of proteins
The Pico assay guide, provided by the manufacturer, was adapted for labeling the antibody. The labeling buffer used was 0.5 M sodium bicarbonate (pH 8.0) and contained 1 M N-ethyl maleimide. The antibody to be labeled was diluted to 2 mg/mL in 0.1 M sodium bicarbonate buffer and the fluorescent dye was reconstituted in DMSO as described by the manufacturer (stock concentration was 200 μM). The working dye solution (concentration of 40 μM) was reconstituted in water and mixed with antibody at 1:3 molar ratio (antibody:dye) for 1 h in the dark. The reaction was stopped by adding 1 M ethanolamine in 0.1 M tris pH 7.0 and the antibody then exchanged into 1xPBS using the Amicon ultracentrifugal device.
Photo-induced cross linking of unmodified proteins
The concentration of Ru(bpy) and APS, the amount of antibody cross-linked as well as length of irradiation time can be varied to modify the extent of cross-linking. For this study, a solution of 0.25 mM Ru(bpy) in 10 mM NaH2PO4, pH 7.0 was prepared which was protected from light. In addition, 1.5 mM of APS in 10 mM NaH2PO4, pH 7.0 was also prepared. The antibody to be cross linked in this study was diluted to 40 mg/ml in 10 mM NaH2PO4, pH 7.0. Then 27.5 μL of 0.25 mM Ru(bpy) was added to the antibody which was then immediately followed by addition of 27.5 μL of 1.5 mM APS. The mixture was immediately irradiated for 30 min using a 75 W incandescent light source. After 30 min the reaction was immediately quenched with 50 μL of 0.1 M Tris pH 7.0.
Mass spectrometry
Samples were analyzed on an Agilent QTOF LC/MS (Model 6510A, Agilent Technologies, Santa Clara, CA, USA) coupled to an Agilent 1100 capillary HPLC system (Agilent Technologies, Santa Clara, CA, USA). The samples were introduced into the mass spectrometer and desalted using a polymeric reverse phase protein micro trap from Michrom BioResources (Auburn, CA, USA) The samples were loaded under aqueous conditions (0.02%TFA, 0.08% formic acid in water) to wash out salts for the first five minutes and then eluted under organic conditions (0.02%TFA, 0.08% formic acid in acetonitrile). Samples, at an approximate concentration of 1 mg/mL, were injected at 2 μl for a load of 2 μg. To help simplify the mass spectrum, the samples were first treated with the enzyme n-glycanase to remove glycosylation heterogeneity in the Fc region of the heavy chain. Following deglycosylation, samples were treated with 50 mM dithiothreitol at 37°C for 30 min to reduce the interchain disulfide bonds and separate the light chain and heavy chain subunits. The mass spectrometer was run in positive ion mode with capillary voltage of 4750 and a m/z scan range of 200–3200. Deconvolution of the mass spectra was performed using MassHunter Maximum Entropy software version B.03.01.
Capillary electrophoresis
High-throughput analysis of all samples was performed on the LabChip GXII (Caliper LifeSciences) as described by the manufacturer. Briefly, denatured protein was prepared by mixing a small volume (5 μL) of rat serum sample with 45 μL of the denaturation solution (HT Pico Protein Express Sample Buffer) provided by the manufacturer. The mixture was heated at 75°C for 5 min and the samples run under non-reducing conditions. The denatured protein was electrokinetically loaded directly into the chip from a microtiter plate (Bio-Rad Laboratories). The separation channel of the chip has dimensions of 14 mm in length and 31 μm in width.
Density and viscosity measurements
Buffer density was measured at 25°C using an Anton Paar DMA bench top densitometer. (Anton Paar, Graz, Austria). Viscosity was determined experimentally using an Anton Paar capillary viscometer (Anton Paar, Graz, Austria) at 25°C. Partial specific volume was calculated using the public domain software program SEDNTERP (Sednterp v1.09, database v1.1) taking into account density for each antibody.
Analytical ultracentrifugation
Sedimentation velocity experiments were performed using a Proteome Lab XL-I analytical ultracentrifuge (Beckman Coulter, Fullerton, CA, USA) equipped with absorbance optics. Experiments were performed in 12-mm double sector charcoal filled Epon centerpieces (Spin Analytical, Durham, NH, USA) at 25°C using a rotor speed of 42,000 rpm. Scans were acquired at a wavelength of 280 nm and 0.003 cm radial increments. The sedimentation boundary was analyzed with SEDFIT (version 12.1.0). The continuous c(s) distribution with 90% confidence level was calculated after optimizing baseline, meniscus, and cell bottom positions by nonlinear regression. All s values obtained with the c(s) distribution in PBS were converted to s20,W with SEDNTERP (version 1.09) using the measured density and viscosity of PBS.
Pharmacokinetic study
Studies were conducted in accordance with the Abbott IACUC guidelines.
Male Sprague-Dawley rats (n = 4 for unlabeled antibody and n = 2 for fluorescently tagged mAb or dimer groups) received a single, 4 mg/kg intravenous dose of mAb (1mL/kg). 250 ul blood was collected over a period of approximately a month from the tail vein of each rat, serum samples were prepared at room temperature and stored frozen at -80 until ready for analysis.
Bioanalysis of serum samples
mAb concentrations were measured by an ELISA method using biotinylated antigen for capture and goat anti-human IgG-HRP for detection in a 1% final serum concentration. Serum antibody concentrations were calculated by a four-parameter logistic curve fit using ExcelFit4. PK parameters were calculated with non-compartmental analysis using WinNonlin Professional (version 5.0.1, Pharsight, Mountain View, California, USA). The fluorescently labeled mAb and dimer preparation serum samples were directly analyzed on the LabChip GXII.
| Abbreviations: | ||
| CE | = | capillary electrophoresis |
| FDA | = | Food and Drug Administration |
| IgG | = | Immunoglobulin |
| IV | = | Intravenous |
| MS | = | Mass spectrometry |
| PICUP | = | photo induced cross-linking of unmodified proteins |
| PK | = | pharmacokinetic |
| PTM | = | Post translational modification |
Acknowledgments
We thank Karin Orsi and Christopher Stedman for assistance with the animal studies and Barbara Perilli-Palmer for advice on purification. Gary Welch is gratefully acknowledged for his support of this project.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Hill H. Development of bioanalysis: a short history. Bioanalysis 2009; 1:3 - 7; http://dx.doi.org/10.4155/bio.09.3; PMID: 21083180
- Gomez FA. Bioanalytical applications in microfluidics. Bioanalysis 2010; 2:1661 - 2; http://dx.doi.org/10.4155/bio.10.145; PMID: 21083314
- West J, Becker M, Tombrink S, Manz A. Micro total analysis systems: latest achievements. Anal Chem 2008; 80:4403 - 19; http://dx.doi.org/10.1021/ac800680j; PMID: 18498178
- Baker CA, Duong CT, Grimley A, Roper MG. Recent advances in microfluidic detection systems. Bioanalysis 2009; 1:967 - 75; http://dx.doi.org/10.4155/bio.09.86; PMID: 20414455
- Hoppin J, Orcutt KD, Hesterman JY, Silva MD, Cheng D, Lackas C, et al. Assessing antibody pharmacokinetics in mice with in vivo imaging. J Pharmacol Exp Ther 2011; 337:350 - 8; http://dx.doi.org/10.1124/jpet.110.172916; PMID: 21317355
- Kostal V, Katzenmeyer J, Arriaga EA. Capillary electrophoresis in bioanalysis. Anal Chem 2008; 80:4533 - 50; http://dx.doi.org/10.1021/ac8007384; PMID: 18484738
- Chen X. Micro total analysis systems in biopharmaceutical process development. Bioanalysis 2009; 1:1183 - 6; http://dx.doi.org/10.4155/bio.09.114; PMID: 21083042
- Chen X, Flynn GC. A high throughput dimer screening assay for monoclonal antibodies using chemical cross-linking and microchip electrophoresis. J Chromatogr B Analyt Technol Biomed Life Sci 2009; 877:3012 - 8; http://dx.doi.org/10.1016/j.jchromb.2009.07.020; PMID: 19646934
- Chen X, Tang K, Lee M, Flynn GC. Microchip assays for screening monoclonal antibody product quality. Electrophoresis 2008; 29:4993 - 5002; http://dx.doi.org/10.1002/elps.200800324; PMID: 19130579
- Correia IR. Stability of IgG isotypes in serum. MAbs 2010; 2:221 - 32; http://dx.doi.org/10.4161/mabs.2.3.11788; PMID: 20404539
- Fancy DA, Kodadek T. Chemistry for the analysis of protein-protein interactions: rapid and efficient cross-linking triggered by long wavelength light. Proc Natl Acad Sci U S A 1999; 96:6020 - 4; http://dx.doi.org/10.1073/pnas.96.11.6020; PMID: 10339534
- Reichert JM. Antibody-based therapeutics to watch in 2011. MAbs 2011; 3:76 - 99; http://dx.doi.org/10.4161/mabs.3.1.13895; PMID: 21051951
- Reichert JM. Metrics for antibody therapeutics development. MAbs 2010; 2:695 - 700; http://dx.doi.org/10.4161/mabs.2.6.13603; PMID: 20930555
- Reichert JM. Antibodies to watch in 2010. MAbs 2010; 2:84 - 100; http://dx.doi.org/10.4161/mabs.2.1.10677; PMID: 20065640
- Sánchez LM, Penny DM, Bjorkman PJ. Stoichiometry of the interaction between the major histocompatibility complex-related Fc receptor and its Fc ligand. Biochemistry 1999; 38:9471 - 6; http://dx.doi.org/10.1021/bi9907330; PMID: 10413524
- West AP Jr., Bjorkman PJ. Crystal structure and immunoglobulin G binding properties of the human major histocompatibility complex-related Fc receptor(,). Biochemistry 2000; 39:9698 - 708; http://dx.doi.org/10.1021/bi000749m; PMID: 10933786
- Leblond F, Davis SC, Valdés PA, Pogue BW. Pre-clinical whole-body fluorescence imaging: Review of instruments, methods and applications. J Photochem Photobiol B 2010; 98:77 - 94; http://dx.doi.org/10.1016/j.jphotobiol.2009.11.007; PMID: 20031443