Abstract
Therapeutic monoclonal antibodies have several advantages over small molecule drugs and small proteins and peptides, including a long serum half-life. The long serum half-life of IgG is due, in part, to its molecular weight (150kDa) and its ability to bind FcRn. Both the CH2 and CH3 domains of Fc are involved in FcRn binding. Antibody fragments and antibody-like scaffolds have improved penetration into tissues due to their small size, yet suffer from a short serum half-life of less than one hour. The human CH2 domain (CH2D) of IgG1 retains a portion of the FcRn binding site, is amenable to modification for target binding, and may represent the smallest antibody-like scaffold retaining a relatively long serum half-life. Here we describe the generation of a dimeric CH2D (dCH2D) and determination of its pharmacokinetics (PK), as well as the PK of wild-type monomeric CH2D (mCH2D) and a short stabilized CH2D variant (ssCH2D) in normal B6 mice, human FcRn transgenic mice and cynomolgus macaques. The elimination half-life of dCH2D was 9.9, 10.4 and 11.2 hours, and that of ssCH2D was 13.1, 9.9 and 11.4 hours, in B6 mice, hFcRn mice and cynomolgus macaques, respectively. These half-lives were slightly longer than that of mCH2D (6.9 and 8.8 hours) in B6 and hFcRn mice, respectively. These data demonstrate that engineered CH2D-based variants have relatively long serum half-lives, making them a unique scaffold suitable for development of targeted therapeutics.
Therapeutic monoclonal antibodies have several advantages over small molecule drugs and small proteins and peptides, including a long serum half-life. The long serum half-life of IgG is due, in part, to its molecular weight (150kDa) and its ability to bind FcRn. Both the CH2 and CH3 domains of Fc are involved in FcRn binding. Antibody fragments and antibody-like scaffolds have improved penetration into tissues due to their small size, but they commonly have serum half-lifes of less than one hour. The human CH2 domain (CH2D) of IgG1 retains a portion of the FcRn binding site, is amenable to modification for target binding, and may represent the smallest antibody-like scaffold retaining a relatively long serum half-life. We describe here the generation of a dimeric CH2D (dCH2D) and determination of its pharmacokinetics (PK), as well as the PK of wild-type monomeric CH2D (mCH2D) and a short stabilized CH2D variant (ssCH2D) in normal B6 mice, human FcRn transgenic mice and cynomolgus macaques. The elimination half-life of dCH2D was 9.9, 10.4 and 11.2 h, and that of ssCH2D was 13.1, 9.9 and 11.4 h, in B6 mice, hFcRn mice and cynomolgus macaques, respectively. These half-lives were slightly longer than that of mCH2D (6.9 and 8.8 h) in B6 and hFcRn mice, respectively. These data demonstrate that engineered CH2D-based variants have relatively long serum half-lives, making them a unique scaffold suitable for development of targeted therapeutics.
Introduction
Over 30 monoclonal antibodies including three antibody-derived (Fab) fragments have been approved in the United States and Europe for the treatment of various diseases.Citation1 Although these products demonstrate significant clinical benefit in certain indications, a fundamental limitation of such large molecules is their poor penetration into tissues (e.g., solid tumors) and poor or absent binding to epitopes on the surface of some molecules that are accessible only by molecules of smaller size.Citation2-Citation4 Therefore, developing novel scaffolds of much smaller size with greater stability may have distinct advantages over antibodies against certain targets. Scaffolds under development are based on various human and non-human molecules and can be divided into two major groups; antibody-derived and others.
Antibody-derived scaffolds include the VH domain, camelids (nanobodies), single chain variable fragments (scFv), antigen-binding fragments (Fab), Avibodies, minibodies, CH2D, and Fcabs.Citation5-Citation8 Non-antibody-derived scaffolds include the 10th fibronectin type III subunit (Adnexins), tenacsin type III subunit (Centryns), and designed ankyrin repeat proteins (DARPins).Citation4,Citation9-Citation11 These scaffolds are attractive as platforms for developing novel therapeutics due to their smaller size (12–50 kDa) compared with IgG (150 kDa). The small size leads to greater and more rapid tissue accumulation and the ability to potentially recognize epitopes in protein targets not accessible to full size antibodies.Citation2,Citation4,Citation9,Citation12,Citation13 This could be particularly important for the development of therapeutics targeting rapidly mutating viruses, solid tumors, or cell-cell synapses. Typically, these small scaffolds are monomeric, of high solubility and do not significantly aggregate or can be engineered to reduce aggregation. Moreover, two or more scaffolds may be linked to form multi-specific and multifunctional molecules. Such small domains are well expressed in bacterial, yeast, and mammalian cell systems.Citation14-Citation16
The major disadvantage of the smaller protein scaffolds is their rapid renal clearance and thus short circulating half-life. Typically, proteins smaller than 50–60kDa have short serum half-lives of less than one hour.Citation4,Citation6,Citation9,Citation10,Citation13 A number of approaches are used to increase the serum half-life of the various scaffolds, including fusion to albumin or Fc, hijacking serum albumin with albumin-binding peptides or addition of a second scaffold that binds to FcRn or albumin, adding half-life extension peptides or using pegylation.Citation13,Citation17,Citation18 The advantage of the small size, however, is potentially lost because such modifications increase the molecule’s size to avoid renal clearance.
To address the short half-life disadvantage of the smaller antibody-like scaffolds, we are currently developing variants of the isolated human CH2 domain (IgG1 constant domain 2, CH2D, ) as a new antibody-like scaffold platform. The human CH2D is small (~12 kDa), is amenable to loop and β sheet modifications that can be used to generate large libraries of binders to target molecules, and yet retains a portion of the native FcRn binding site. Earlier reports demonstrated that murine CH2D had a serum elimination phase half-life of 26 h in mice;Citation19 however, the human counterpart has never been tested in vivo. Several variants of the CH2D have been described, and more recently, it was reported that a stabilized and shortened version (ssCH2D) binds to human FcRn.Citation20 We report herein preparation of several CH2D variants and evaluation of their pharmacokinetics (PK) in normal B6 mice, human FcRn transgenic mice and cynomolgus macaques. These data demonstrate that the CH2D has a longer serum half-life compared with other unmodified and published scaffolds of a similar molecular weight.
Figure 1. Diagram of IgG [PDB: 1HZH (reference: Saphire EO, Parren PW, Pantophlet R, Zwick MB, Morris GM, Rudd PM, et al. Crystal structure of a neutralizing human IGG against HIV-1: a template for vaccine design. Science. 2001; 293:1155–9.)], CH2D [PDB 3DJ9 (reference: Prabakaran P, Vu BK, Gan J, Feng Y, Dimitrov DS, Ji X. Structure of an isolated unglycosylated antibody C(H)2 domain. Acta Crystallogr D Biol Crystallogr. 2008; 64:1062–7.)] and ssCH2D (modified based on PDB 3DJ9). The crystal structure was rendered by was rendered by PyMOL (The PyMOLMolecular Graphics System, Version 1.2, Schrödinger, LLC).
![Figure 1. Diagram of IgG [PDB: 1HZH (reference: Saphire EO, Parren PW, Pantophlet R, Zwick MB, Morris GM, Rudd PM, et al. Crystal structure of a neutralizing human IGG against HIV-1: a template for vaccine design. Science. 2001; 293:1155–9.)], CH2D [PDB 3DJ9 (reference: Prabakaran P, Vu BK, Gan J, Feng Y, Dimitrov DS, Ji X. Structure of an isolated unglycosylated antibody C(H)2 domain. Acta Crystallogr D Biol Crystallogr. 2008; 64:1062–7.)] and ssCH2D (modified based on PDB 3DJ9). The crystal structure was rendered by was rendered by PyMOL (The PyMOLMolecular Graphics System, Version 1.2, Schrödinger, LLC).](/cms/asset/0093d549-80ca-4513-9b0e-51e7aa1d5537/kmab_a_10920652_f0001.gif)
Results
Design of CH2D Variants
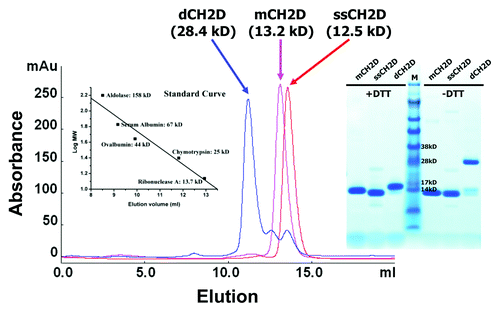
Our laboratory previously reported on the characteristics of a short stabilized variant of CH2D (ssCH2D) that has enhanced binding to FcRn.Citation21 The ssCH2D (12.5 kD), the wild-type monomeric human (mCH2D, 13.2 kD) and a natural hinge CH2D dimer (dCH2D, 28.4 kD) were recombinantly produced for these PK studies. The dCH2D was formed by adding a portion of the natural IgG hinge sequence GSGSCDKTHT to the N-terminus of mCH2D, which created a disulfide bond between monomers generating intact and stable dimers (). To decrease or eliminate the FcRn binding, a variant form of ssCH2D was generated by mutating two amino acids, I253A and S254A (, bold and underlined). It has been reported that the histidine at position 310 in CH2D and the histidine at position 435 in CH3D are critical for the pH-dependent binding to FcRn. Histidine-containing tags are commonly used to facilitate the purification of recombinant proteins; to determine if the addition of HIS-tags to CH2D could influence FcRn binding and half-life, several CH2Ds with and without HIS-tags were made and tested only in the murine studies.
Table 1. Amino acid sequences of test proteins
Expression, Purification and In Vitro Characterization of CH2D Variants
The CH2D protein variants were produced in E. coli and endotoxin was removed.In some cases, the CH2D proteins included HIS-tags on either the N-terminus, the C-terminus, or were produced without HIS-tags to determine the role, if any, for HIS-tags on FcRn binding or serum half-life. The short stabilized version, ssCH2D, has two additional cysteines (, underlined and bold) to form a second disulfide bond and is shortened on the N-terminus by seven amino acids.Citation21 The size exclusion chromatography and SDS-PAGE gel of the isolated and purified test proteins are represented in .
Figure 2. Size exclusion chromatography of mCH2D, ssCH2D and dCH2D. mCH2D and ssCH2D are monomeric and dCH2D is dimeric. Left insert: standard curve; right insert: SDS-PAGE under reducing (+DTT) and non-reducing (-DTT) conditions.
Pharmacokinetics in B6 Mice
The PK data for the variant CH2Ds in normal B6 mice are summarized in . The mCH2D, dCH2D, and ssCH2D variants had an elimination phase (β-phase) half-lives of 6.9, 9.9 and 13.1 h, respectively. Making two mutations in the putative FcRn binding region of ssCH2D (I253A, S254A) reduced the elimination phase half-life from 13.1 h to 8.7 h. The ssCH2D variant lacking a HIS-tag had an elimination phase half-life of 16.6 h. Modifying the FcRn binding portion of this ssCH2D variant lacking a HIS-tag reduced the elimination phase half-life from 16.6 h to 7.4 h. Switching the HIS-tag to the C-terminus on the ssCH2D variant produced an elimination phase half-life of 12.4 h. The bacterially-produced human Fc control had an elimination phase half-life of 21.1 h.
Table 2. Pharmacokinetics in B6 mice
Pharmacokinetics in hFcRn Mice
The half-life data for the variant CH2Ds in the hFcRn transgenic mouse are presented in . mCH2D, dCH2D, and ssCH2D variants had elimination phase half-lives of 8.8, 10.4 and 9.9 h, respectively. Making two mutations in the putative FcRn binding region of ssCH2D reduced the elimination phase half-life from 9.9 h to 3.9 h. ssCH2D lacking a HIS-tag had an elimination phase half-life of 8.4 h, while the ssCH2D lacking a HIS-tag but including the two mutations in the FcRn binding site had a reduced elimination phase half-life of 3.2 h. Switching the HIS-tag to the C-terminus on ssCH2D resulted in an elimination phase half-life of 9.5 h. In this model, the human Fc control had an elimination half-life of 15.0 h.
Table 3. Pharmacokinetics in the hFcRn mouse
Pharmacokinetics of CH2D in Cynomolgus Macaques
Representative time-dependant concentration curves for the various test CH2Ds are shown in – and summary data are presented in . The elimination phase half-life for dCH2D was 11.2 h for the animals in the 10 mg/kg dosing group and 8.8 h for those in the 20 mg/kg dosing group. For ssCH2D, the elimination phase half-life was 11.4 h for the 10 mg/kg group and 12.7 h for the 20 mg/kg group. Using individual animal data from two independent analyses, the mean elimination half-life for ssCH2D in the 10 mg/kg group of animals was 10.7 h, which compares to 11.4 h for the pooled serum data. For the 20 mg/kg group, the mean for the individual ssCH2D animals was 11.7 h, which compares to 12.7 h for the pooled samples.
Figure 3. Serum concentration-time course plot of ssCH2D, 10 mg/kg dose group, of individual primates numbered 316, 429, 918 (left) and for pooled serum samples of the same individuals (□) (right). The experimental points are connected by solid linear lines and represented as mean ± standard deviation (n = 2).
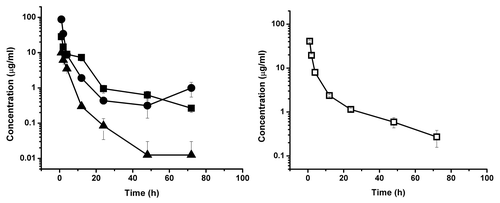
Figure 4. Serum concentration-time course plot of ssCH2D, 20 mg/kg dose group, of individual primates numbered 450, 009, 239 (left) and for pooled serum sample of the same individuals (□) (right). The experimental points are connected by solid linear lines and is represented as mean ± standard deviation (n = 2).
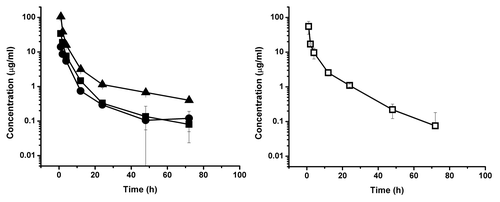
Figure 5. Serum concentration-time course plot of dCH2D, 10 mg/kg dose group, of individual primates numbered 411, 896, 977 (left) and for pooled serum sample of the same individuals (□) (right). The experimental points are connected by solid linear lines and is represented as mean ± standard deviation (n = 2).
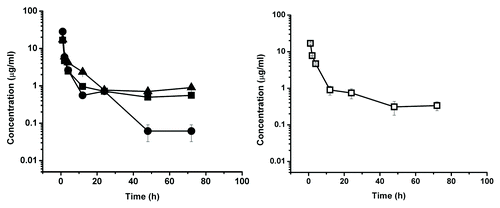
Figure 6. Serum concentration-time course plot of dCH2D, 20 mg/kg dose group, of individual primates numbered 549, 559, 644 (left) and for pooled serum sample of the same individuals (□) (right). The experimental points are connected by solid linear lines and is represented as mean ± standard deviation (n = 2).
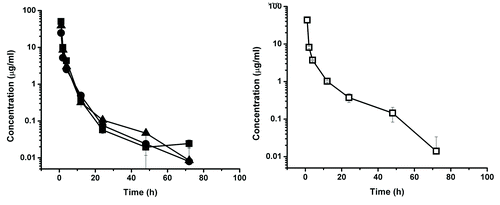
Table 4. Pharmacokinetics in Cynomolgus macaques (10mg/kg and 20mg/kg)
Discussion
The PK studies reported herein support the hypothesis that the human CH2D scaffold has a longer serum half-life compared with other antibody-derived fragments of the same molecular weight. VH domains and other related fragments in the 12–25kDa molecular weight range have reported elimination phase half-life in mice and primates of less than one hour.Citation6,Citation14,Citation15 The CH2D variants tested in this study demonstrated serum half-lives ranging from 7–13 h in B6 mice, 3–10 h in hFcRn mice and 8–13 h in cynomolgus macaques. These data are supported by published work suggesting that isolated murine CH2D has a serum half-life in mice of 25 h.Citation19 The relatively small differences in half-life for monomers observed in this study compared with the published data using murine CH2D could be due to several factors, however, without a direct comparison in a single study, it is not possible to properly compare the two studies and results.
The observed serum half-life for the human CH2D variants tested is likely, in large part, due to the binding of CH2D to the FcRn receptor because these CH2Ds had no target binding specificity, although the dimer may have retained some FcγR binding. This conclusion is further supported by the reduction in half-life that occurred when CH2D was mutated in two of the residues (I253A, S254A) reported to be involved in FcRn binding.Citation22-Citation24 It has been reported that the histidine residue at position 310 is also important for FcRn binding at pH 6.0; therefore, it is possible that an additional mutation in H310 could further reduce FcRn binding and shorten the half-life to that reported for a VH domain.Citation22-Citation24 The observed reduction in half-life from these two mutations in the ssCH2D variants, including those variants lacking HIS-tags, further supports the notion that the isolated CH2D binds to FcRn and that this action contributes to the longer serum half-life. That CH2D binds to FcRn is further supported by published work demonstrating that ssCH2D, reported previously as “m01s,” binds to soluble, recombinant hFcRn in a flow cytometry assay and that the interaction can be blocked by added human IgG1.Citation20
Serum persistence of antibodies and derived fragments is dependent on several factors, including molecular weight, overall charge, renal clearance, binding target, oxidation under the conditions for storage and formulation, and binding to the neonatal Fc receptor (FcRn).Citation25-Citation27 Most antibody fragments that are less than 50 kDa will be rapidly cleared by the kidneys. It has been reported that the murine FcRn is a promiscuous receptor in that it binds to human IgG with a higher affinity than it does to murine IgG.Citation22,Citation28,Citation29 This higher affinity of human IgG for the mFcRn results in a prolonged serum half-life in normal mice that is only limited by immune responses. We also observed in this study that the CH2D variants tested in the B6 mouse had slightly longer half-lives compared with those data obtained from the hFcRn mouse and macaques. The transgenic hFcRn mouse model used in our experiments was hemizygous for the FcRn receptor in that it contains one copy of the human FcRn heavy chain and associates with the murine β1-microglobulin.Citation24,Citation30,Citation31 It has been reported that human IgG tested in the hemizygous transgenic mouse has a shorter half-life than the same IgG tested in the homozygous version (two copies of the hFcRn gene) of the transgenic hFcRn mouse.Citation24,Citation30,Citation31 However, the shorter half-life for our CH2Ds in the hFcRn transgenic mouse tested compared with that in the normal B6 mouse is consistent with other reports on antibody half-life and our control Fc protein (21 h in the B6 mouse and 15 h in the hFcRn mouse).Citation22,Citation32
Both the ssCH2D and dCH2D at 10 mg/kg had an approximately 11 h half in the cynomolgus macaques with similar clearance (CL), volume of distribution (Vd) and area-under-the-curve (AUC) values. The half-life was shorter for the dCH2D in the 20 mg/kg group and a moderate increase in the Vd was calculated for the ssCH2D in the 20 mg/kg group. This variance in Vd from the 10 mg/kg dosing group to the 20 mg/kg dosing group could be due to the combination of the treatment for the Shigella infection (only in the 20 mg group), the volume loading of 20 ml/kg of normal saline 24 h prior to dosing and the larger CH2D dosing volume.
Taken together, these data strongly suggest that CH2D has a circulating half-life that is significantly longer than that reported for other unmodified small scaffolds of similar size. Interestingly, the CH2Ds reported herein did not have any target binding other than possibly the dCH2D may have retained binding to FcγRs, but this interaction has not been confirmed. It should be mentioned that the half-life of CH2Ds engineered to bind to specific targets could be altered, either longer or shorter, due to possible structural changes in the scaffold that could affect binding to FcRn or the effects of target binding and target location. The dimer also did not appear to have a longer half-life than a monomer although it was twice the molecular weight, suggesting that there is no advantage of the dimer over the monomer in terms of serum half-life. Renal clearance, urine concentration and specific tissue distribution were not examined as part of the present study.
There are several other ways by which small scaffolds can be modified to increase the circulating half-life, including the use of albumin-binding peptides, pegylation, fusion to Fc, and the addition of other carrier proteins. Although these may improve the serum half-life, they also increase the size and alter other characteristics that can influence their affinity, PK and pharmacodynamics. We have constructed large, diverse DNA and phage-displayed libraries of targeting CH2Ds based on novel loop designs. These libraries are available for screening against targets of therapeutic interest and studies using targeting CH2Ds will be reported in the future.
Materials and Methods
Protein Production
All the proteins used in this study were produced in E. coli HB2151. Briefly, 50 mL seed culture (SB media w/ 2% glucose + kanamycin) was incubated at 37°C for 16 h and was used to inoculate 1 L of pre-warmed SB media containing 100 μg/mL ampicillin and 0.2% glucose at a 1:100 dilution. Cell cultures were allowed to incubate at 37°C until OD600 ≈0.8 at which point the culture was induced with 1 mM isopropyl-β-D-thiogalactopyranoside (IPTG). The culture was then allowed to incubate at 30°C for 18 h. Cells were harvested by centrifugation and stored at -80°C. Pre-induction and post-induction samples were analyzed by SDS-PAGE and western blot.
For purification of CH2 domains with HIS-tag, cell paste was resuspended in 10 ml of Buffer A (50 mM TRIS-HCl, and 450 mM NaCl, pH 8.0) and Polymyxin B sulfate was added to the suspension at 0.5 mu/ml and gently rotated for 1 h at room temperature. The resulting lysate was centrifuged at 20,000 × g for 45 min. Clarified lysate was loaded on to a Ni-Sepharose column (IMAC resin from Qiagen) pre-equilibrated with Buffer A (2.5 ml of resin used per 1 L expression scale). The column was washed with 10 column volumes of Buffer A and 10 column volumes of Buffer A + 30 nM imidazole, and bound protein was eluted with Buffer A + 200 mM imidazole. Protein-containing fractions were analyzed by Coomassie-stained SDS-PAGE and western blotting (anti-His antibody).
For purification of CH2 domains without HIS-tag, a two-step method was used. The first step was cation exchange chromatography. Cell pellet was resuspend in 50 ml of Buffer A (50 mM MES, pH 6.0), and Polymyxin B sulfate was added to the suspension at 0.5 mu/ml and gently rotated for 1 h at room temperature. The resulting lysate was centrifuged at 20,000 × g for 45 min. Clarified lysate was loaded on SP column GE healthcare) pre-equilibrated with Buffer A. The column was washed with 10-column volumes of Buffer A and bound protein was gradually eluted with Buffer B (50 mM MES, pH 6.0 with 0.5 M NaCl). Protein-containing fractions were analyzed by Coomassie-stained SDS-PAGE. The fractions with target protein were collected, and concentrated to about 5 ml. The second step was size exclusion chromatography. The concentrated impure protein was loaded to 16/60 Sephacryl S-100 column (GE Healthcare). The protein-containing fractions were analyzed by Coomassie-stained SDS-PAGE and confirmed by ELISA as described previously.Citation21
The Fc without any tag was purified by protein G. The cell pellet was resuspended in 50 ml PBS, and Polymyxin B sulfate was added to the suspension at 0.5 mu/ml and gently rotated for 1 h at room temperature. The resulting lysate was centrifuged at 20,000 × g for 45 min. Clarified lysate was loaded on to Protein G column (protein G resin from GE healthcare) pre-equilibrated with PBS. The column was washed with 10 column volumes of PBS and bound protein was gradually eluted with 0.5M acetic acid, neutralizing by 1M TRIS-HCl, pH 9.0. Protein-containing fractions were analyzed by Coomassie-stained SDS-PAGE.
The fractions containing prominent target protein were pooled, dialyzed against PBS and the pool was concentrated. Endotoxin of the proteins used for mouse study was removed by Detoxi-Gel Endotoxin Removing Gel (Thermo Scientific) according to the manufactory instructions and its level was determined by the EndoSafe PTS kit (Charles River Labs). The endotoxin level of the proteins used for the monkey study was reduced by the De-toxTM process (Blue Sky). The final formulation was in PBS at pH 7.4.
Murine Animal Models
Studies were performed at Jackson Laboratories and approved by the Animal Use and Care Committee. Twenty four (24) female B6 mice or 24 (24) female transgenic hFcRn mice (Tg276 hemizygous, Jackson Laboratories) were housed in individually in positively ventilated polycarbonate cages with HEPA filtered air at a density of 4 mice per cage. The animal room was lighted entirely with artificial fluorescent lighting, with a controlled 12 h light/dark cycle (6 a.m. to 6 p.m. light). The normal temperature and relative humidity ranges in the animal rooms were 22 ± 4°C and 50 ± 15%, respectively. The animal rooms were set to have 15 air exchanges per hour. Filtered tap water, acidified to a pH of 2.5 to 3.0, and a diet was provided ad libitum. After 1 week of acclimation, the mice each received a single IV injection (100 ug/mouse) of one of three CH2Ds (n = 8 for each CH2D); tail vein injections (50 ul) were performed with CH2Ds at a concentration of 2 mg/ml.
Mice were bled, either orbitally or submandibularly, (50 μl) at pre-dose, and at various time-points. All mice received a baseline bleed, then for the remaining bleeds subsets of 4 mice were bled at alternating time-points. All mice were bled at the last time-point. Blood was pooled for each group, processed to serum and frozen at -80°C. Samples were analyzed by enzyme-linked immunosorbent assay (ELISA) (see below). All PK analyses were performed using ELISA concentration/timepoint data using the PK Solutions 2.0, noncompartmental PK data analysis software from Summit Research Services. For data points below the level of detection or below 0.01ug/ml, the software assigns a zero for that value and it is not used in the analysis.
Primate Animal Models
Studies were performed at Texas Biological Research Institute and approved by the Institutional Animal Care and Use Committee. Only the dCH2D and ssCH2D variants were tested in cynomolgus primates. Animals on average weighed 5.5 kg and the CH2Ds were dosed as a single IV administration at either 10 mg/kg or 20 mg/kg in 3 animals per test article (12 total). Animals in the 10 mg/kg group were administered approximately 16 ml at 2–3 ml/min of ssCH2D and 11 ml at 2–3 ml/min of dCH2D. Animals in the 20 mg/kg group received 31 ml at 1 ml/min of ssCH2D and 22 ml at 1 ml/min for dCH2D. In addition, animals in the 20 mg/kg group developed a shigella infection and were treated and cured with Bytril® for one week with one week washout before starting the study. Finally, all animals in the 20 mg/kg group received 20 ml/kg (avg. 90 ml) of normal saline subcutaneously to expand their blood volume 24 h prior to dosing. Blood draws were timed following administration. Purified CH2D protein was provided in PBS. Animals were individually caged for the duration of the study and observed daily for clinical signs and symptoms. 3–5 ml of blood was drawn at baseline (t0), 1, 2, 4, 12, 24, 48, and 72 h after test article administration. Serum was prepared for ELISA standards. For all ELISAs the material used for injection was used to make the standard curves. ELISAs were performed on individual animal samples and on pooled samples. The data are reported from both pooled serum samples and from individual animals for each group. All PK analyses were performed using ELISA concentration/time point data running the PK Solutions 2.0, noncompartmental PK data analysis software from Summit Research Services.
Measurements of CH2D Levels in Sera
A mouse monoclonal antibody to specific human IgG1 Fc CH2 domain was purchased from AdB serotec (Oxford, UK; clone# 8A4) and used for capturing CH2D from serum. To measure the concentration of CH2Ds in mouse sera, 96-well plates were coated with mouse IgG 8A4 on half area ELISA plate wells at 100 ng/well in 50 ul PBS and incubated at 4°C overnight. Plates were washed 3 times with PBST (PBS+0.05% Tween 20), 100 µl blocking buffer (3% non-fat dry milk in PBST) was added and the plates were incubated at 37°C for 1 h. After washing, the diluted serum/PBS samples at different time points were added and incubated for 2 h at 37°C. The purified CH2D-proteins serially diluted in pre-dose serum/PBS were used to make standard curves. Each dilution was tested in duplicate. The ELISA plates were then washed four times with PBST and secondary antibody, anti-human IgG (Fc) peroxidase conjugate (Sigma) used at 1:1000 or 1:2000 in blocking buffer, was added. Plates were incubated at 37°C for 1 h and washed four times with PBST. HRP substrate ABTS was added and plates were read in a 96-well plate reader at 405 nm wavelength.
Because the macaque IgG was recognized by the mouse IgG 8A4 (see above), it was depleted from serum before the CH2D-containing serum was applied for capture ELISA. Serum was clarified by centrifugation at 20,000 g for 10 min and diluted in PBS at 1:1 ratio (named serum/PBS). The serum/PBS sample was incubated with protein G resin at 4°C for 1 h (CH2D does not bind to protein A or G). After incubation, the sample was spun at 5,000 g for 2 min, the supernatant was recovered and tested as described above.
| Abbreviations: | ||
| PK | = | pharmacokinetics |
| CH2D | = | human CH2 domain |
| mCH2D | = | wild-type monomeric CH2D |
| ssCH2D | = | short stable CH2D |
| dCH2D | = | CH2D dimer, hFcRn, human FcRn |
| mFcRn | = | murine FcRn |
| Fc | = | IgG Fc fragment, FcγR, Fc gamma receptor |
| TBRI | = | Texas Bioslogical Research Institute |
Acknowledgments
This project was supported by a CRADA between NCI and RCT, and the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
Disclosure of Potential Conflicts of Interest
K.R.G., D.B., D.A.W., and S.A.K are employed at Research Corporation Technologies, which provided financial support for this study.
Notes
† These authors contributed equally to this work
References
- Reichert JM. Marketed therapeutic antibodies compendium. MAbs 2012; 4:413 - 5; http://dx.doi.org/10.4161/mabs.19931; PMID: 22531442
- Labrijn AF, Poignard P, Raja A, Zwick MB, Delgado K, Franti M, et al. Access of antibody molecules to the conserved coreceptor binding site on glycoprotein gp120 is sterically restricted on primary human immunodeficiency virus type 1. J Virol 2003; 77:10557 - 65; http://dx.doi.org/10.1128/JVI.77.19.10557-10565.2003; PMID: 12970440
- Chen W, Zhu Z, Feng Y, Dimitrov DS. Human domain antibodies to conserved sterically restricted regions on gp120 as exceptionally potent cross-reactive HIV-1 neutralizers. Proc Natl Acad Sci U S A 2008; 105:17121 - 6; http://dx.doi.org/10.1073/pnas.0805297105; PMID: 18957538
- Binz HK, Amstutz P, Plückthun A. Engineering novel binding proteins from nonimmunoglobulin domains. Nat Biotechnol 2005; 23:1257 - 68; http://dx.doi.org/10.1038/nbt1127; PMID: 16211069
- Dimitrov DS. Engineered CH2 domains (nanoantibodies). MAbs 2009; 1:26 - 8; http://dx.doi.org/10.4161/mabs.1.1.7480; PMID: 20046570
- Pavlinkova G, Beresford GW, Booth BJ, Batra SK, Colcher D. Pharmacokinetics and biodistribution of engineered single-chain antibody constructs of MAb CC49 in colon carcinoma xenografts. J Nucl Med 1999; 40:1536 - 46; PMID: 10492377
- Emanuel SL, Engle LJ, Chao G, Zhu RR, Cao C, Lin Z, et al. A fibronectin scaffold approach to bispecific inhibitors of epidermal growth factor receptor and insulin-like growth factor-I receptor. MAbs 2011; 3:38 - 48; http://dx.doi.org/10.4161/mabs.3.1.14168; PMID: 21099371
- Ulrichts H, Silence K, Schoolmeester A, de Jaegere P, Rossenu S, Roodt J, et al. Antithrombotic drug candidate ALX-0081 shows superior preclinical efficacy and safety compared with currently marketed antiplatelet drugs. Blood 2011; 118:757 - 65; http://dx.doi.org/10.1182/blood-2010-11-317859; PMID: 21576702
- Holliger P, Hudson PJ. Engineered antibody fragments and the rise of single domains. Nat Biotechnol 2005; 23:1126 - 36; http://dx.doi.org/10.1038/nbt1142; PMID: 16151406
- Wörn A, Plückthun A. Stability engineering of antibody single-chain Fv fragments. J Mol Biol 2001; 305:989 - 1010; http://dx.doi.org/10.1006/jmbi.2000.4265; PMID: 11162109
- Wikman M, Steffen AC, Gunneriusson E, Tolmachev V, Adams GP, Carlsson J, et al. Selection and characterization of HER2/neu-binding affibody ligands. Protein Eng Des Sel 2004; 17:455 - 62; http://dx.doi.org/10.1093/protein/gzh053; PMID: 15208403
- Schmidt MM, Wittrup KD. A modeling analysis of the effects of molecular size and binding affinity on tumor targeting. Mol Cancer Ther 2009; 8:2861 - 71; http://dx.doi.org/10.1158/1535-7163.MCT-09-0195; PMID: 19825804
- Zahnd C, Kawe M, Stumpp MT, de Pasquale C, Tamaskovic R, Nagy-Davidescu G, et al. Efficient tumor targeting with high-affinity designed ankyrin repeat proteins: effects of affinity and molecular size. Cancer Res 2010; 70:1595 - 605; http://dx.doi.org/10.1158/0008-5472.CAN-09-2724; PMID: 20124480
- Chapman AP. PEGylated antibodies and antibody fragments for improved therapy: a review. Adv Drug Deliv Rev 2002; 54:531 - 45; http://dx.doi.org/10.1016/S0169-409X(02)00026-1; PMID: 12052713
- Graff CP, Wittrup KD. Theoretical analysis of antibody targeting of tumor spheroids: importance of dosage for penetration, and affinity for retention. Cancer Res 2003; 63:1288 - 96; PMID: 12649189
- Williams A, Baird LG. DX-88 and HAE: a developmental perspective. Transfus Apher Sci 2003; 29:255 - 8; http://dx.doi.org/10.1016/S1473-0502(03)00170-8; PMID: 14572818
- Fishburn CS. The pharmacology of PEGylation: balancing PD with PK to generate novel therapeutics. J Pharm Sci 2008; 97:4167 - 83; http://dx.doi.org/10.1002/jps.21278; PMID: 18200508
- Schellenberger V, Wang CW, Geething NC, Spink BJ, Campbell A, To W, et al. A recombinant polypeptide extends the in vivo half-life of peptides and proteins in a tunable manner. Nat Biotechnol 2009; 27:1186 - 90; http://dx.doi.org/10.1038/nbt.1588; PMID: 19915550
- Kim JK, Tsen MF, Ghetie V, Ward ES. Catabolism of the murine IgG1 molecule: evidence that both CH2-CH3 domain interfaces are required for persistence of IgG1 in the circulation of mice. Scand J Immunol 1994; 40:457 - 65; http://dx.doi.org/10.1111/j.1365-3083.1994.tb03488.x; PMID: 7939418
- Gong R, Wang Y, Feng Y, Zhao Q, Dimitrov DS. Shortened engineered human antibody CH2 domains: increased stability and binding to the human neonatal Fc receptor. J Biol Chem 2011; 286:27288 - 93; http://dx.doi.org/10.1074/jbc.M111.254219; PMID: 21669873
- Gong R, Vu BK, Feng Y, Prieto DA, Dyba MA, Walsh JD, et al. Engineered human antibody constant domains with increased stability. J Biol Chem 2009; 284:14203 - 10; http://dx.doi.org/10.1074/jbc.M900769200; PMID: 19307178
- Ober RJ, Radu CG, Ghetie V, Ward ES. Differences in promiscuity for antibody-FcRn interactions across species: implications for therapeutic antibodies. Int Immunol 2001; 13:1551 - 9; http://dx.doi.org/10.1093/intimm/13.12.1551; PMID: 11717196
- Oganesyan V, Damschroder MM, Woods RM, Cook KE, Wu H, Dall’acqua WF. Structural characterization of a human Fc fragment engineered for extended serum half-life. Mol Immunol 2009; 46:1750 - 5; http://dx.doi.org/10.1016/j.molimm.2009.01.026; PMID: 19250681
- Roopenian DC, Akilesh S. FcRn: the neonatal Fc receptor comes of age. Nat Rev Immunol 2007; 7:715 - 25; http://dx.doi.org/10.1038/nri2155; PMID: 17703228
- Boswell CA, Tesar DB, Mukhyala K, Theil FP, Fielder PJ, Khawli LA. Effects of charge on antibody tissue distribution and pharmacokinetics. Bioconjug Chem 2010; 21:2153 - 63; http://dx.doi.org/10.1021/bc100261d; PMID: 21053952
- Suzuki T, Ishii-Watabe A, Tada M, Kobayashi T, Kanayasu-Toyoda T, Kawanishi T, et al. Importance of neonatal FcR in regulating the serum half-life of therapeutic proteins containing the Fc domain of human IgG1: a comparative study of the affinity of monoclonal antibodies and Fc-fusion proteins to human neonatal FcR. J Immunol 2010; 184:1968 - 76; http://dx.doi.org/10.4049/jimmunol.0903296; PMID: 20083659
- Wang W, Wang EQ, Balthasar JP. Monoclonal antibody pharmacokinetics and pharmacodynamics. Clin Pharmacol Ther 2008; 84:548 - 58; http://dx.doi.org/10.1038/clpt.2008.170; PMID: 18784655
- Andersen JT, Daba MB, Berntzen G, Michaelsen TE, Sandlie I. Cross-species binding analyses of mouse and human neonatal Fc receptor show dramatic differences in immunoglobulin G and albumin binding. J Biol Chem 2010; 285:4826 - 36; http://dx.doi.org/10.1074/jbc.M109.081828; PMID: 20018855
- Kim JK, Tsen MF, Ghetie V, Ward ES. Identifying amino acid residues that influence plasma clearance of murine IgG1 fragments by site-directed mutagenesis. Eur J Immunol 1994; 24:542 - 8; http://dx.doi.org/10.1002/eji.1830240308; PMID: 8125126
- Roopenian DC, Christianson GJ, Sproule TJ. Human FcRn transgenic mice for pharmacokinetic evaluation of therapeutic antibodies. Methods Mol Biol 2010; 602:93 - 104; http://dx.doi.org/10.1007/978-1-60761-058-8_6; PMID: 20012394
- Roopenian DC, Christianson GJ, Sproule TJ, Brown AC, Akilesh S, Jung N, et al. The MHC class I-like IgG receptor controls perinatal IgG transport, IgG homeostasis, and fate of IgG-Fc-coupled drugs. J Immunol 2003; 170:3528 - 33; PMID: 12646614
- Petkova SB, Akilesh S, Sproule TJ, Christianson GJ, Al Khabbaz H, Brown AC, et al. Enhanced half-life of genetically engineered human IgG1 antibodies in a humanized FcRn mouse model: potential application in humorally mediated autoimmune disease. Int Immunol 2006; 18:1759 - 69; http://dx.doi.org/10.1093/intimm/dxl110; PMID: 17077181