Abstract
The serine protease chymase (EC = 3.4.21.39) is expressed in the secretory granules of mast cells, which are important in allergic reactions. Fynomers, which are binding proteins derived from the Fyn SH3 domain, were generated against human chymase to produce binding partners to facilitate crystallization, structure determination and structure-based drug discovery, and to provide inhibitors of chymase for therapeutic applications. The best Fynomer was found to bind chymase with a KD of 0.9 nM and koff of 6.6x10−4 s−1, and to selectively inhibit chymase activity with an IC50 value of 2 nM. Three different Fynomers were co-crystallized with chymase in 6 different crystal forms overall, with diffraction quality in the range of 2.25 to 1.4 Å resolution, which is suitable for drug design efforts. The X-ray structures show that all Fynomers bind to the active site of chymase. The conserved residues Arg15-Trp16-Thr17 in the RT-loop of the chymase binding Fynomers provide a tight interaction, with Trp16 pointing deep into the S1 pocket of chymase. These results confirm the suitability of Fynomers as research tools to facilitate protein crystallization, as well as for the development of assays to investigate the biological mechanism of targets. Finally, their highly specific inhibitory activity and favorable molecular properties support the use of Fynomers as potential therapeutic agents.
Introduction
Mast cells have been considered for many years to participate specifically in allergic reactions through the release of cytokines, chemokines, proteases, leukotrienes, and bio-active polyamines. A variety of roles for mast cells have been recently identified, highlighting their relevance in both innate and adaptive immunity, as well as pathological inflammatory conditions.Citation1
The serine protease chymase (EC = 3.4.21.39) is a chymotrypsin-like enzyme that is expressed in the secretory granules of mast cells.Citation2 Upon release, it has been described to degrade the extracellular matrix (e.g., proteoglycans, collagen, elastin, fibronectin), induce leukocyte migration and cytokine production, activate TGF-β and MMP-9, and promote tissue remodeling.Citation3,Citation4 Consequently, chymase is a potential new drug target, and chymase inhibitors have been generated and tested in a number of in vivo models demonstrating pathological responses related to allergic asthma and/or COPD: (1) mast-cell mediated inflammation in ovalbumin sensitized rats, (2) allergen-induced bronchoconstriction and airway hyperresponsiveness in allergic sheep, (3) tobacco smoke induced neutrophilia in mice,Citation5 (4) vascular proliferation after balloon catheter injury in a dog modelCitation6 and (5) various other studies.Citation7,Citation8
The search for small molecules as protease inhibitors with high affinity and specificity by either applying high throughput screening, screening of small focused libraries, fragment based screening or virtual screening is well established. All of these approaches work, but the last two require the three dimensional structure of the target protein. Many proteases crystallize well and high resolution structures can be obtained, facilitating the process of chemical lead generation. Some proteins, however, have proven to be more difficult or even impossible to crystallize, and for such examples a number of ways have been developed to improve the chances for successful crystallization. Genetic engineering of the target protein, either by the introduction of surface mutations or by the variation of protein construct length, is often effective. Co-crystallization of proteins with ligands may be a good alternative. Recently Fab fragments of monoclonal antibodies have become popular as crystallization aids, and have been observed to stabilize flexible proteins in a specific conformation.Citation9 The structure determination of cytochrome C oxidase was one of the first successful examples of the use of this strategy.Citation10
Antibodies and antibody fragments are routinely employed for analytical, purification, diagnostic and therapeutic purposes because of their high affinity and specificity to virtually any desired target antigen, but they still have a number of serious drawbacks, such as the need for complex mammalian cell production systems, a dependency on disulfide bonds for stability, and the tendency of some antibody fragments to aggregate, which limits solubility. As a consequence, novel classes of versatile binding proteins using small globular proteins as scaffolds have been generated.Citation11 Typically, surface components (e.g., extracellular loops) of a protein framework with suitable biophysical properties are combinatorially mutated to produce a diverse protein library to be screened for specific binding to the target of interest.Citation12 More than 50 different protein scaffolds have been proposed over the past 10 to 15 y, the most advancedCitation13 of which are affibodies (based on the Z- domain of staphylococcal protein A), Kunitz type domains, AdnectinsTM (based on the 10th domain of human fibronectin), Anticalins (derived from lipocalins), DARPins (derived from ankyrin repeat proteins), avimers (based on multimerized LDLR-A) and Fynomers, which are derived from the human Fyn SH3 domain.Citation14 The Fyn SH3 domain is a particularly attractive scaffold for the generation of binding proteins because the resulting “Fynomer” (1) can be expressed in bacteria in soluble form in high amounts, (2) is monomeric and does not aggregate when stored in solution, (3) is very stable (Tm ~70°C), (4) lacks cysteine residues, and (5) is of human origin featuring an amino acid sequence completely conserved from mouse to man and, hence, is considered to be non-immunogenic.Citation15
Here, we introduce Fynomers as binding reagents for the drug target chymase, and report their generation, characterization and use in crystallization. All the Fynomers generated are potent and selective inhibitors of the enzymatic activity of chymase, suggesting their potential use in functional studies of chymase in vivo. The X-ray structures of the complexes give insight into the mode of inhibition and into key interactions that drive potency and selectivity of inhibitors of this serine protease. Taken together, our results qualify Fynomers as excellent molecules to facilitate research in drug discovery and diagnostics and as excellent inhibitors of chymase.
Results
Generation of naïve Fynomer phage display libraries
The SH3 domain of the human Fyn kinase was successfully used as a scaffold to engineer proteins (Fynomers) that bind with high affinity and specificity to different target proteins.Citation15,Citation16 Originally, a library consisting of 1.2x109 clones was constructed in which six positions each in the RT- and the n-src-loop of the human Fyn SH3 domain were combinatorially mutated.Citation15 To expand the diversity of the Fynomer libraries, three large novel libraries were generated (). Library 0 shares its design with the previously reported Fynomer library,Citation15 but additionally includes n-src-loops of variable length, ranging from four to six residues. Library 8 has a novel design in the RT-loop, whereas library 9 combines mutations in the RT-loop with mutations at positions in the interspace between the RT- and the n-src-loop. In total, these three novel Fynomer libraries comprise 8.5x1010 independent clones, of which more than 98% contained a Fynomer insert as shown by PCR screening, and of which 63% expressed detectable amounts of soluble Fynomer as determined by dot blotCitation15,Citation20 analysis (data not shown).
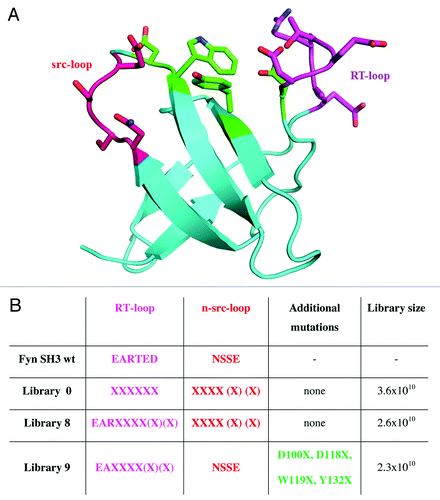
Figure 1. Fyn SH3 wild-type protein structure and randomized positions for library construction. A. Structure of the Fyn SH3 domain (Protein Data Bank entry 1M27). Colored amino acid residues represented as sticks indicate randomized positions in the RT-loop (magenta), n-src-loop (red)Citation17 and between the loops (green). B. The design of the different Fynomer libraries 0, 8 and 9 and the library sizes obtained are shown. For comparison, the amino acid sequences in the loop regions of the native Fyn SH3 domain are also shown (amino acid residues are numbered according to the sequence reported by Refs.Citation18,Citation19 X designates positions at which random mutations were introduced, allowing any of the 20 natural L-amino acids.
Isolation, affinity maturation and production of chymase-specific Fynomers
Fynomers specific to human chymase were isolated by phage display. After three rounds of panning on biotinylated chymase as the target, several Fynomers were found to bind to chymase as determined by phage ELISA. These Fynomers isolated from naïve libraries were used as templates for affinity maturation strategies: new randomizations were introduced in either the RT- or n-src-loop, and after affinity maturation selections, several high-affinity Fynomers binding to chymase were isolated ().
Table 1. Anti-chymase Fynomers. Equilibrium binding constants (KD) and kinetic rate constants kon and koff of the 8 different chymase-specific Fynomers are indicated as determined by SPR measurements. Inhibitory activity of the Fynomers was measured in a fluorescence quenching assay. The variable sequences of the RT- and the n-src-loops are given in comparison to the sequence of the SH3 domain of the human Fyn kinase
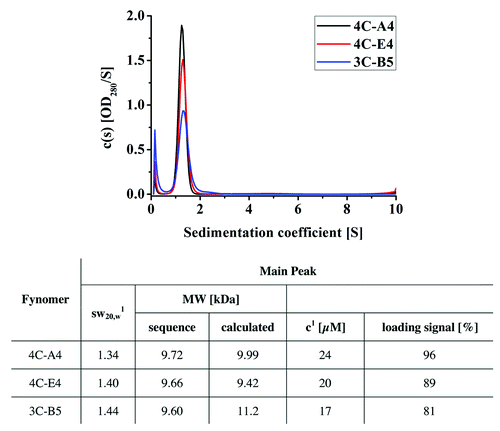
A total of eight Fynomers were subcloned into a cytosolic expression vector carrying a myc- and a hexahistidine tag. All Fynomers could be expressed in E. coli with high yield. Purification yields ranged between 24 and 78 mg/l of purified proteins under non-optimized conditions in shake flasks (). All purified Fynomers were > 90% pure and monomeric as determined by SDS PAGE, size exclusion chromatography and analytical ultracentrifugation (AUC). The results of the AUC runs for the three most potent Fynomers, 4C-A4, 4C-E4 and 3C-B5, that were finally used for the co-crystallization with chymase are shown in . The Fynomers are monodisperse at the concentrations investigated. The main species in all samples is the monomeric Fynomer. The samples had frictional coefficients between 1.34–1.44, which indicates that the expected globular shape of the Fynomer domains was well preserved in the buffer used. All samples contained, to different degrees, an impurity that sedimented with S ≈0.2.
Figure 2. Sedimentation coefficient distribution c(s) for the Fynomers 4C-E4, 4C-A4 and 3C-B5. Analysis of sedimentation coefficient was performed using analytical ultracentrifugation. The difference in the ratios of peak height to peak width can be explained by the different molar loading concentrations (the loading signal was OD280, 1.0 cm = 0.5 for all samples, but due to the mutations the constructs have different extinction coefficients). 1Signal-weighted sedimentation coefficient corrected for buffer density and viscosity determined by manual integration in Sedfit.
Characterization of the Fynomers using Biacore
Affinities and kinetic data from all the Fynomers are summarized in . These data were obtained by analyzing the response curves that could be fitted with a 1/1 kinetic model. The response curves of 4C-A4, 4C-E4 and 3C-B5, the three Fynomers that were also used for crystallization, are shown in . All Fynomers were found to be potent chymase binders with KD values ranging from 0.9–17.2 nM. Differences in affinity are mainly due to variability in the dissociation constant koff as often observed for antibodies.
Figure 3. Surface plasmon resonance measurement results. A. Binding kinetics and affinities of Fynomers differ significantly. Dose response curve are shown as determined in the kinetic titration assay for the three crystallized Fynomers 4C-E4, 4C-A4 and 3C-B5. Dilution series started at 10 nM, 20 nM and 60 nM for the Fynomers 4C-E4, 4C-A4 and 3C-B5. The dilution factor between the concentrations was 2. B. All Fynomers share the same binding site. Competition experiments were performed using Fynomer 3C-D7 as competitor molecule. The Fynomer concentration in all experiments was approximately 15 times higher than the corresponding dissociation constant (KD). The first bar in gray shows the sensor response in response units [RU] obtained for the binding of Fynomer 3C-D7 (c = 150 nM) to chymase alone, the bar in black represents the response of the Fynomer 4C-E4 (c = 10 nM) alone. The hatched bar represents the experimental sensor signal obtained when a mixture of 3C-D7 (c = 150 nM) and 4C-E4 (c = 10 nM) was added. The white bar shows the calculated sum (calc.) of the responses of the two Fynomers. The measurements were repeated in the same way as described above for a number of Fynomers, with the signals being measured at the end of the association phase.
![Figure 3. Surface plasmon resonance measurement results. A. Binding kinetics and affinities of Fynomers differ significantly. Dose response curve are shown as determined in the kinetic titration assay for the three crystallized Fynomers 4C-E4, 4C-A4 and 3C-B5. Dilution series started at 10 nM, 20 nM and 60 nM for the Fynomers 4C-E4, 4C-A4 and 3C-B5. The dilution factor between the concentrations was 2. B. All Fynomers share the same binding site. Competition experiments were performed using Fynomer 3C-D7 as competitor molecule. The Fynomer concentration in all experiments was approximately 15 times higher than the corresponding dissociation constant (KD). The first bar in gray shows the sensor response in response units [RU] obtained for the binding of Fynomer 3C-D7 (c = 150 nM) to chymase alone, the bar in black represents the response of the Fynomer 4C-E4 (c = 10 nM) alone. The hatched bar represents the experimental sensor signal obtained when a mixture of 3C-D7 (c = 150 nM) and 4C-E4 (c = 10 nM) was added. The white bar shows the calculated sum (calc.) of the responses of the two Fynomers. The measurements were repeated in the same way as described above for a number of Fynomers, with the signals being measured at the end of the association phase.](/cms/asset/d2faf785-cd3a-4f7c-9a44-e1d5e943c712/kmab_a_10920452_f0003.gif)
Competition experiments were performed with the three Fynomers 3C-B5, 3C-D7 and 3C-H2 as the reference proteins. The lower affinity of these selected Fynomers allowed efficient regeneration of the sensor surface between experiments. In a first approximation, for non-competitive binding, a response for the mixture is expected that corresponds to the sum of the responses of the two individual components. In the case of competition, the response of the mixture is close to the signal of a single Fynomer. All Fynomers showed competitive binding on chymase when measured against the three reference Fynomers. This indicates that for all Fynomers the binding epitopes are identical, or at least strongly overlapping. The results obtained with Fynomer 3C-D7 are shown in .
Binding studies were also performed with the four arbitrarily selected Fynomers 4C-A4, 4C-E4, C-G2 and C-F12 on the immobilized zymogen form of chymase. None of these Fynomers showed binding to this precursor of the active chymase. Since the main difference between the active chymase and the inactive zymogen is the blocked active site, we conclude that the mutual binding epitope of the Fynomers is the active site itself or at least lies very close to the active site of the enzyme.
Inhibitory activity
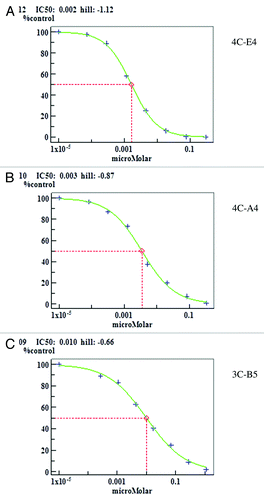
The biochemical assay identifies Fynomers as potent inhibitors of chymase with activities in the range of nanomolar IC50 values ( and ). In addition, we tested the specificity of the most potent Fynomer 4C-E4 for inhibitory activity on other serine proteases: cathepsin G, dipeptidyl protease-4 (DPP-4), kallikrein-1 (KLK-1), neutrophil elastase and trypsin. At 4 µM concentration of Fynomer 4C-E4, no inhibition of the enzymatic activity for any of these five proteases could be observed.
Figure 4. Fynomers are potent chymase inhibitors. IC50 values (in µM) are shown of the three crystallized Fynomers 4C-E4, 4C-A4 and 3C-B5 as determined in the biochemical chymase assay.
X-ray crystal structure determination
Three different anti-chymase Fynomers, 4C-A4, 4C-E4 and 3C-B5 were co-crystallized as complexes with chymase. A variety of crystal forms were obtained under different conditions (). For six different crystal forms, X-ray diffraction data were collected and the structures solved at resolutions between 2.25 Å and 1.40 Å (Table S1). Inspection of these structures ( and ) shows an important element for the interaction between the Fynomers and chymase to be the ‘RWT’ sequence motif (Arg15-Trp16-Thr17) of the RT-loop, which reaches deeply into the active site, and appears to be a major contributor to affinity. Although the tryptophan side chain inserts into the primary chymase substrate recognition pocket (S1), the atoms forming the backbone of the Fynomer are not positioned as required for a substrate: the carbonyl carbon of Trp16 is some 5 Å away from the correct location for catalysis. The Fynomers completely block substrate access to the active site, are not cleaved, and are thus very efficient and specific chymase inhibitors. All six complex structures are very similar. Slight differences in orientation of the Fynomers relative to chymase come from the sequence differences of the n-src-loop and from crystal packing effects, and are approximately a rigid body rotation about Arg15-Trp16 (). There are small changes in the chymase structure, namely in the loop containing amino acid 24 (24 loop), correlated with changes in the loop structure containing amino acid 40 of the Fynomers (40 loop) and there are also minor changes in the hydration pattern within the chymase-Fynomer interface. In structure 1, there are 49 atom pairs in the interface with a chymase-Fynomer distance cut-off of 3.5 Å. Of these, 11 pairs make hydrogen bonds at a distance cut-off of 3.2 Å (9 of these are shown in ). There are also ~12 water molecules in the interface (of which 8 are conserved in all 6 structures), so the interface interactions are predominantly hydrophilic, with only Trp16 and Thr17 making hydrophobic interactions.
Table 2. Crystallization conditions for the 6 crystal forms solved. All crystallization conditions contained 20–25% PEG and 0.1–0.2 M salt in a wide pH range of 3.5 to 8.5. All conditions are from primary screens and were not optimized
Figure 5. Structure of the complex of Fynomer 4C-A4 (ribbon diagram) with human chymase (space-filling model). For the Fynomer, the RT-loop is shown in magenta and the n-src-loop in red.
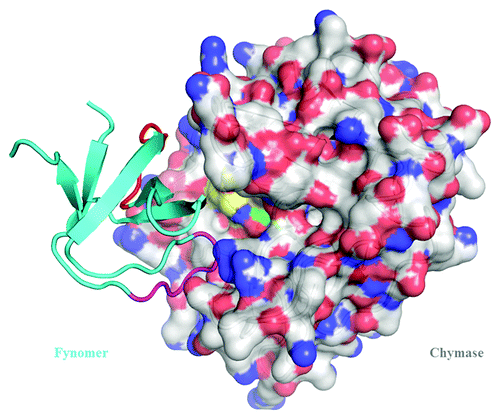
Figure 6. Close-up of the Fynomer 4C-A4: chymase interaction. Colors: RT-loop, magenta; src-loop, red; catalytic His/Ser, yellow/green. The RT-loop conformation is stabilized by an internal salt bridge between Arg15 and Asp18 as well as the β-bend 15–18 and Thr17OG – Asp18OD1 hydrogen bonds (blue dashed lines) and stacking of the Arg15 side chain on Trp38. Interface hydrogen bond interactions (green dashed lines) are between residues (Fynomer-chymase) Ala13-Ser20, Asp14-Ser20, Arg15-Gly199, Arg15-Thr83, Asp31-Arg77, Ser33-Arg77, Pro35-Thr83, Trp38-Thr83, Tyr51-Tyr81 (the Fynomer numbering is arbitrary, chymase is numbered sequentially from 1). The interaction between Trp16:NE1 and Ser182:OG of the catalytic triad is shown in red: with a length of 3.3 Å it is only a weak hydrogen bond in this structure, and in some other structures, a bridging water molecule is present. At the top left, Arg77 of chymase can be seen to bind to Asp31 and Ser33 of the n-src-loop that is pre-organized by Fynomer internal hydrogen bonds. Thr83 of chymase (highlighted in pink) is an important epitope residue as it makes three hydrogen bonds to the Fynomer. Despite both acidic and basic residues playing important roles in binding, there are no direct salt bridges between 4C-A4 and chymase.
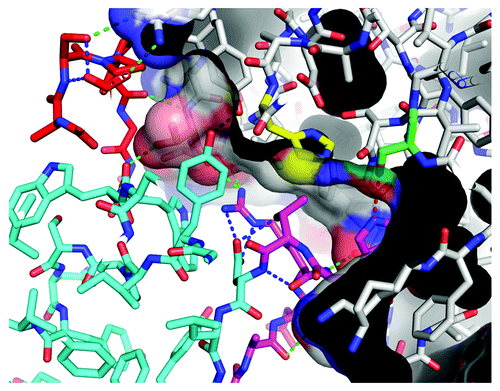
Figure 7. Overlay of all six complexes of chymase (top) and Fynomers (bottom). The complexes were overlaid by matching chymase molecules of stuctures 2–6 onto structure 1. All Fynomers share the same binding mode, with slight rotational differences around the fulcrum of the Arg15-Trp16 motif. The conformations of both, the RT-loop and the ’40 loop’ are strictly conserved, whereas the trace of the n-src-loop differs partly due to sequence differences and partly due to crystal packing effects. In contrast, somewhat larger conformational differences can be seen in the ’24 loop’ of chymase as a response to the differences among the Fynomer sequences. The independently determined Fynomer-chymase complexes have very similar binding interfaces, with a mean buried surface area on complex formation of 1915 +/− 40 Å2 as determined with AREAIMOL from the CCP4 program suite.
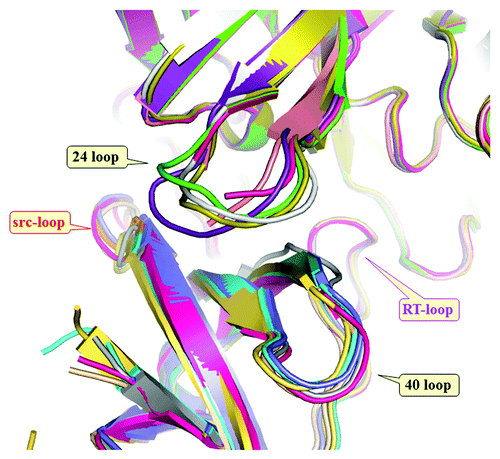
Discussion
We successfully used the Fyn SH3 domain as a scaffold for the generation of potent inhibitors of the pharmaceutically relevant drug target chymase. Moreover, the crystal structures of three Fynomers in complex with chymase were solved. To make best use of the suitable properties of the Fyn SH3 domain, the main scaffold was kept constant and the RT- and n-src-loops were randomized to introduce mutations, as well as variation of loop-length. This strategy yielded libraries comprising 8x1010 Fynomer variants that were sufficient to generate molecules with a range of binding properties. From these Fynomer-phage libraries, high affinity binding proteins could be isolated after one round of affinity maturation. Two of the advantages of Fynomers are that they can be expressed in E. coli in soluble form and can be purified by a rapid and simple two step procedure. This resulted in a purification yield of 45 mg/l under non-optimized conditions in shake flasks, on average, for the eight Fynomers used in this study (). The ease of expression and purification facilitated further characterization of the Fynomers using a wide range of biochemical and biophysical methods, including crystallization and subsequent structural investigation. Analysis by analytical ultracentrifugation () confirmed that the Fynomers are monomeric and show no tendency to form aggregates over a wide range of protein concentrations. A minor impurity of unknown origin that sediments with S ≈0.2 was identified in all purified samples, but was not further characterized. The narrow and symmetric peak shape observed in analytical size exclusion chromatography (data not shown) suggests a high conformational stability, which is advantageous for crystallization and formation of crystals giving high resolution X-ray diffraction.
The RT-loop sequences of the chymase binding Fynomers are closely related: sequence diversity in the RT-loop can be seen to be rather low. Seven out of eight RT-loop sequences share the (N/Q)A(T/D)RWT motif: all have a conserved RWT motif. This sequence conservation is a result of the selection strategy applied in this particular case, where affinity was given priority over diversity. The first library generated yielded the Arg-Trp-Thr motif, which appeared to give a particularly strong interaction between the Fynomer and the target protein. The RT-loop sequence was then kept constant and further mutation of the n-src-loop was performed in a second maturation cycle. This additional cycle had less effect on the binding affinity, but gave more diverse n-src-loop sequences with length from four to six amino acids.
Investigation of the complex formation behavior of the Fynomers showed a number of interesting properties. Binding affinities in the low nanomolar, and in one case even subnanomolar range, confirmed the capability of the Fynomer selection strategy to isolate strong binding molecules. Kinetic data indicated a similar on-rate for all Fynomers of which the RT-loop contains the NATRWT or QADRWT sequence motif, in contrast to Fynomer C-F12 with the sequence RAERST for which slower kon values were measured. The X-ray structures of the complexes containing a Fynomer with the RWT motif give an explanation for this result. The residues of the RWT motif (Arg15-Trp16-Thr17) make four hydrogen bonds to Asp18 and a stacking interaction with the side chain of Trp38 (). These Fynomer residues thus form a highly stable structural motif that fits perfectly into the chymase active site. Fast recognition and initial binding are further facilitated by a network of hydrogen bonds and by the n-src-loops that, although differing in sequence, also exhibit internal hydrogen bonds that stabilize the local structure.
The koff values vary between 1.1 and 26x10−3 s−1, and no correlation to loop length or sequence of the n-src-loop can be recognized. All Fynomers investigated in the SPR experiments show competitive binding and so must share the same binding site or at least have overlapping binding sites (). The X-ray structures show very clearly that the Fynomers bind at the active site of chymase. The well-defined primary substrate specificity pocket may be a hot spot that can be used for tight molecular interactions as reported for lysozyme.Citation21 Inhibition of chymase with an IC50 in the low nanomolar range was observed in the biochemical assay for all Fynomers investigated. Specificity of inhibition relative to five other related serine proteases was determined for one Fynomer to be at least 1000-fold, consistent with the rigid structure of the Fynomer and the high complementarity of the binding paratope with the epitope on human chymase.
It has been reported that stable protein complexes tend to give crystals of higher diffraction resolution compared with the individual proteins alone.Citation9,Citation22-Citation25 Three different Fynomer-chymase complexes were readily crystallized in overall 6 different crystal forms giving X-ray data between 2.25 and 1.4 Å resolution, suggesting that Fynomers may provide a viable alternative to Fabs or other binding proteins for co-crystallization. The high resolution information of the interaction of the Fynomer with the active site of chymase can readily be used for the design of small molecule inhibitors. On the other hand, all the crystal forms are unsuitable for generation of complexes with small molecule inhibitors, as the active site is blocked by the Fynomer. In order to generate a system that enables small molecule soaking or co-crystallization, a different selection protocol would be required, directed toward the discovery of Fynomers with non-competitive binding to active site ligands. This could be achieved, for example, by using as the target protein the zymogen, or the enzyme with a covalently blocked active site, or with an active site mutant, or indeed by using one of the tight complexes described above as the target for a further round of Fynomer generation. In all cases, a counter screen against the active enzyme would be required.
The strong and selective inhibitory activity of the Fynomers identified in this study suggests their use as tools for the investigation of biological mechanisms in in vitro or in vivo experiments to investigate the biological function of chymase. It is suggested that the expression level of chymase can be used as marker for inflammation and, consequently, labeled Fynomers might be useful in the development of biomarker assays. As the generation of Fynomers against chymase was straightforward, and since Fynomers can be generated against virtually any protease and other pharmaceutically relevant target of interest, the findings presented here encourage us to extend the use of Fynomers as tool molecules in drug discovery, as well as to explore possibilities for their use as therapeutic agents.
Materials and Methods
Construction and characterization of naïve Fynomer phage display libraries
Using the gene encoding the SH3 domain of the human Fyn kinase (amino acid residues are numbered according to the sequence reported by Refs. Citation18,Citation19) cloned in the vector pQE12 (Qiagen) as a template, targeted mutations were introduced by PCR using partially degenerate primers (all primers were purchased from Eurofins MWG Operon, Table S2). For construction of library 0, a PCR fragment comprising a RT-loop repertoire was created using primer 14.fo and primer 1.ba. PCR fragments comprising an n-src-loop repertoire were generated using each of the primers 16.fo, 67.fo and 68.fo with primer 13.ba. The Fynomer gene at the 3′ end of the n-src-loop was amplified using primer 15.ba and 2.fo. The resulting DNA fragments were assembled by PCR.
For library 8, PCR fragments comprising a RT-loop repertoire were created using each of the primers 60.fo, 65.fo and 66.fo with primer 1.ba. PCR fragments comprising an n-src-loop repertoire and the fragment 3′ of the n-src-loop were generated as described for library 0, see above. The resulting DNA fragments were assembled by PCR. For library 9, PCR fragments comprising a RT-loop repertoire together with mutations at position D100 were produced using each of the primers 69.fo, 70.fo and 71.fo with primer 1.ba. A PCR fragment comprising mutations at positions D118 and W119 was generated using primers 73.fo and 72.ba. A PCR fragment comprising the 3′ end of the Fynomer gene including a mutation at position Y132 was generated using primer 33.ba and primer 2.fo. The resulting DNA fragments were assembled by PCR.
The assembled DNA fragments were further amplified by PCR using primers 20b.ba and 21.fo to introduce NcoI and NotI restriction sites, thus allowing for ligation into the phagemid pHEN1.Citation26 Ligation reactions were purified by ethanol precipitation using Pellet Paint (EMD Millipore) and electroporated into E. coli TG1 bacteria. The libraries were rescued with helper phage VCS M13 and used for phage production according to standard protocols.Citation17
PCR reactions with primers LMB3long.ba (5′- CAG GAA ACA GCT ATG ACC ATG ATT AC -3′) and fdseqlong.fo (5′- GAC GTT AGT AAA TGA ATT TTC TGT ATG AGG -3′) on several randomly selected clones from all Fynomer libraries were performed in order to verify the correct size of the inserts. The resulting PCR fragments were sequenced using LMB3long.ba as sequencing primer. The percentage of bacterial clones of the library that expressed soluble Fynomer into bacterial culture supernatant was determined by dot blot as described previously.Citation15,Citation20
Preparation of chymase
Human chymase was prepared as described earlier.Citation27 Like many other proteases, chymase was expressed as an inactive zymogen. The construct used carried an additional His-tag attached N-terminally to the propeptide. Activation of the chymase was performed by cleaving off both the His-tag and the propeptide using enterokinase.
Biotinylated chymase was prepared using EZ-Link NHS-Chromogenic-Biotin (Thermo Scientific), and was made according to the instructions of the manufacturer. The labeling ratio was determined after desalting of the protein preparation and was calculated to be 0.75 biotin/chymase.
Isolation of Fynomers by phage display selections and screening by ELISA
200 nM biotinylated chymase was captured on maxisorp immunotubes (Thermo Scientific) coated with 25 µg/ml NeutrAvidin (Thermo Scientific) overnight. After blocking with 1% BSA in PBS, the naïve Fynomer phage libraries 0, 8 and 9 were combined, and 4 ml of a solution containing 8x1012 transforming units of phage in 1% BSA was added to the immobilized antigen. Unbound phage was washed off with 0.1% Tween-20 in PBS. After rinsing with PBS, bound phage was eluted in 100 mM triethylamine. After collection of the triethylamine eluate, a second elution step with 100 mM glycine buffer pH 2.8 was made. The pH of the eluate was neutralized with 33% v/v 1 M Tris pH 7.4 immediately after collection. The eluates were combined and phage were amplified and purified for subsequent rounds of selection as described previously.Citation17
The second and third rounds of selection were performed with 1 ml phage from the previous round of selection, containing approximately 1012 transforming units of phage. 100 nM biotinylated chymase was captured on 8 wells of a maxisorp microwell plate (Thermo Scientific) coated with 100 µg/ml streptavidin (Thermo Scientific) for the second round, and with 25 µg/ml NeutrAvidin for the third round of selection.
For ELISA screening, DNA fragments coding for Fynomers were amplified by PCR with primers hFyn SH3 domain ba (5′- ATC GCG GAT CCG GAG TGA CAC TCT TTG TGG CCC TTT AT -3′) and 51.fo (5′- ATC CCA AGC TTA GTG ATG GTG ATG GTG ATG CAG ATC CTC TTC TGA GAT GAG TTT TTG TTC ACC CTG GAT AGA GTC AAC TGG AGC CAC -3′), digested with HindIII and BamHI, and ligated into the expression vector pQE12 (Qiagen) for cytosolic expression in E. coli TG1 bacteria. Cleared lysates containing soluble Fynomer with a C-terminal myc- and hexahistidine peptide tag were added to 100 nM biotinylated chymase coated onto high-bind StreptaWell 96-well plates (Roche Applied Science). Bound Fynomer was detected by means of a murine monoclonal anti-myc tag antibody, clone 9E10 (Roche Applied Science), followed by an anti-mouse IgG-horse radish peroxidase conjugate (Sigma-Aldrich).
Affinity maturation of the Fynomers
For Fynomers originating from either the naïve Fynomer libraries 0 or 8, the selected n-src-loop sequences were combined with the same RT-loop repertoire as used for the construction of the naïve libraries, and vice versa. Clones that were isolated from the naïve Fynomer library 9 were affinity matured by introducing mutations in the n-src-loop. The process of library generation and the primers used were essentially the same as described above for cloning of the naïve libraries. For affinity maturation, a library of 9.6x108 mutant clones was obtained.
For the first round of selection, the affinity maturation libraries were pooled, phage was blocked with 1% BSA, incubated with 10 nM biotinylated chymase and phage-antigen complex captured using Streptavidin-coated dynabeads M280 (Life Technologies). Unbound phage was washed off with 0.1% Tween-20 in PBS. After rinsing with PBS, bound phage was eluted in 100 mM triethylamine and 100 mM glycine buffer pH 2.8 as described above. The second round of selection was performed with 1 nM biotinylated chymase immobilized on NeutrAvidin-coated maxisorp plates. After the second round of selection, bacterial lysates containing Fynomer mutants were screened by ELISA as described above using 50 nM biotinylated chymase as target antigen.
Expression and purification of Fynomers
Fynomers were subcloned from the phage display vector into the bacterial expression vector pQE12. The resulting constructs encode Fynomers endowed with a C-terminal myc-hexahistidine tag sequence. Fynomers were expressed in E. coli TG1 cells by inoculation of an overnight pre-culture in 50 ml LB medium supplemented with 100 μg/ml ampicillin. This overnight culture was grown at 37°C and then used as inoculum (1:25) for a 1-L culture of LB medium containing 100 μg/ml ampicillin. Again the culture was incubated with shaking at 37°C. Once the culture reached the log phase at OD600nm of 0.7 heterologous protein expression was induced by addition of IPTG to a final concentration of 0.5 mM. Growth of the cells was continued overnight at 30°C. The biomass was harvested by centrifugation.
Cytosolic proteins were extracted with 40 ml B-PER reagent (Thermo Scientific) from pelleted cells (ca. 4 g). The supernatant was centrifuged at 28’000 x g for 30 min and the clear lysate applied on a 3 ml Talon column in 50 mM sodium phosphate pH 8.0, 300 mM NaCl. After washing the column with several column volumes of buffer, the protein was eluted with 200 mM imidazole. Protein containing fractions were pooled, concentrated to 2 ml using an Amicon cell (1 kDa cutoff) and then chromatographed on a Superdex 75 16/60 column in 20 mM Tris/HCl pH 8.2, 150 mM NaCl. The purity of the Fynomers was assessed by SDS-PAGE. The dispersity of the isolated Fynomers was examined by analytical ultracentrifugation in sedimentation velocity mode (20°C, 200’000 x g). For this, the Fynomers were prepared as 500 µl/OD280 nm, 1 cm = 0.5 aliquots. The buffer was 20 mM Tris/HCl pH 8.2, 150 mM NaCl (ρ = 1.0056 g/cm3; η = 1.19 mPas).
In vitro binding assays using surface plasmon resonance
Affinity measurements were performed on a Biacore S51 instrument (GE Healthcare). HEPES buffer (10 mM HEPES pH 7.4, 150 mM NaCl, 3 mM EDTA, 0.01% P20) was used as the running buffer during immobilization and binding experiments. Acetate buffer (10 mM sodium acetate pH 5.5) was used as the coupling buffer for the immobilization of the protein. All the experiments, the immobilization of protein and the binding studies, were performed at 25°C. Both non-biotinylated active chymase and its zymogen form were immobilized separately on a CM5 sensor chip by amine coupling. Carboxyl groups of the dextran matrix were activated using a mixture of N-hydroxysuccinimide (NHS) and N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide (EDC). Chymase was diluted with coupling buffer pH 5.5 to a concentration of 0.03 μg/ml prior to immobilization. The activated sensor surface was then contacted with the protein solutions for 5 min. After the protein was covalently bound to the chip, the remaining reactive esters were quenched with 1 M ethanolamine pH 8.0. Finally, approximately 600 RU of chymase were immobilized. For the determination of the kinetic constants, a kinetic titration assay was performed. Five solutions with increasing concentrations (dilution factor 2) were successively injected without full regeneration between the concentrations. The sensograms were fitted with a 1/1 kinetic titration model using the software provided by the instrument manufacturer. Competition assays for two Fynomers were performed in the following manner. Three solutions were prepared: the first with the reference Fynomer at a concentration c = 10xKD, the second with another Fynomer at c = 1xKD, the third with a mixture of the two Fynomers using again the same concentrations. Equilibrium sensor responses were measured for all 3 solutions.
In vitro functional FRET assay
The kinetic chymase assay using MR121-CAAPFW as the quenched fluorescent substrate was performed in triplicate at room temperature in 384-well microtiter plates in a final volume of 100 μl. All reagents were diluted in the assay buffer composed of 100 mM HEPES pH 7.4, 0.01% Triton X-100, 80 μg/ml heparin. Each well contained 100 μl assay solution with 1 nM chymase, 1 μM unlabeled and 100 nM MR121 peptide. Fynomers were serially diluted in the assay buffer and added to the reaction solution. The enzymatic reaction was followed for 20 min in a plate reader in a kinetic measuring mode with excitation at 612 nm and emission at 670 nm such as to detect the increase of the MR121 fluorescence during the reaction time. The slope in the linear range of kinetics was calculated, and the IC50 values of the test compounds were determined using a four parameter equation for curve fitting.
Inhibition assay with a panel of proteases
The inhibitory activity of solutions containing Fynomer 4C-E4 at different concentrations (4 µM to 0.04 nM) was tested with the following panel of different serine proteases: cathepsin G, dipeptidyl protease-4 (DPP-4), kallikrein-1 (KLK-1), neutrophil elastase and trypsin. All inhibition assays were performed by the company Genscript (Piscataway, USA). Briefly, serine proteases were incubated with 4C-E4 and the corresponding substrate for each enzyme was added (cathepsin G: Suc-AAPF-pNA; DPP-4: Gly-Pro-AMC; KLK-1: D-VLR-AFC; neutrophil elastase: MeOSuc-AAPV-AMC; trypsin: Z-GPR-AMC). Absorption of the cleaved substrate (colorimetric or fluorometric read-out) was read using either a kinetic model (cathepsin G, DPP-4, neutrophil elastase) or an endpoint model (KLK-1, trypsin).
Preparation of chymase/Fynomer complexes, crystallization and crystal structure determination
Chymase and Fynomer were pooled using a 1.5 molar excess of Fynomer. After 60 min incubation at RT, the slightly turbid solution was centrifuged at 3′000 x g for 10 min. The chymase/Fynomer complex was then separated from the free Fynomer by chromatography on a Superdex 75 16/60 column in 50 mM sodium phosphate pH 6.0, 300 mM NaCl, 0.3% CHAPS. Prior to crystallization experiments, the Fynomer-chymase complexes were concentrated to 15 mg/ml. Crystallization screening against an INDEX screen (Hampton Research) was performed at 21°C, either in sitting drops by vapor diffusion, or in microbatch experiments. Crystals appeared within five days out of several conditions with polyethylene glycol as precipitating agent, and grew to their final size within two weeks after setup.
For data collection, crystals were flash frozen at 100°K with dried paraffin oil as cryoprotectant. Diffraction data were collected at a wavelength of 1.0 Å using a PILATUS 6M detector at the beam line X10SA of the Swiss Light Source (Villigen, Switzerland). In all cases, data were processed with XDSCitation28 and scaled with SADABS (Bruker AXS). The structures were solved by molecular replacement with PHASERCitation29 using as search models an in-house chymase structure and the SH3 domain of 1fyn.pdb with the RT- and n-src-loops deleted. Refinement was performed with Refmac5Citation30 from the CCP4 suiteCitation31 and BUSTERCitation32-Citation34 or PHENIXCitation35 and model building done with COOT.Citation36
Accession numbers
The PDB coordinates for the six Fynomer-chymase complexes have been deposited into PDB under the IDs 4afq, 4afs, 4afu, 4afz, 4ag1, 4ag2.
| Abbreviations: | ||
| AUC | = | analytical ultracentrifugation |
| SPR | = | surface plasmon resonance |
| FRET | = | fluorescence resonance energy transfer |
Additional material
Download Zip (110.7 KB)Acknowledgments
We thank Beat Wipf and Christian Miscenic for supporting the project with microbial fermentation, Eric Kusznir for performing the experiments with the analytical ultracentrifuge and Markus G. Rudolph for refining the twinned complex structure.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Note
Supplemental materials can be found at: http://www.landesbioscience.com/journals/mabs/article/20452/
References
- Shea-Donohue T, Stiltz J, Zhao A, Notari L. Mast cells. Curr Gastroenterol Rep 2010; 12:349 - 57; http://dx.doi.org/10.1007/s11894-010-0132-1; PMID: 20711694
- Miller HRP, Pemberton AD. Tissue-specific expression of mast cell granule serine proteinases and their role in inflammation in the lung and gut. Immunology 2002; 105:375 - 90; http://dx.doi.org/10.1046/j.1365-2567.2002.01375.x; PMID: 11985658
- de Garavilla L, Greco MN, Sukumar N, Chen Z-W, Pineda AO, Mathews FS, et al. A novel, potent dual inhibitor of the leukocyte proteases cathepsin G and chymase: molecular mechanisms and anti-inflammatory activity in vivo. J Biol Chem 2005; 280:18001 - 7; http://dx.doi.org/10.1074/jbc.M501302200; PMID: 15741158
- Takai S, Jin D, Miyazaki M. New approaches to blockade of the renin-angiotensin-aldosterone system: chymase as an important target to prevent organ damage. J Pharmacol Sci 2010; 113:301 - 9; http://dx.doi.org/10.1254/jphs.10R05FM; PMID: 20675958
- Maryanoff BE, de Garavilla L, Greco MN, Haertlein BJ, Wells GI, Andrade-Gordon P, et al. Dual inhibition of cathepsin G and chymase is effective in animal models of pulmonary inflammation. Am J Respir Crit Care Med 2010; 181:247 - 53; http://dx.doi.org/10.1164/rccm.200904-0627OC; PMID: 19875688
- Takai S, Jin D, Sakaguchi M, Katayama S, Muramatsu M, Sakaguchi M, et al. A novel chymase inhibitor, 4-[1-([bis-(4-methyl-phenyl)-methyl]-carbamoyl)3-(2-ethoxy-benzyl)-4-oxo-azetidine-2-yloxy]-benzoic acid (BCEAB), suppressed cardiac fibrosis in cardiomyopathic hamsters. J Pharmacol Exp Ther 2003; 305:17 - 23; http://dx.doi.org/10.1124/jpet.102.045179; PMID: 12649348
- Doggrell SA, Wanstall JC. Cardiac chymase: pathophysiological role and therapeutic potential of chymase inhibitors. Can J Physiol Pharmacol 2005; 83:123 - 30; http://dx.doi.org/10.1139/y04-136; PMID: 15791285
- Diaconu NC, Rummukainen J, Naukkarinen A, Mättö M, Harvima RJ, Pelkonen J, et al. Mast cell chymase is present in uterine cervical carcinoma and it detaches viable and growing cervical squamous carcinoma cells from substratum in vitro. Arch Dermatol Res 2011; 303:499 - 512; http://dx.doi.org/10.1007/s00403-011-1121-4; PMID: 21274549
- Griffin L, Lawson A. Antibody fragments as tools in crystallography. Clin Exp Immunol 2011; 165:285 - 91; http://dx.doi.org/10.1111/j.1365-2249.2011.04427.x; PMID: 21649648
- Iwata S, Ostermeier C, Ludwig B, Michel H. Structure at 2.8 A resolution of cytochrome c oxidase from Paracoccus denitrificans. Nature 1995; 376:660 - 9; http://dx.doi.org/10.1038/376660a0; PMID: 7651515
- Skerra A. Engineered protein scaffolds for molecular recognition. J Mol Recognit 2000; 13:167 - 87; http://dx.doi.org/10.1002/1099-1352(200007/08)13:4<167::AID-JMR502>3.0.CO;2-9; PMID: 10931555
- Binz HK, Plückthun A. Engineered proteins as specific binding reagents. Curr Opin Biotechnol 2005; 16:459 - 69; http://dx.doi.org/10.1016/j.copbio.2005.06.005; PMID: 16005204
- Gebauer M, Skerra A. Engineered protein scaffolds as next-generation antibody therapeutics. Curr Opin Chem Biol 2009; 13:245 - 55; http://dx.doi.org/10.1016/j.cbpa.2009.04.627; PMID: 19501012
- Musacchio A, Wilmanns M, Saraste M. Structure and function of the SH3 domain. Prog Biophys Mol Biol 1994; 61:283 - 97; http://dx.doi.org/10.1016/0079-6107(94)90003-5; PMID: 8073124
- Grabulovski D, Kaspar M, Neri DA. A novel, non-immunogenic Fyn SH3-derived binding protein with tumor vascular targeting properties. J Biol Chem 2007; 282:3196 - 204; http://dx.doi.org/10.1074/jbc.M609211200; PMID: 17130124
- Bertschinger J, Grabulovski D, Neri D. Selection of single domain binding proteins by covalent DNA display. Protein Eng Des Sel 2007; 20:57 - 68; http://dx.doi.org/10.1093/protein/gzl055; PMID: 17242027
- Viti F, Nilsson F, Demartis S, Huber A, Neri D. Design and use of phage display libraries for the selection of antibodies and enzymes. Methods Enzymol 2000; 326:480 - 505; http://dx.doi.org/10.1016/S0076-6879(00)26071-0; PMID: 11036659
- Semba K, Nishizawa M, Miyajima N, Yoshida MC, Sukegawa J, Yamanashi Y, et al. yes-related protooncogene, syn, belongs to the protein-tyrosine kinase family. Proc Natl Acad Sci U S A 1986; 83:5459 - 63; http://dx.doi.org/10.1073/pnas.83.15.5459; PMID: 3526330
- Kawakami T, Pennington CY, Robbins KC. Isolation and oncogenic potential of a novel human src-like gene. Mol Cell Biol 1986; 6:4195 - 201; PMID: 3099169
- Silacci M, Brack S, Schirru G, Mårlind J, Ettorre A, Merlo A, et al. Design, construction, and characterization of a large synthetic human antibody phage display library. Proteomics 2005; 5:2340 - 50; http://dx.doi.org/10.1002/pmic.200401273; PMID: 15880779
- De Genst E, Silence K, Decanniere K, Conrath K, Loris R, Kinne J, et al. Molecular basis for the preferential cleft recognition by dromedary heavy-chain antibodies. Proc Natl Acad Sci U S A 2006; 103:4586 - 91; http://dx.doi.org/10.1073/pnas.0505379103; PMID: 16537393
- Sennhauser G, Grütter MG. Chaperone-assisted crystallography with DARPins. Structure 2008; 16:1443 - 53; http://dx.doi.org/10.1016/j.str.2008.08.010; PMID: 18940601
- Monroe N, Sennhauser G, Seeger MA, Briand C, Grütter MG. Designed ankyrin repeat protein binders for the crystallization of AcrB: plasticity of the dominant interface. J Struct Biol 2011; 174:269 - 81; http://dx.doi.org/10.1016/j.jsb.2011.01.014; PMID: 21296164
- Abskharon RNN, Soror SH, Pardon E, El Hassan H, Legname G, Steyaert J, et al. Combining in-situ proteolysis and microseed matrix screening to promote crystallization of PrPc-nanobody complexes. Protein Eng Des Sel 2011; 24:737 - 41; http://dx.doi.org/10.1093/protein/gzr017; PMID: 21536542
- Steyaert J, Kobilka BK. Nanobody stabilization of G protein-coupled receptor conformational states. Curr Opin Struct Biol 2011; 21:567 - 72; http://dx.doi.org/10.1016/j.sbi.2011.06.011; PMID: 21782416
- Hoogenboom HR, Griffiths AD, Johnson KS, Chiswell DJ, Hudson P, Winter G. Multi-subunit proteins on the surface of filamentous phage: methodologies for displaying antibody (Fab) heavy and light chains. Nucleic Acids Res 1991; 19:4133 - 7; http://dx.doi.org/10.1093/nar/19.15.4133; PMID: 1908075
- Perspicace S, Banner D, Benz J, Müller F, Schlatter D, Huber W. Fragment-based screening using surface plasmon resonance technology. J Biomol Screen 2009; 14:337 - 49; http://dx.doi.org/10.1177/1087057109332595; PMID: 19403917
- Kabsch W. XDS. Acta Crystallogr D Biol Crystallogr 2010; 66:125 - 32; http://dx.doi.org/10.1107/S0907444909047337; PMID: 20124692
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr 2007; 40:658 - 74; http://dx.doi.org/10.1107/S0021889807021206; PMID: 19461840
- Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr 1997; 53:240 - 55; http://dx.doi.org/10.1107/S0907444996012255; PMID: 15299926
- Collaborative Computational Project, Number 4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr 1994; 50:760 - 3; http://dx.doi.org/10.1107/S0907444994003112; PMID: 15299374
- Bricogne G. Direct phase determination by entropy maximization and likelihood ranking: status report and perspectives. Acta Crystallogr D Biol Crystallogr 1993; 49:37 - 60; http://dx.doi.org/10.1107/S0907444992010400; PMID: 15299544
- Roversi P, Blanc E, Vonrhein C, Evans G, Bricogne G. Modelling prior distributions of atoms for macromolecular refinement and completion. Acta Crystallogr D Biol Crystallogr 2000; 56:1316 - 23; http://dx.doi.org/10.1107/S0907444900008490; PMID: 10998628
- Blanc E, Roversi P, Vonrhein C, Flensburg C, Lea SM, Bricogne G. Refinement of severely incomplete structures with maximum likelihood in BUSTER-TNT. Acta Crystallogr D Biol Crystallogr 2004; 60:2210 - 21; http://dx.doi.org/10.1107/S0907444904016427; PMID: 15572774
- Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 2010; 66:213 - 21; http://dx.doi.org/10.1107/S0907444909052925; PMID: 20124702
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 2004; 60:2126 - 32; http://dx.doi.org/10.1107/S0907444904019158; PMID: 15572765