Abstract
During the past ten years, monoclonal antibodies (mAbs) have taken center stage in the field of targeted therapy and diagnosis. This increased interest in mAbs is due to their binding accuracy (affinity and specificity) together with the original molecular and structural rules that govern interactions with their cognate antigen. In addition, the effector properties of antibodies constitute a second major advantage associated with their clinical use. The development of molecular and structural engineering and more recently of in vitro evolution of antibodies has opened up new perspectives in the de novo design of antibodies more adapted to clinical and diagnostic use. Thus, efforts are regularly made by researchers to improve or modulate antibody recognition properties, to adapt their pharmacokinetics, engineer their stability, and control their immunogenicity. This review presents the latest molecular engineering results on mAbs with therapeutic and diagnostic applications.
Introduction
In vitro molecular engineering aims at modifying the biochemical and biophysical characteristics or the functional properties of peptides and proteins to render them more suitable for use in research, clinical science or industry. These modifications are often subtle and target a small subset of the amino acids that form the protein of interest. There are two strategies of molecular engineering: targeted vs. random. The first (structure-guided) is based on structural knowledge derived from X-ray crystallography, NMR, and in silico molecular modeling or docking of the molecule alone or in interaction with its partner. Such knowledge is of great help in identifying the amino acid residues that are appropriate to modify and in predicting the nature of the substitutions to make. Different strategies of mutagenesis are possible. Site-directed mutagenesis enables accurate amino acid substitutions at particular positions. Alternatively, or in addition to site-directed mutagenesis, there is semi-rational engineering, which involves multiple amino acid substitutions at contiguous or non-contiguous positions are designed, yielding libraries of mutants that are recombinantly expressed and then screened to identify the best variants. Based on the degeneracy of the genetic code, a large part of the natural repertoire of amino acids can be explored.Citation1,Citation2
In the absence of structural information or as a complement to it, random mutagenesis can be performed. In this case, the DNA encoding the whole protein, a structural domain or a restricted region thereof, is amplified by error-prone PCR (ep-PCR).Citation3,Citation4 The randomly mutated DNA is then sub-cloned into an appropriate recombinant expression vector before selection or screening. It is noteworthy that mutagenesis by ep-PCR does not allow exploration of the whole repertoire of natural amino acids. Indeed, ep-PCR induces individual base substitutions that, owing to the start codon and the position of the substitution within the codon, will only result in a limited number of amino acid mutations.Citation5 Despite this limitation, random mutagenesis is very useful in identifying relevant amino acid positions associated with function, activity or biochemistry of the protein of interest. Such key positions can then be explored more exhaustively by site-directed mutagenesis. Also, ep-PCR identifies “long-distance” key residues that contribute indirectly to the catalytic activity of enzymes or to the recognition properties of antibodies (Abs).Citation6,Citation7
Strategies vary according to the number of variants to be expressed and then selected in vivo or screened in vitro. All are designed to determine the amino acid sequence of the selected variants and thus to establish the precise nature of the substitutions responsible for their selection. In the case of limited molecular diversity (< 102-103), each mutant can be individually expressed, tested and characterized. However in many instances, larger libraries of mutants are designed (103-109 variants). They need large-scale expression and screening strategies mostly via display technologies that ensure a physical link between the recombinant mutated protein and its coding information (DNA or RNA). 8–9.
Molecular engineering is commonly applied to monoclonal antibodies (mAbs). Different types of mAbs of various origins or structures can be generated, including murine, chimeric, humanized or human mAbs. Recently mAbs from immunized non-human primates were also described.Citation10,Citation11 Despite ethic constrains regarding the use of chimpanzees in routine toxicology studies, this alternative strategy appears promising especially in obtaining high affinity neutralizing mAbs against microbial toxins. For in vitro diagnosis and in vivo imaging, murine mAbs produced by the fusion cell strategy are classically usedCitation12,Citation13 On the other hand, for therapeutic purposes, a larger spectrum of Abs is used. Of the ≈30 mAbs approved to date, as well as the hundreds under clinical investigation, most are humanized, followed by human and chimeric mAbs.Citation14 Although the first therapeutic mAb approved (1986) was murine (Orthoclone OKT3®/muromomab anti-CD3), few such mAbs are now used or developed.
Whatever their use, mAbs often need to be engineered at the molecular level to modulate, decrease or increase aspects of their properties. These modifications are designed to circumvent some limitations of non-engineered (natural) mAbs ().
Table 1. mAb properties and functions that can be modified by molecular engineering. “Diagnostic purposes” includes both in vitro diagnosis (EIA) and in vivo imaging
Engineering may target variable domains (VH or VL) or modify the constant domains, including the crystallizable fragment (Fc). Engineering of the variable domains is mainly used to modulate the antigen-binding properties of the antibody (Ab): affinity and specificity, and in some instances stabilization of the Ab in different denaturation or proteolytic conditions. Modulation of Ab binding properties has in vitro applications in developing more sensitive and specific EIAs. It also allows the design of more potent and specific therapeutic tools, and the production of labeled Abs for in vivo imaging. The most frequent aim of molecular engineering of Abs is to reduce the immunogenicity of therapeutic Abs of murine origin to reduce or avoid the human anti-mouse antibody (HAMA) response when injected into human subjects.Citation15 Different strategies have been developed to attenuate the human immune response, resulting in mAbs with reduced levels of murine sequences. One consists in the fusion of the murine variable domains to human constant regions resulting in chimeric Abs. About 15% of FDA-approved and Phase 3 therapeutic mAbs are chimeric.Citation14 Grafting of mouse complementarity-determining residues (CDRs) onto human acceptor Ab frameworks yields to the so-called “humanized” mAbs, which to date constitute the largest group of therapeutic mAbs used and developed (about 45% of FDA-approved and Phase 3 mAbs). However, humanization is more difficult to manage since there are often subtle structural relationships between CDRs and framework regions (FRs) that may result in humanized Abs with modified binding properties.Citation16 To overcome this problem in vitro molecular engineering is often used.Citation17 Finally, fully human Abs have recently been obtained. They are obtained from natural or synthetic recombinant repertoiresCitation18 or directly from transgenic mice whose murine immune repertoire has been replaced by the human one.Citation19 This new generation of Abs currently accounts for about 40% of FDA-approved and Phase 3 therapeutic mAbs.
Here we review the latest original results on in vitro molecular engineering of Abs for diagnostic or therapeutic purposes.
Immunogenicity Engineering: Humanization, De-immunization
Most therapeutic proteins that are clinically approved or undergoing clinical trials are, to a variable extent, immunogenic. Immunogenicity of therapeutic proteins in general and of Abs in particular has various causes.Citation20-Citation24 Extrinsic and intrinsic factors may cause or influence the immunogenicity of Abs. The presence of aggregates, adjuvant-like contaminants, co-medication of the patient, immunological status of the patient, but also in some instances cytokine release are known as extrinsic factors. Major intrinsic factors that affect or induce an immune response against therapeutic Abs include the presence via glycosylation sites of particular carbohydrate side chains both in variable and constant regions. Immunogenicity is also affected by other post-translational modifications of Abs such as glycation, deamidation and oxidation of amino acid side chains. Finally, the presence of CD4+ T helper cell epitopes within an injected molecule is one of the factors correlated with the raising of immune responses to therapeutic proteins.Citation25 T cell epitopes are linear sequential peptide fragments derived from the injected protein that are processed by antigen-presenting cells (APCs). These T cell epitopes are presented in the context of the donor’s HLA class II molecules and are recognized by their corresponding T cell receptor present at the surface of CD4+ T helper cells.
Thus, one major set of engineering actions performed on Ab molecules aims to limit their immunogenicity by reduction of their non-human sequence content (chimerization and humanization) or by de-immunization, which consists in identifying and then removing T cell epitopes.
Humanization
Historically, chimerizationCitation26 then humanizationCitation27 were the first two molecular engineering processes described and applied to mAbs intended for therapeutic purposes. Although they were developed in the 1980s, these techniques are still usedCitation14 because they reduce the potential for a HAMA response by the host organism toward non-human injected Abs, which greatly reduces therapeutic efficacy.Citation28 Despite the reduction in immunogenicity compared with their murine equivalents, chimeric and to a lesser extent humanized mAbs are still associated with a risk of eliciting an immune response.Citation29 To address this problem, in addition to murine complementarity-determining residues (CDR) grafting onto acceptor human frameworks, a variety of other humanization methods () have been reported that ensure more accurate grafting of the residues associated with the binding properties and also respect of the structural and functional relationships that exist between CDR and FR residues.Citation16
Table 2. Strategies for humanization of therapeutic antibodies
Recently, Bernett et al. developed a novel and sophisticated method of humanization that results in fully human mAbs from parent murine sequences.Citation42 Three mAbs targeting three different human antigens (CD25, VEGF and TNFα) were used to validate their molecular engineering, resulting in the introduction of 59, 46, and 45 substitutions in the parent murine sequences, respectively. The crucial and central point of their strategy is the rational molecular engineering of residues within and proximal to CDRs, together with the optimization of the variable domain interface. This was achieved by successive and iterative explorations of the human germline repertoire using semi-automated computational methods, to progressively select functional humanized mAbs with the highest level of humanness. The resulting fully human Abs retain the potency of the corresponding chimeric mAbs and have in vitro activity comparable to that of their respective marketed drugs, i.e., daclizumab, bevacizumab, and infliximab.
As mentioned previously, humanization of mAbs has been widely developed and several companies offer this service. However, there are different strategies possible. One of the difficulties stems from the choice of the method, which should be the most adapted and efficient for obtaining a functional humanized form of the Ab of interest. It is also noteworthy that, in many cases some “hidden” structural or functional key residues within the variable domains may be omitted during the humanization process. In this case an additional step of random mutagenesis of the variable domains coupled to appropriate screening may be necessary to recover a fully active humanized mAb.
De-immunization
There are many methods that allow identification of CD4+ T cell epitopes present within the amino acid sequences of proteins. Over the last decade, several algorithms have been constructed that map and identify HLA class II binding motifs within the molecule of interest. These in silico predictive methods tend to over-predict the number of epitopes in a given sequence, and subsequent in vitro confirmation of their activity is needed.Citation43 Particular attention has been paid to the study of the immunogenic potential of therapeutic proteins in general and Abs in particular. Thus, and as already noted, engineered forms of therapeutic Abs are designed to minimize their immunogenicity, using methods including humanization and selection of fully human Abs. It is noteworthy, however, that owing to their intrinsic sequence variability within the variable domains, even fully human Ab therapeutics can be immunogenic and thus can induce a marked immune response.Citation44-Citation46
A very interesting study of the immunogenicity of humanized Abs was recently published by Harding FA et al.Citation24 These authors analyzed the CD4+ helper T cell epitope contents of a set of 8 humanized Abs representing different VH and VL regions from different genomic segments and affinity maturation processes. They clearly establish that prominent CD4+ T cell epitopes are found only in CDR sequence−containing regions and never in FRs. This limited distribution strongly suggests that the former could be removed by appropriate engineering, hopefully without major if any effect upon the binding properties of the Ab of interest. Thus, they reduced the immunogenic potential while maintaining native-like binding properties by incorporating up to two amino acid substitutions in a single epitope. Together, these observations and results indicate that in humanized Abs but also in fully human Abs, i) CDRs are the only segments likely to contain CD4+ T cell epitopes, ii) most of the CDRs are not immunogenic, and iii) immunogenic CDRs can be de-immunized by site-directed engineering while retaining full biological function.
Clearly, there are various strategies to select and design appropriate forms of potent and low immunogenic therapeutic Abs, some of which involve a step of targeted molecular engineering of the molecule of interest. Thus, different mAbs specifically termed ‘deimmunized’ mAbs have been evaluated in clinical studies. ETI-204 is a therapeutic humanized and de-immunized monoclonal antibody specific for the protective antigen (PA) from Bacillus anthracis (Anthin®/Elusys Therapeutics).Citation47 Therapeutic assays have been performed with MLN2704 (Millennium Pharmaceuticals Inc.), a de-immunized mAb conjugated to drug maytansinoid 1 (DM1) specific for the prostate-specific membrane antigen (PSMA).Citation48 Also, PanGenetics B.V. as recently initiated two clinical trials. The first one consists in a double blind, placebo controlled study using a humanized anti-NGF antibody (PG110) in patients with chronic pain. The second study concerns their anti-CD40 antibody (PG102) in a phase 1 study in psoriatic arthritis patients
Variable Domain Engineering
Variable domains in Abs contain the totality of the antigen-binding site whose molecular determinants are mainly located within the CDRs (3 CDRs in the VH and 3 in the VL). Each CDR is surrounded by two FRs, called FR1, FR2, FR3 and FR4. Although in some instances residues belonging to FRs participate directly in antigen recognition and interaction, the main role of FRs is structural—to ensure the correctness of the three-dimensional functional topography of CDRs. FRs are responsible for the stability of the Fv region of Abs through the VH/VL interface contacts that they establish. Consequently, molecular engineering of the variable domains is likely to modulate the binding parameters (affinity or specificity), the overall stability of the Fv region, and also the pharmacokinetics of IgGs. Classically, libraries of variants are generated by targeted or random mutagenesis, VH/VL or CDR shuffling, prior expression and in vitro screening using display technologies.Citation49-Citation51 Finally, it is important to keep in mind that the molecular engineering processes described below, are susceptible to induce immunogenicity modulation of the resulting antibodies. In consequence and accordingly to the strategies described above, CD4+ T cell epitopes identification will be performed on the de novo engineered antibodies.
Molecular Engineering of Antigen-binding Properties
If in antibodies the residues involved in antigen recognition are mainly found within the CDRs, blanket targeting of these areas would require the construction of huge and multiple libraries. However, the screened sequence space can be reduced using several alternative strategies. X-ray crystallographic structure determination of the antibody-antigen complex, provides an accurate knowledge of the paratope-epitope interface opening up two engineering approaches: site-specific random mutagenesisCitation49 or structure-based computational design.Citation52 Structure-based design techniques sample in silico a large number of molecular designs whose properties would be tested. With these structure-guided approaches, site-specific or random mutagenesis is used to experimentally validate the predicted variants. Alternatively, in the absence of a crystallographic structure, given the large number of antibody coordinates currently available, a three-dimensional model of the paratope can be constructed relatively easily.Citation53 While such model can provide a good guide for mutagenesis, without detailed knowledge of the paratope-epitope interface, the mutagenesis approach will result in exploring a larger number of positions.
Affinity Maturation
In numerous cases natural (e.g., murine, human) mAbs do not display the binding properties most appropriate to their use in diagnosis or therapy. Thus, many mAbs suffer from a lack of affinity for their antigen (low picomolar affinity), a characteristic that may greatly hamper their development in therapy or their use in immunoassays. This appears to be a consequence of the affinity ceiling that characterizes the mammalian immune system and B cell responses.Citation54,Citation55 Also, the chemical and structural nature of the targeted antigens such as haptens, peptides, or proteins with complex three-dimensional structure (receptor), may affect the affinity improvement. Finally, compounds that display an intrinsic structural flexibility result in a heterogeneous mixture of immunogens unfavorable to the raising of high-affinity Abs. Increasing the affinity of an Ab can lead to: 1) a reduction in the amount needed for treatment; 2) an increase in therapeutic efficacy (diminution of the minimum effective plasma concentration, reduction of the dose or dosing intervals during the treatment, greater pharmacological activity in the case of antagonist Abs); 3) stronger indirect Fc-mediated effector-response; or 4), in the field of in vitro diagnosis, development of highly sensitive assays. Different approaches, tools and strategies are available and have been validated through engineering of Abs directed against various antigens.
Considerable effort has been made to produce Abs specific for small molecules to enable development of sensitive and high-throughput immunosensing.Citation2 Despite the difficulties associated with the assay of small molecules, successful affinity maturation of several anti-hapten Abs has been achieved.Citation56 The greatest affinity improvement described to date was by Boder and colleagues.Citation7 These authors selected by yeast display, after four rounds of affinity mutagenesis and screening, an anti-fluorescein variant with a 10,000-fold affinity increase compared with the starting Ab. Ten consensus mutations were retained, among which 6 were in CDRs and 4 in FRs. Interestingly, only one of them was in direct contact with fluorescein, indicating that long-distance effects can affect affinity. We noted a similar effect in the case of the engineering of the specificity of an anti-steroid Ab.Citation57 This result emphasizes the potential difficulty of predicting the key residues associated with the binding properties of an Ab, an identification that only random mutagenesis can allow. Again using yeast display, the same group engineered both the affinity and the thermal stability of an Ab fragment specific for the carcinoembryonic antigen.Citation31 In both examples the affinity increase was the result of the improvement of the off-rate (dissociation half-time), which lasts several days (4–7 d) at 37°C vs. a few minutes for the parent Abs, a parameter that may increase the retention time of the Ab in the tumor.Citation58,Citation59
Using ribosome display, Andreas Plückthun and colleaguesCitation60 have improved 500 times the affinity of a peptide-binding scFv via error-prone PCR, DNA shuffling and an off-rate selection procedure. Very few mutations were necessary, and mainly affected pre-existing interactions via subtle changes in the FRs rather than through creation of de novo contacts in CDRs. Also, DNA shuffling was performed during the evolution process, allowing identification of beneficial combinations. This result confirms the additivity and independence of particular point mutations, which is a way to improve affinity further by looking for positive synergistic effects.Citation61-Citation64 Here again only one mutation of the four finally selected made direct contact with the targeted peptide; the others were close together at the VH/VL interface, a location that indirectly modulates affinity, probably by affecting domain orientation and spacing as well as CDR loop flexibility.
An alternative to increasing Ab affinity is mimicking of the in vivo somatic hypermutation of Ab genes by targeting and randomizing only certain CDR codons.Citation65 These codons are tagged with short base sequence motifs, referred to as natural DNA mutational hot spots, which are the preferred sites naturally mutated at high frequency during in vivo somatic hypermutation events.Citation66 This targeted hot-spot strategy has been successfully used to increase the affinity of three distinct conventional Abs engineered as immunotoxins for cancer therapy purposesCitation61,Citation62,Citation67 and one single-domain lama-derived VHH Ab.Citation63
Recently, we affinity engineered a murine mAb specific for human prostate-specific antigen (PSA).Citation68,Citation69 In this work, we developed in parallel two distinct strategies based on hot-spot mutations and structure-guided molecular engineering. Interestingly, we selected in both cases differently mutated scFvs with improved affinity for human PSA that we were unable to combine. This indicates that, despite the fact that they concern different CDRs, mutations from the two strategies provide alternative but non-complementary solutions for affinity enhancement of the parent Ab. Structural analysis of the complex formed between the Ab and PSA suggests that this incompatibility might reflect the difficulty of preserving the sophisticated structural relationships that exist between the different CDRs in the Ab, and its capacity to recognize a structurally flexible antigen. The gain of sensitivity associated with the affinity increase allowed the clinical assay (63 patients) of a particular molecular form of PSA present in serum that allowed successful discrimination between prostate cancer and benign prostatic hyperplasia.
Alternatively, Ab affinity can also be greatly improved by using combinatorial libraries. Rajpal et al.Citation70 developed and applied a new and powerful method called look-through mutagenesis to optimize the affinity of an anti-TNFα Ab. This multidimensional exploring mutagenesis method simultaneously assesses and optimizes combinatorial mutations of selected amino acids. Thus, a subset of nine different amino acids representative of the major chemical functionalities (small, nucleophilic, hydrophobic, aromatic, acidic, amine and basic) was systematically introduced at all the CDR positions, which is a total of 57 positions along the six CDRs. This gave 41 different libraries: 6 single CDR mutated libraries (only one CDR of the 6 is mutated each time), 15 double (two CDRs simultaneously mutated) and 20 triple libraries (three CDRs simultaneously mutated). Following a first step of expression and screening (positive binding selection) by yeast display, 38 mutations spread over 21 CDR positions were selected since they resulted in higher affinity for TNFα. These beneficial mutations located in VH or VL domains were then combined to design a new combinatorial library, which after a second round of expression led to the selection of variants with a 500- to 870-fold affinity increase.
Simultaneous mutagenesis of the 6 hypervariable loops of a non-human primate Fab that neutralizes anthrax toxin was also successfully performed using phage-display technology.Citation71 A 5x108 variant library was built, with each variant containing an average of four mutations, and screened in vitro by combining the progressive lowering of the biotinylated antigen with an increase in elution times (off-rate selection criteria). The best variant thus selected displays an affinity enhanced 19-fold (180 pM) and an IC50 value decreased by 40%.
Finally, affinity maturation of Abs can also be achieved by structure-based computational design.Citation52,Citation72,Citation73 The procedure described was applied to several Abs recognizing antigens of different natures: hapten, globular and soluble macro-protein, receptor, and receptor ligand. Thus, the binding affinity of an antibody to the I-domain of the integrin VLA1 was increased by an order of magnitude.Citation52 More than 80 variants were designed targeting mainly antigen-contacting residues using suggestions from computational methods. Single and combined mutants were expressed and tested. The highest-affinity variant was a combination of four mutations: two in the light chain and two in the heavy chain. Alternatively, Lippow and colleagues have developed a method based on an iterative computational design that focuses on electrostatic binding contributions and the design of single mutants.Citation72,Citation73 By combining multiple designed mutations, affinity of antibodies specific for various antigens was improved: 10-fold for the anti-epidermal growth factor receptor, cetuximab (Erbitux®), and 140-fold in the case of an anti-lysozyme Ab.
Based on these successes, it is likely that computational design methods will occupy in the next future an increasing importance for the optimization of antibody binding affinity properties. One of the advantages of computational design approaches is the possibility to design a small set of mutations through exploration of a larger space than is possible experimentally. The following experimental phases: mutagenesis, expression and functional assays, being made faster and thus less expensively.
Specificity Modulation
Alteration of the specificity of recognition of Abs constitutes another important issue both in diagnosis and therapy. It can be achieved by random or targeted mutagenesis strategies using expression (via display technologies) similar to that in the case of affinity maturation. The main differences concern the choice and design of an appropriate screening strategy to eliminate the variants that still have undesirable cross-reactivity and to keep those that display the desired specificity of recognition. Screening strategies based on competition are often used. Reduction of cross-reactivity with different antigens, extension of the specificity of recognition to related allo-antigens, and enlargement of the cross-reactivity toward xeno-antigens have been achieved.
It is noteworthy that most specificity improvements concern Abs directed against small molecules, especially haptens. This probably reflects the challenge of generating high-quality anti-hapten Abs useful for the development of sensitive and specific immunoassays. Among haptens, steroid hormones are involved in many normal cellular processes (e.g., reproduction, metabolism), but they are also markers of diseases whose clinical symptoms are reflected by steroid level variations. Thus, accurate determination of their concentration in blood or urine is of great importance for clinical diagnosis and for monitoring the efficacy of a therapeutic process. Several metabolites, chemically and structurally related to the targeted steroid, may co-exist in the analyzed fluid, emphasizing the importance of designing highly specific Abs.
Owing to the peculiar mode of recognition of small molecules by Abs—classically haptens interact with Abs in cavities rich in aromatic and hydrophobic residues formed at the interface of the variable domains—it can be difficult to engineer only the specificity of recognition without modulation of the affinity. Thus, by combining phage display technology with molecular modeling and site-specific randomization of an anti-cortisol Ab, Chames et al. increased affinity and specificity 8- and 5-fold, respectively.Citation74 A second example of double engineering concerned an anti-17β-estradiol mAb.Citation75 Based on the potential additivity of mutations, Lamminmaki and colleagues systematically explored the binding properties of all the possible combinations between three light-chain point mutations and a four-amino acid heavy-chain insertion previously selected by phage display. They selected one triple combined mutant that displayed a 17-fold affinity increase together with 400-fold reduced cross-reactivity with testosterone. Interestingly, some of these beneficial mutations do not directly contact the antigen, confirming that long-distance structural or functional modulators exist. Also, some previously identified beneficial point mutations appear to be neutral or deleterious when combined, indicating that variations in binding properties are likely to occur depending on their structural environment.
We have improved the specificity of recognition of an anti-progesterone mAb by combining site-directed saturation mutagenesis and error-prone PCR.Citation57 The starting anti-progesterone displayed a subnanomolar affinity (KD = 20 pM) for progesterone, together with strong cross-reactivity for two hepatic and structurally different metabolites 5α and 5β-progesterone. Based on an accurate molecular model, we identified five first sphere amino acids that were mutated by saturation and combined, forming a 3.2 106 mutant library expressed and screened by phage display. Following a competitive screening procedure, we selected only triple-mutated, more specific variants with two of the five pre-selected positions being systematically excluded from that selection. With the objective of further improving the specificity of recognition of our Ab, we randomized the VH domain, keeping the light chain unchanged. Three point mutations found transversally in different selected variants were thus identified and combined with the previously selected best triple-mutants. Finally, a penta-mutant was selected that combined a sub-nanomolar affinity for progesterone (KD = 75 pM) with a double improvement of specificity: 54- and 85-fold decrease in affinity for 5α and 5β-progesterone, respectively. This constitutes the first description of such discriminating engineering of an Ab vs. three structurally different ligands that establish contacts via two alternative binding sites.
For diagnosis or neutralization of compounds it can be useful to broaden the specificity of recognition of an Ab. Random mutagenesis DNA-shuffling and phage display were therefore used to produce Abs able to recognize sulphonamides, so-called sulpha antibiotics, which are widely used in veterinary medicine.Citation76 The cross-reaction profile of the best mutant revealed a capacity to recognize 10 different sulphonamides within a narrow concentration range, which is useful for identification of animals contaminated with sulpha residues. Interestingly, the authors of this study predicted, using molecular modeling, that most of the amino acid changes are located on the surface of the Ab, and not in proximity to the binding cavity. Another example of enlargement of cross-reactivity was described in the case of the neutralization of two subtypes of type A botulinum neurotoxin.Citation77 Starting from an Ab fragment that binds BoNT/A1 subtype with high affinity (KD = 136 pM) and the A2 subtype with a lower affinity (KD = 109 nM), A2 affinity was increased 1250-fold (KD = 87 pM) while maintaining a subnanomolar affinity for the A1 subtype (KD = 115 pM). To reach that goal, different and successive yeast display libraries were built, expressed and co-screened for both neurotoxin A1 and A2 subtypes. The targeted amino acid residues selected for mutagenesis were all solvent-accessible and located in CDR1H, 2H, 3H or in CDR1L. Finally, the best variant contains six mutations all located in the VH domain, with three mutations in solvent-accessible residues of CDR1H. Surprisingly, the three other important mutations were located in FRs; these mutations were not anticipated because they were fortuitously introduced during the PCR amplification of the targeted mutated CDRs. Finally, a 14-fold improvement of the affinity of for the mouse ortholog (epithin) of the cancer-associated human serine protease MT-SP1 was achieved.Citation78 To this goal, the authors used a computational design strategy that resulted in several in silico predictions. Six positions were chosen owing to their proximity to any atom of the three residues that differ between MT-SP1 and epithin. Following optimization of side chain rotamers, energy minimization, and free energy calculations, eight amino-acid substitutions were tested experimentally. Substitution Thr98Arg in CDR3H displayed the greatest effect on protease inhibition. Based on the MT-SP1/Ab complex crystal structure, it was shown that the arginine side chain make two additional hydrogen bounds: one intramolecular within CDR3H (with the backbone carbonyl oxygen of ValH100h), and one intermolecular contact with the carboxylate group of Glu217 on the protease.
Fragment Variable Overall Stability
Natural Abs are highly soluble and stable molecules, characteristics that are well-suited to their development and use as biopharmaceuticals. However, the design of new Ab formats as well as the results of the different engineering processes used to confer on Abs novel activities or to enhance their clinical utility can affect their biophysical properties, including stability and solubility.Citation79,Citation80 Thus, several Ab fragments e.g., single-chain variable fragments (scFvs), diabody, Ab domains, and bi-specific Ab-fragments are commonly designed, engineered, expressed and used for the discovery of new therapeutics and for optimization. However, the absence of the constant domains in such Ab fragments and also the engineering of the variable regions (e.g., humanization, modulation of binding properties), may have detrimental effects on the biophysical properties of the resulting molecules.Citation81 Thus, variable regions appear involved in the thermal and chemical stability and also the solubility of the Ab moiety. For instance, weak thermal stability can induce aggregation, result in a low expression yield, affect biological activity, or increase the immunogenicity of the molecule of interest. Improvement of the melting temperature of Abs can be achieved by optimizing, via rational engineering or a random approach, different Ab structural characteristics, e.g., hydrophobic core or surface residues, charged cluster amino acids, residues that determine framework subclasses or VH/VL interface residues.
Stability Engineering
Andreas Plückthun’s group pioneered the development of the molecular engineering of immunoglobulin domains or fragments to improve their biophysical properties, especially by exploring and analyzing the complex relationship that may exist between CDRs and FRs.Citation82,Citation83 By exchanging particular amino acid residues (6 in total alone or in combination) between the VH domains of human germline families 2, 4 and 6, they improved the stability by 20.9 kJ/mol and the expression yield 4-fold of an anti-peptide scFv.Citation84,Citation85 As an alternative to rational structure-based strategies, it is possible to stabilize Abs via the design of libraries of mutants that will be screened owing to the correlation that exists between stability and biological activity.Citation86 Expression of such libraries can be done by phage or ribosome display, and the selection of more stable molecules is achieved by using stringent conditions, including high temperature, presence of denaturing agents such as guanidine hydrochloride, addition of proteases or the use of reducing conditions such as dithiothreitol.Citation87,Citation88
Improvement of the stability of a bispecific Ab for TNF family member receptors, TRAIL-R2 (TNF-related apoptosis-inducing ligand receptor-2) and LTβR (lymphotoxin-β receptor), was recently reported.Citation89 Focused libraries of mutated scFvs designed by combining sequence- and structure-based knowledge, were expressed in E. coli and screened via a thermal challenge prior to antigen-binding assay. According to criteria such as residue frequency, covariation, and computational prediction, small focused libraries were designed targeting 4 and 6 VL and VH positions, respectively. Fifteen single more stable variants with a superior T50 (°C) ranging from 53 to 67°C, compared with that of the corresponding WT scFv (51°C) were selected. Interestingly, combination of some mutations revealed further thermal stabilization with the best combined tetra-variant displaying a T50 of 82°C.
Solubility Engineering
Among biophysical parameters that impair the diagnostic and therapeutic applications of mAbs is the propensity to aggregate, which induces both a reduction of the active fraction of Abs and a potential increase of their immunogenicity. A recent study compared and mutated the sequences of two related Ab domains—one aggregation-prone and its aggregation-resistant counterpart—to identify “aggregation hotspots.”Citation90 By swapping and mutagenesis analysis of CDR loops, these authors were able to convert an aggregation-prone human VH Ab into an aggregation-resistant one. Only CDR1 in the VH segment appeared crucial since it contained the only hotspot identified as regulating the reversible folding of VH variants via the presence or not of charged residues: a triad of charged mutations was thus identified. This result clearly emphasizes the importance of developing Ab selection methods based on both affinity and solubility criteria, a strategy that will allow a posteriori identification of new aggregation-resistant Abs and also improve our understanding of the role played by charged residues within CDRs on binding to antigens with high affinity. Finally, it was demonstrated that aggregation resistance was decoupled from the conformational stability of the Abs studied. This addresses the question of the sophisticated equilibrium that exists between the high affinity, conformational stability and solubility of Abs, and thus of the prediction and engineering strategies used to design Abs in a more rational and controlled manner.Citation91
Fragment Variable Antibody pH Engineering
It was recently demonstrated that engineering of the Ab variable region can improve the half-life of an Ab by lowering its isoelectric point (pI).Citation92,Citation93 Classically, reduction of the elimination of IgG Abs is mediated by engineering of the Fc to increase their binding affinity to FcRn. A similar result, however, can be obtained by non-FcRn dependent engineering of the variable domains. Igawa et al. have studied the pharmacokinetic properties of an anti-human IL6 receptor IgG4 humanized Ab.Citation92 The pI of this Ab was modified by site-directed mutagenesis of specific residues at the surface of the variable heavy chain domain while maintaining unchanged both the variable light chain and the constant region. Thus, they designed four variants with pIs ranging from 7.2 to 9.2 and established that the pharmacokinetic parameters (half-life and clearance) correlate with the pI, suggesting that lowering the pI will reduce the elimination rates of Abs. In a second phase of this work, the authors extended and applied their results to a humanized anti-IL6 receptor IgG1 Ab that had an initial pI of 9.3. Based on a molecular model of this Ab, they selected multiple substitutions within the surface of its variable domains in both CDRs and FRs. Two multiple variants with 23 (pI 6.9) and 35 mutations (pI 5.5) were selected, a large majority of the substitutions being located within the frameworks. The two mutated Abs had unchanged affinities for human soluble IL-6 receptor and an overall progressive reduction in clearance of 2.0- and 3.1-fold in cynomolgus monkey for the Abs with a pI of 6.9 and 5.5, respectively.
In a second articleCitation93 the same authors reported the engineering of tocilizumab (Actemra®), an Ab that targets the IL-6 receptor (Il-6R) and is approved for the treatment of moderate to severe rheumatoid arthritis. They aimed to hasten the dissociation of the Ab within the acidic compartment of the endosome (pH 6.0) while keeping unchanged its capacity to bind to IL-6R in plasma at pH 7.4. To achieve this they used histidine scanning of all the CDR residues as well as some of the FRs important for antigen binding. They identified several individual mutations that confer pH-dependent binding on IL-6R. The combination of some of these single mutations showed additive effects, leading to two multiple variants containing 6 and 5 substitutions, respectively. Interestingly, these two mutants bind IL-6R 2.3 and 2.8 times better at pH 7.4 concomitantly with an affinity decrease of 3.8 and 3.7 at pH 6.0. Also, they showed that these modifications are associated with a reduction of Ab clearance owing to back recycling from the acidic endosome to the plasma where the Ab can bind another IL-6R molecule. These results establish that it is possible to modulate in a controlled manner the lifetime in the plasma of Abs by engineering their variable domains via modulation of their pH-dependent antigen-binding properties. From a clinical point of view, this opens up the possibility of delivering therapeutic Abs both less frequently and at lower doses.
Constant Domain Engineering
Within the constant domains of Abs, CH1 and CL are parts of the Fab or Fab’ fragments that are distinguished from the Fc region, which is formed by CH2 and CH3 in IgG. Published data on the engineering of Fab constant domains is scarce, despite the fact that antigen binding is controlled by the constant domains.Citation94 Furthermore, it is suggested that CH1 and CL domains might be involved in the adjustment of the Ab binding site by ensuring the stabilization of the variable domains.Citation95
Fab Constant Domain Engineering
The first engineering work targeting the CH1 domain was performed to create new glycosylation sites compatible with CHO expression cells.Citation96 Different humanized Ab mutants were designed by introducing an Asn-linked glycosylation acceptor sequence into the CH1 and Cκ domains of an anti-CD22 humanized Ab.Citation97 From 10 potential glycosylation mutants, two different CH1 mutants were finally selected after modification of their sequences at position Asn-162 or Asn-198. This targeted engineering of Fab constant domains was performed to validate the possibility of improving the efficiency of site-specific conjugation of drugs to Abs, and to minimize the incidence of immunoreactivity perturbation of the resulting immunoconjugates.
The second study of CH1/CL engineering aimed to improve the intrinsic thermodynamic stability of an anti-testosterone Fab fragment.Citation98 Different engineering approaches were explored targeting variable sites within the Fab constant domains. First, the mobile loop (residues 128–133) in the CH1 domain was subjected to rational mutagenesis to restrict its natural mobility, without however beneficial stabilizing properties. Second, site-directed mutagenesis was used to produce mutations that increase the hydrophobic potential of the interface between the CH1 and CL domains. This was achieved by substitution of weakly hydrophobic or nucleophilic amino acid residues for more hydrophobic ones such as Val, Leu, Ile, Met, or Phe, to exclude water molecules accommodated in two cavities at the CH1/CL interface. Fourteen single or combined mutants were thus designed and their stability was determined. The highest thermodynamic stability was reached by the replacement of the hydrophilic Thr178 in the CL domain by a valine or an isoleucine.
Fc Engineering
The biological activities of Abs within the immune system of vertebrates reflect the effector properties mediated by their Fc.Citation99 The Fc domain is responsible for immune cell binding via interaction with different human Fcγ activating (hFcγRI, RIIA, RIIC and RIIIA) or inhibitory (hFcγRIIB) receptors (FcRs).Citation100,Citation101 It also determines binding to complement factors.Citation102 Fc is also the region of Abs where glycosylation occurs. Consequently, Fc engineering aims to improve the efficacy of IgGs by modulating their effector properties, such as Ab-dependent cell-mediated toxicity (ADCC), Ab-dependent cellular phagocytosis (ADCP), complement-dependent cytotoxicity (CDC), but also their pharmacokinetics.Citation103-Citation105
Engineering of IgG Effector Properties
Based on mutagenesis, domain swapping, and 3D structure studies of Fc/FcRs complexesCitation106 it is possible to modulate the effector functionality of Fc-containing molecules (e.g., Abs, Fc-fusion peptides or proteins). Effector properties of Abs depend on amino acid residues in the CH2 constant domain and the hinge region, and all successful modulations of ADCC, ADCP or CDC of therapeutic Abs have targeted these two sections of the Fc.Citation103,Citation104 Molecular engineering of IgGs (mostly IgG1) results in either an increase or a decrease in effector functions, both approaches being motivated by the need for optimization of therapeutic Abs. The main examples of Fc engineering have been reviewed.Citation104 Effector function increases may be achieved by single substitutions, but more positive and pronounced effects are obtained via multiple mutations. In addition, residues outside the Fc/FcR binding interface, such as Thr256, Lys290, Glu333, Lys334, and Ala339, are also appropriate targets for modulating Fc properties.Citation103-Citation105,Citation107 Positions that when mutated, alone or in combination, increase ADCC, ADCP or CDC are: Gly236, Ser239, Phe243, Pro247, Asp280, Lys290, Arg292, Ser298, Thr299, Tyr300, Val305, Lys326, Ala330, Ile332, Glu333, Lys334, Ala339, and Pro396.
For reasons of safety, in the special case of therapeutic Abs that target cell-surface markers on immune cells, it may be advisable in some instances to silence their Fc functionality. Different single or multiple mutants have been developed to decrease complement activation or Fc-mediated cellular responses.Citation104 Strong and simultaneous decrease of ADCC and ADCP are possible through multiple substitutions in the lower hinge region (229–239), when normal ADCC can be maintained together with a strongly reduced CDC via mutations within the hinge region (218–229). Also, simultaneous decreases of ADCC, ADCP and CDC were achieved by designing different multiple variants e.g., a triple IgG1 mutant Leu234Phe, Leu235Glu and Pro331Ser (MedImmune); a tetra IgG2 variant His268Gln, Val309Leu, Ala330Ser, and Pro331Ser (Merck); a penta IgG1 mutant Cys226Ser, Cys229Ser, Glu233Pro, Leu234Val, and Leu235Ala (Seattle Genetics). Also, by combining computational design algorithms and high-throughput expression and screening, the company Xencor has been able to modulate the FcγR binding capacity of different therapeutic Abs.108. Thus, ADCC and ADCP functions were increased via Ser239Asp and Ile332Glu, when addition of the mutation Ala330Leu resulted in a triple variant with increased ADCC, ADCP and removed CDC. On his hand, the triple mutant Gly236Ala, Ser239Asp, Ile332Glu results in the increase of ADCP only. Finally, the double variant Ser267Glu, Leu328Phe of an anti-CD19 Ab (Xencor) displays a selective inhibitory binding capacity of the FcγRIIb receptor.
We recently demonstrated that mutation of histidine residues 310 and 435 at the CH2-CH3 interface of the Fc portion of an anti-Rhesus D human IgG1 has differential long-distance effects on its capacity to bind Fcγ receptors and on the functions of these receptors.Citation109 We showed that His310 and His435 can coordinate a Zn2+ cation which results in control of the CH2-CH2 interdomain opening. Structures obtained in the absence of Zn2+ have a reduced interdomain gap that likely hampers FcγR binding. This closed conformation of the Fc is stabilized by inter-CH2 domain sugar contacts. Zinc appears to counteract the sugar-mediated constriction, suggesting that zinc could be an important control factor in IgG1/FcγR interactions. The results of binding studies performed in the presence of EDTA on FcγR-expressing cells support this hypothesis. When a mutated Fc fragment, in which histidines 310 and 435 have been substituted by lysines (Fc H/K), was compared with the wild-type Fc in crystallographic studies, we found that the mutations leave the interface unaltered but have a long-range effect on the CH2 interdomain separation. Moreover, these substitutions have a differential effect on the binding of IgG1 to Fcγ receptors and on the functions of these receptors. Interaction with the inhibitory FcγRIIB is strongly perturbed () by the mutations and mutant IgG1 H/K only weakly engages this receptor. By contrast, higher affinity FcγR are mostly unaffected ().
Figure 1. Differential effects of His310 and His435 mutations on FcgammaRIIIA and FcgammaRIIB engagement.
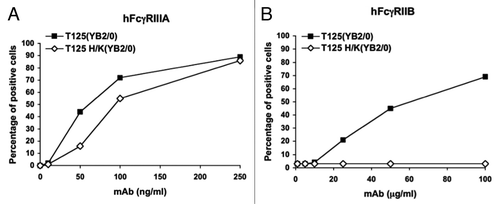
It has not been demonstrated that other metallic cations such as Ca2+ or Mg2+ affect FcγR binding, but our results suggest that manipulating zinc concentrations in vivo should help to clarify the role of this metallic cation in the regulation of Ab functions. Together, these results reveal how accurate and sophisticated are the molecular and structural rules that govern antibody effector functions.
1) Engineering of IgG Pharmacokinetics
Fc engineering also aims to modulate (increase or decrease) the pharmacokinetic parameters of therapeutic Abs. The pharmacokinetics of an IgG are related to its capacity to bind to the neonatal Fc receptor (FcRn), which ensures transport of IgGs across the placenta and plays a crucial role in controlling the persistence of therapeutic Abs in the serum.Citation110 Interaction with FcRn appears to be influenced by pH.Citation111 In many cases, Abs that bind at pH 6.0 and not at pH 7.4 exhibit a stronger affinity for the receptor and display a longer half-life; IgG molecules that do not bind to FcRns are degraded by the lysosomal pathway.Citation112,Citation113 The critical amino acid residues that mediate IgG-FcRn complex formation are located at the CH2-CH3 domain interface: Ile253, His310, Gln311, His or Arg435 and Phe or Tyr436. They are distinct from those involved in the FcγRs and C1q binding sites. To increase the half-life of Abs and consequently to reduce both the required amount of therapeutics to be produced and the frequency of administration, residues surrounding these critical positions will be targeted to strengthen the interaction. Most of the variants with longer half-lives combine several mutations: IgG1-Met252Tyr, Ser254Thr, Thr256Glu which has a 4-fold longer half-life (MedImmune)Citation113,Citation114 and IgG1-Thr307Ala, Glu380Ala, Asn434Ala, even if in this case the single substitution Asn434Ala was already efficient by itself (personal communication, Derry Roopenian, Jackson Lab).Citation112
In some cases it may be useful to decrease the half-life of Fc-containing molecules. This is in particularly true of IgG immunoconjugates, which it may be important to quickly eliminate from the circulation to reduce their side effects. To achieve this goal, mutations will target either residues directly involved in the formation of the IgG/FcRn complexes,Citation115 such as Ile253Ala, His310Ala, His435Gln, His435Arg or His310Ala/His435Gln, or neighboring ones such as Pro257Ile/Asn434His or Asp376Val/Asn434His, which will indirectly affect the former.Citation112,Citation116,Citation117
Finally, the Fc region of an IgG1 was submitted to molecular engineering to address the importance of glycosylation regarding FcγR engagement. Dane Wittrup and colleagues constructed different saturation mutagenesis libraries and screened them by yeast display to select tentatively aglycosylated variants still able to engage Fc receptors.Citation118 Three distinct libraries were constructed between positions 296 and 300 whose wild-type template sequence was Tyr296-Asn297-Ser298-Thr299-Tyr300. Screening was performed by fluorescence-activated cell sorting (FACS) for binding of engineered Fc regions using decreasing concentrations of fluorophore-labeled tetramers of a soluble form of the activating hFcγRIIA (50 nM, 2 nM, then 80 pM). Following three rounds of screening, the selected sub-library was dominated by the double-mutant Ser298Gly/Thr299Ala, which lacks the canonical Asn-X-Ser/Thr N-linked glycosylation motif. Site-directed mutagenesis was then performed revealing a synergistic additive effect of the corresponding single mutations. Engagement of FcγRs was maintained in vitro and in vivo, demonstrating that Fc glycosylation is not strictly required to trigger IgG activation of immune cells. These results open up new perspectives in terms of design and the recombinant strategy of expression of therapeutic Abs especially in bacteria, providing significant bioprocessing advantages.Citation119-Citation121
2) Stabilization of Fc Domain
Recent studies of molecular engineering of the IgG1-Fc domain describe two strategies to stabilize the Fc moietyCitation122,Citation123 to compensate for the reduced thermal stability resulting from the engineering of the C-terminal loops of the CH3-domains of IgG1-Fc.Citation124,Citation125
In the first strategy,Citation122 two error-prone IgG1-Fc libraries were constructed and expressed anchored on the surface of the yeast Saccharomyces cerevisiae. Screening was performed by flow sorting (FACS) following a 10 min heat shock at 79°C. Yeast cells presenting on their surface stabilized Fc variants were selected using either CD64 (Fcγ receptor I) or an anti-human IgG CH2-domain Ab. An average of 2.0 DNA mutations were thus introduced, and most of the selected stabilized Fc clones displayed mutations in the CH3 domain. Twelve single mutants, four double and one triple, were selected, all displaying more stabilized CH3-domains compared with Fc-wt. Interestingly, selected multiple variants displayed additive effects of the corresponding single mutants. Moreover, all these more stable IgG1-Fc variants had similar binding kinetics to FcRn, CD16a (FcγRIIIA) and protein A, demonstrating the efficacy of that engineering/screening strategy.
In the second strategy,Citation123 the CH3 domain within the Fc moiety was stabilized through structure-based engineering of de novo disulfide bonds. Among the 29 disulfide bonds predicted by molecular modeling and the analysis of the crystal structure of an Fc fragment, five were chosen, designed, and expressed in the yeast Pichia pastoris, and their thermostability and binding properties were studied. Only two of these constructs show increased thermostability: the disulfides between Pro343 and Ala431 and between Ser375 and Pro396, which connect different loops and/or strands. These two newly engineered disulfide bonds increased the thermostability by 5 or 10°C as compared with the CH3 in the wild-type Fc region. Furthermore, once combined these two disulphide bonds showed an additive thermostabilizing effect of 15°C of the CH3 domain, without affecting the CH2 domain. Also, it is noteworthy that the binding to the neonatal Fc fragment was not influenced by the introduced mutations. Finally, once introduced into the Fc fragment in which the C-terminal loop of the CH3 domain that was engineered for binding to Her2/neu, a 19°C increase in thermostability was measured. These two articles demonstrate how Fc can be engineered in a stable manner to display new binding properties, using two different molecular engineering strategies.
Antibody Labeling
Abs constitute a family of tools that are particularly powerful in the biomedical domain. We have discussed here how any domain of these proteins can be engineered to improve, adapt, or customize them for better use. As shown in many reviews, Abs are also very attractive because of the modulation of structural organization that makes them particularly suited to the design of a large number of functional Ab fragments, fusion, or chimeric Ab-based compounds.Citation124-Citation127
Despite their natural or engineered binding and biophysical properties, full-length Abs, owing to their size, do not appear to be particularly well-suited to drug delivery (radionuclides, enzymes, drugs, cytokines, toxins) or molecular imaging, whereas Ab fragments do.Citation128-Citation130 Labeled Ab fragments, minibodies or nanobodies combine better pharmacokinetics (fast blood clearance) together with a high tumor to blood ratio in a shorter time, resulting in high-quality contrasting imaging. Clearly, the challenge in the years ahead will be to design and improve control of Ab fragment labeling. Binding properties (affinity, specificity) of Abs, their valency (scFv vs. diabodies, multivalent scFv formats), and their pharmacokinetics can be kept unchanged or improved. Thus, a labeled (124I) and engineered (scFv)2-Fc His310Ala/His435Gln specific for carcinoembryonic antigen gives a significantly better molecular image compared with the wild-type format of the same Fc-Ab fragment fusion protein.Citation131 One way to direct and control the labeling is to introduce by site-directed mutagenesis additional amino acid residues within de novo designed Ab fragments or formats, in a region that will not interfere with Ab properties.
Conclusion
Ab engineering occupies a central and crucial position in the design and development of appropriate and adapted new Ab therapeutics, as well as new in vitro diagnostic and in vivo imaging tools. Together with the development of strategies of in vitro molecular evolution during the last decade, their field of use and application has enlarged. More and more Abs are being approved for marketing and hundreds are under clinical evaluation, making this category of proteins one of the more promising therapeutic molecules. It is thus not surprising to see the efforts made by researchers and clinicians both to identify relevant markers to target and to improve their properties, in particular by molecular engineering.Citation132,Citation133
Furthermore, most of the results obtained with intact immunoglobulins are extended and applied to Ab fragments, fusion Ab-derived compounds, and also to alternative scaffolds, or intrabodies, providing researchers and clinicians with a large panel of new compounds responding to needs in therapy, diagnosis and imaging.
Substantial efforts are, however, still needed to make progress in our capacity to control the molecular variability of these new therapeutic candidates, which appear more heterogeneous in solution than expected. Many challenges have to be faced to design more accurately defined and affordable therapeutic Ab-based compounds. Despite the potential of molecular engineering in developing better therapeutics, this family of proteins will expand further into the clinical market only when the recombinant strategies of expression are more controlled and less costly. However, it is important to keep in mind that mAb glycosylation is deeply linked to the recombinant expression system used, modulations that may impact both on their intrinsic immunogenicity and their effector functions.Citation134
| Abbreviations: | ||
| mAbs | = | monoclonal antibodies |
| Ab | = | antibody |
| DNA | = | DeoxyriboNucleic Acid |
| RNA | = | RiboNucleic Acid |
| EIA | = | Enzyme ImmunoAssay |
| ep-PCR | = | error-prone PCR |
| ADCC | = | Antibody-Dependent Cell-mediated Cytotoxicity |
| ADCP | = | Antibody-Dependent Cellular Phagocytosis |
| CDC | = | Complement-Dependent Cytotoxicity |
| FcγR | = | Fc gamma receptors |
| HLA | = | Human Leukocyte Antigen |
| FcRn | = | neonatal Fc receptor |
| VEGF | = | Vascular Endothelial Growth Factor |
| TNFα | = | Tumour Necrosis Factor alpha |
| Fv | = | variable Fragment |
| scFv | = | single-chain variable Fragment |
| VL | = | light chain variable domain |
| VH | = | heavy chain variable domain |
| CDR | = | Complementarity Determining Region |
| FR | = | Framework Region |
| IgG | = | immunoglobulin type G |
| PSA | = | Prostate-Specific Antigen |
| FACS | = | Fluorescence-Activated Cell Sorting |
| NMR | = | Nuclear Magnetic Resonance |
References
- Balint RF, Larrick JW. Antibody engineering by parsimonious mutagenesis. Gene 1993; 137:109 - 18; http://dx.doi.org/10.1016/0378-1119(93)90258-5; PMID: 7506686
- Sheedy C, MacKenzie CR, Hall JC. Isolation and affinity maturation of hapten-specific antibodies. Biotechnol Adv 2007; 25:333 - 52; http://dx.doi.org/10.1016/j.biotechadv.2007.02.003; PMID: 17383141
- Labrou NE. Random mutagenesis methods for in vitro directed enzyme evolution. Curr Protein Pept Sci 2010; 11:91 - 100; http://dx.doi.org/10.2174/138920310790274617; PMID: 20201809
- Lewis L, Lloyd C. Optimisation of antibody affinity by ribosome display using error-prone or site-directed mutagenesis. Methods Mol Biol 2012; 805:139 - 61; http://dx.doi.org/10.1007/978-1-61779-379-0_9; PMID: 22094805
- Volles MJ, Lansbury PT Jr.. A computer program for the estimation of protein and nucleic acid sequence diversity in random point mutagenesis libraries. Nucleic Acids Res 2005; 33:3667 - 77; http://dx.doi.org/10.1093/nar/gki669; PMID: 15990391
- Arkin MR, Wells JA. Probing the importance of second sphere residues in an esterolytic antibody by phage display. J Mol Biol 1998; 284:1083 - 94; http://dx.doi.org/10.1006/jmbi.1998.2234; PMID: 9837728
- Boder ET, Midelfort KS, Wittrup KD. Directed evolution of antibody fragments with monovalent femtomolar antigen-binding affinity. Proc Natl Acad Sci U S A 2000; 97:10701 - 5; http://dx.doi.org/10.1073/pnas.170297297; PMID: 10984501
- Löfblom J. Bacterial display in combinatorial protein engineering. Biotechnol J 2011; 6:1115 - 29; http://dx.doi.org/10.1002/biot.201100129; PMID: 21786423
- Lipovsek D, Plückthun A. In-vitro protein evolution by ribosome display and mRNA display. J Immunol Methods 2004; 290:51 - 67; http://dx.doi.org/10.1016/j.jim.2004.04.008; PMID: 15261571
- Pelat T, Hust M, Thullier P. Obtention and engineering of non-human primate (NHP) antibodies for therapeutics. Mini Rev Med Chem 2009; 9:1633 - 8; http://dx.doi.org/10.2174/138955709791012283; PMID: 20105119
- Thullier P, Chahboun S, Pelat T. A comparison of human and macaque (Macaca mulatta) immunoglobulin germline V regions and its applications for antibody engineering. MAbs 2010; 5:528 - 38; http://dx.doi.org/10.4161/mabs.2.5.12545
- Köhler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975; 256:495 - 7; http://dx.doi.org/10.1038/256495a0; PMID: 1172191
- Wu AHB. A selected history and future of immunoassay development and applications in clinical chemistry. Clin Chim Acta 2006; 369:119 - 24; http://dx.doi.org/10.1016/j.cca.2006.02.045; PMID: 16701599
- Reichert JM. Antibody-based therapeutics to watch in 2011. MAbs 2011; 3:76 - 99; http://dx.doi.org/10.4161/mabs.3.1.13895; PMID: 21051951
- Klee GG. Human anti-mouse antibodies. Arch Pathol Lab Med 2000; 124:921 - 3; PMID: 10835540
- Baca M, Presta LG, O’Connor SJ, Wells JA. Antibody humanization using monovalent phage display. J Biol Chem 1997; 272:10678 - 84; http://dx.doi.org/10.1074/jbc.272.16.10678; PMID: 9099717
- Schlapschy M, Gruber H, Gresch O, Schäfer C, Renner C, Pfreundschuh M, et al. Functional humanization of an anti-CD30 Fab fragment for the immunotherapy of Hodgkin’s lymphoma using an in vitro evolution approach. Protein Eng Des Sel 2004; 17:847 - 60; http://dx.doi.org/10.1093/protein/gzh098; PMID: 15708864
- Hoogenboom HR. Selecting and screening recombinant antibody libraries. Nat Biotechnol 2005; 23:1105 - 16; http://dx.doi.org/10.1038/nbt1126; PMID: 16151404
- Lonberg N. Human antibodies from transgenic animals. Nat Biotechnol 2005; 23:1117 - 25; http://dx.doi.org/10.1038/nbt1135; PMID: 16151405
- Van Walle I, Gansemans Y, Parren PW, Stas P, Lasters I. Immunogenicity screening in protein drug development. Expert Opin Biol Ther 2007; 7:405 - 18; http://dx.doi.org/10.1517/14712598.7.3.405; PMID: 17309332
- Schellekens H. How to predict and prevent the immunogenicity of therapeutic proteins. Biotechnol Annu Rev 2008; 14:191 - 202; http://dx.doi.org/10.1016/S1387-2656(08)00007-0; PMID: 18606364
- De Groot AS, McMurry J, Moise L. Prediction of immunogenicity: in silico paradigms, ex vivo and in vivo correlates. Curr Opin Pharmacol 2008; 8:620 - 6; http://dx.doi.org/10.1016/j.coph.2008.08.002; PMID: 18775515
- Holgate RG, Baker MP. Circumventing immunogenicity in the development of therapeutic antibodies. IDrugs 2009; 12:233 - 7; PMID: 19350467
- Harding FA, Stickler MM, Razo J, DuBridge RB. The immunogenicity of humanized and fully human antibodies: residual immunogenicity resides in the CDR regions. MAbs 2010; 2:256 - 65; http://dx.doi.org/10.4161/mabs.2.3.11641; PMID: 20400861
- Weber CA, Mehta PJ, Ardito M, Moise L, Martin B, De Groot AS. T cell epitope: friend or foe? Immunogenicity of biologics in context. Adv Drug Deliv Rev 2009; 61:965 - 76; http://dx.doi.org/10.1016/j.addr.2009.07.001; PMID: 19619593
- Morrison SL, Johnson MJ, Herzenberg LA, Oi VT. Chimeric human antibody molecules: mouse antigen-binding domains with human constant region domains. Proc Natl Acad Sci U S A 1984; 81:6851 - 5; http://dx.doi.org/10.1073/pnas.81.21.6851; PMID: 6436822
- Jones PT, Dear PH, Foote J, Neuberger MS, Winter G. Replacing the complementarity-determining regions in a human antibody with those from a mouse. Nature 1986; 321:522 - 5; http://dx.doi.org/10.1038/321522a0; PMID: 3713831
- Schroff RW, Foon KA, Beatty SM, Oldham RK, Morgan AC Jr.. Human anti-murine immunoglobulin responses in patients receiving monoclonal antibody therapy. Cancer Res 1985; 45:879 - 85; PMID: 3871353
- Hwang WYK, Foote J. Immunogenicity of engineered antibodies. Methods 2005; 36:3 - 10; http://dx.doi.org/10.1016/j.ymeth.2005.01.001; PMID: 15848070
- Roguska MA, Pedersen JT, Keddy CA, Henry AH, Searle SJ, Lambert JM, et al. Humanization of murine monoclonal antibodies through variable domain resurfacing. Proc Natl Acad Sci U S A 1994; 91:969 - 73; http://dx.doi.org/10.1073/pnas.91.3.969; PMID: 8302875
- Graff CP, Chester K, Begent R, Wittrup KD. Directed evolution of an anti-carcinoembryonic antigen scFv with a 4-day monovalent dissociation half-time at 37 degrees C. Protein Eng Des Sel 2004; 17:293 - 304; http://dx.doi.org/10.1093/protein/gzh038; PMID: 15115853
- Tamura M, Milenic DE, Iwahashi M, Padlan E, Schlom J, Kashmiri SV. Structural correlates of an anticarcinoma antibody: identification of specificity-determining residues (SDRs) and development of a minimally immunogenic antibody variant by retention of SDRs only. J Immunol 2000; 164:1432 - 41; PMID: 10640759
- Gonzales NR, Padlan EA, De Pascalis R, Schuck P, Schlom J, Kashmiri SV. SDR grafting of a murine antibody using multiple human germline templates to minimize its immunogenicity. Mol Immunol 2004; 41:863 - 72; http://dx.doi.org/10.1016/j.molimm.2004.03.041; PMID: 15261458
- Kashmiri SV, De Pascalis R, Gonzales NR, Schlom J. SDR grafting--a new approach to antibody humanization. Methods 2005; 36:25 - 34; http://dx.doi.org/10.1016/j.ymeth.2005.01.003; PMID: 15848072
- Dall’Acqua WF, Damschroder MM, Zhang J, Woods RM, Widjaja L, Yu J, et al. Antibody humanization by framework shuffling. Methods 2005; 36:43 - 60; http://dx.doi.org/10.1016/j.ymeth.2005.01.005; PMID: 15848074
- Damschroder MM, Widjaja L, Gill PS, Krasnoperov V, Jiang W, Dall’Acqua WF, et al. Framework shuffling of antibodies to reduce immunogenicity and manipulate functional and biophysical properties. Mol Immunol 2007; 44:3049 - 60; http://dx.doi.org/10.1016/j.molimm.2006.12.019; PMID: 17241664
- Lazar GA, Desjarlais JR, Jacinto J, Karki S, Hammond PW. A molecular immunology approach to antibody humanization and functional optimization. Mol Immunol 2007; 44:1986 - 98; http://dx.doi.org/10.1016/j.molimm.2006.09.029; PMID: 17079018
- Hwang WY, Almagro JC, Buss TN, Tan P, Foote J. Use of human germline genes in a CDR homology-based approach to antibody humanization. Methods 2005; 36:35 - 42; http://dx.doi.org/10.1016/j.ymeth.2005.01.004; PMID: 15848073
- Huse WD, Sastry L, Iverson SA, Kang AS, Alting-Mees M, Burton DR, et al. Generation of a large combinatorial library of the immunoglobulin repertoire in phage lambda. Science 1989; 246:1275 - 81; http://dx.doi.org/10.1126/science.2531466; PMID: 2531466
- Rosok MJ, Yelton DE, Harris LJ, Bajorath J, Hellström KE, Hellström I, et al. A combinatorial library strategy for the rapid humanization of anticarcinoma BR96 Fab. J Biol Chem 1996; 271:22611 - 8; http://dx.doi.org/10.1074/jbc.271.37.22611; PMID: 8798431
- Rader C, Cheresh DA, Barbas CF 3rd. A phage display approach for rapid antibody humanization: designed combinatorial V gene libraries. Proc Natl Acad Sci U S A 1998; 95:8910 - 5; http://dx.doi.org/10.1073/pnas.95.15.8910; PMID: 9671778
- Bernett MJ, Karki S, Moore GL, Leung IW, Chen H, Pong E, et al. Engineering fully human monoclonal antibodies from murine variable regions. J Mol Biol 2010; 396:1474 - 90; http://dx.doi.org/10.1016/j.jmb.2009.12.046; PMID: 20045416
- De Groot AS, Moise L. Prediction of immunogenicity for therapeutic proteins: state of the art. Curr Opin Drug Discov Devel 2007; 10:332 - 40; PMID: 17554860
- Bender NK, Heilig CE, Dröll B, Wohlgemuth J, Armbruster FP, Heilig B. Immunogenicity, efficacy and adverse events of adalimumab in RA patients. Rheumatol Int 2007; 27:269 - 74; http://dx.doi.org/10.1007/s00296-006-0183-7; PMID: 17006705
- West RL, Zelinkova Z, Wolbink GJ, Kuipers EJ, Stokkers PC, van der Woude CJ. Immunogenicity negatively influences the outcome of adalimumab treatment in Crohn’s disease. Aliment Pharmacol Ther 2008; 28:1122 - 6; http://dx.doi.org/10.1111/j.1365-2036.2008.03828.x; PMID: 18691349
- Inman RD, Davis JC Jr., Heijde D, Diekman L, Sieper J, Kim SI, et al. Efficacy and safety of golimumab in patients with ankylosing spondylitis: results of a randomized, double-blind, placebo-controlled, phase III trial. Arthritis Rheum 2008; 58:3402 - 12; http://dx.doi.org/10.1002/art.23969; PMID: 18975305
- Hulmes JD, Hantash J, Snow M, O’Dell M, Bowsher RR, Kirman I. Immunogenicity assessment of humanized deimmunized therapeutic monoclonal antibody ETI-204 (Anthin®) in phase I clinical study with a validated electrochemiluminescence immunoassay. AAPS Biotechnology Conference, 2010; May 16-19, San Francisco CA, USA.
- Henry MD, Wen S, Silva MD, Chandra S, Milton M, Worland PJ. A prostate-specific membrane antigen-targeted monoclonal antibody-chemotherapeutic conjugate designed for the treatment of prostate cancer. Cancer Res 2004; 64:7995 - 8001; http://dx.doi.org/10.1158/0008-5472.CAN-04-1722; PMID: 15520207
- Hudson PJ, Souriau C. Engineered antibodies. Nat Med 2003; 9:129 - 34; http://dx.doi.org/10.1038/nm0103-129; PMID: 12514726
- Carter PJ. Introduction to current and future protein therapeutics: a protein engineering perspective. Exp Cell Res 2011; 317:1261 - 9; http://dx.doi.org/10.1016/j.yexcr.2011.02.013; PMID: 21371474
- Igawa T, Tsunoda H, Kuramochi T, Sampei Z, Ishii S, Hattori K. Engineering the variable region of therapeutic IgG antibodies. MAbs 2011; 3:243 - 52; http://dx.doi.org/10.4161/mabs.3.3.15234; PMID: 21406966
- Clark LA, Boriack-Sjodin PA, Eldredge J, Fitch C, Friedman B, Hanf KJM, et al. Affinity enhancement of an in vivo matured therapeutic antibody using structure-based computational design. Protein Sci 2006; 15:949 - 60; http://dx.doi.org/10.1110/ps.052030506; PMID: 16597831
- Riechmann L, Weill M. Phage display and selection of a site-directed randomized single-chain antibody Fv fragment for its affinity improvement. Biochemistry 1993; 32:8848 - 55; http://dx.doi.org/10.1021/bi00085a016; PMID: 8364031
- Foote J, Eisen HN. Kinetic and affinity limits on antibodies produced during immune responses. Proc Natl Acad Sci U S A 1995; 92:1254 - 6; http://dx.doi.org/10.1073/pnas.92.5.1254; PMID: 7877964
- Batista FD, Neuberger MS. Affinity dependence of the B cell response to antigen: a threshold, a ceiling, and the importance of off-rate. Immunity 1998; 8:751 - 9; http://dx.doi.org/10.1016/S1074-7613(00)80580-4; PMID: 9655489
- Kobayashi N, Oyama H. Antibody engineering toward high-sensitive high-throughput immunosensing of small molecules. Analyst (Lond) 2011; 136:642 - 51; http://dx.doi.org/10.1039/c0an00603c
- Dubreuil O, Bossus M, Graille M, Bilous M, Savatier A, Jolivet M, et al. Fine tuning of the specificity of an anti-progesterone antibody by first and second sphere residue engineering. J Biol Chem 2005; 280:24880 - 7; http://dx.doi.org/10.1074/jbc.M500048200; PMID: 15878862
- Rudnick SI, Adams GP. Affinity and avidity in antibody-based tumor targeting. Cancer Biother Radiopharm 2009; 24:155 - 61; http://dx.doi.org/10.1089/cbr.2009.0627; PMID: 19409036
- Graff CP, Wittrup KD. Theoretical analysis of antibody targeting of tumor spheroids: importance of dosage for penetration, and affinity for retention. Cancer Res 2003; 63:1288 - 96; PMID: 12649189
- Zahnd C, Spinelli S, Luginbühl B, Amstutz P, Cambillau C, Plückthun A. Directed in vitro evolution and crystallographic analysis of a peptide-binding single chain antibody fragment (scFv) with low picomolar affinity. J Biol Chem 2004; 279:18870 - 7; http://dx.doi.org/10.1074/jbc.M309169200; PMID: 14754898
- Beers R, Chowdhury P, Bigner D, Pastan I. Immunotoxins with increased activity against epidermal growth factor receptor vIII-expressing cells produced by antibody phage display. Clin Cancer Res 2000; 6:2835 - 43; PMID: 10914732
- Ho M, Kreitman RJ, Onda M, Pastan I. In vitro antibody evolution targeting germline hot spots to increase activity of an anti-CD22 immunotoxin. J Biol Chem 2005; 280:607 - 17; PMID: 15491997
- Yau KY, Dubuc G, Li S, Hirama T, Mackenzie CR, Jermutus L, et al. Affinity maturation of a V(H)H by mutational hotspot randomization. J Immunol Methods 2005; 297:213 - 24; http://dx.doi.org/10.1016/j.jim.2004.12.005; PMID: 15777944
- Johannes TW, Woodyer RD, Zhao H. Directed evolution of a thermostable phosphite dehydrogenase for NAD(P)H regeneration. Appl Environ Microbiol 2005; 71:5728 - 34; http://dx.doi.org/10.1128/AEM.71.10.5728-5734.2005; PMID: 16204481
- Chowdhury PS, Pastan I. Improving antibody affinity by mimicking somatic hypermutation in vitro.. Nat Biotechnol 1999; 17:568 - 72; http://dx.doi.org/10.1038/9872; PMID: 10385321
- Neuberger MS, Milstein C. Somatic hypermutation. Curr Opin Immunol 1995; 7:248 - 54; http://dx.doi.org/10.1016/0952-7915(95)80010-7; PMID: 7546385
- Salvatore G, Beers R, Margulies I, Kreitman RJ, Pastan I. Improved cytotoxic activity toward cell lines and fresh leukemia cells of a mutant anti-CD22 immunotoxin obtained by antibody phage display. Clin Cancer Res 2002; 8:995 - 1002; PMID: 11948105
- Muller BH, Savatier A, L’Hostis G, Costa N, Bossus M, Michel S, et al. In vitro affinity maturation of an anti-PSA antibody for prostate cancer diagnostic assay. J Mol Biol 2011; 414:545 - 62; http://dx.doi.org/10.1016/j.jmb.2011.10.008; PMID: 22019475
- Stura EA, Muller BH, Bossus M, Michel S, Jolivet-Reynaud C, Ducancel F. Crystal structure of human prostate-specific antigen in a sandwich antibody complex. J Mol Biol 2011; 414:530 - 44; http://dx.doi.org/10.1016/j.jmb.2011.10.007; PMID: 22037582
- Rajpal A, Beyaz N, Haber L, Cappuccilli G, Yee H, Bhatt RR, et al. A general method for greatly improving the affinity of antibodies by using combinatorial libraries. Proc Natl Acad Sci U S A 2005; 102:8466 - 71; http://dx.doi.org/10.1073/pnas.0503543102; PMID: 15939870
- Laffly E, Pelat T, Cédrone F, Blésa S, Bedouelle H, Thullier P. Improvement of an antibody neutralizing the anthrax toxin by simultaneous mutagenesis of its six hypervariable loops. J Mol Biol 2008; 378:1094 - 103; http://dx.doi.org/10.1016/j.jmb.2008.03.045; PMID: 18423488
- Lippow SM, Wittrup KD, Tidor B. Computational design of antibody-affinity improvement beyond in vivo maturation. Nat Biotechnol 2007; 25:1171 - 6; http://dx.doi.org/10.1038/nbt1336; PMID: 17891135
- Lippow SM, Tidor B. Progress in computational protein design. Curr Opin Biotechnol 2007; 18:305 - 11; http://dx.doi.org/10.1016/j.copbio.2007.04.009; PMID: 17644370
- Chames P, Coulon S, Baty D. Improving the affinity and the fine specificity of an anti-cortisol antibody by parsimonious mutagenesis and phage display. J Immunol 1998; 161:5421 - 9; PMID: 9820517
- Lamminmäki U, Westerlund-Karlsson A, Toivola M, Saviranta P. Modulating the binding properties of an anti-17β-estradiol antibody by systematic mutation combinations. Protein Sci 2003; 12:2549 - 58; http://dx.doi.org/10.1110/ps.0353903; PMID: 14573866
- Korpimäki T, Rosenberg J, Virtanen P, Lamminmäki U, Tuomola M, Saviranta P. Further improvement of broad specificity hapten recognition with protein engineering. Protein Eng 2003; 16:37 - 46; http://dx.doi.org/10.1093/proeng/gzg010; PMID: 12646691
- Garcia-Rodriguez C, Levy R, Arndt JW, Forsyth CM, Razai A, Lou J, et al. Molecular evolution of antibody cross-reactivity for two subtypes of type A botulinum neurotoxin. Nat Biotechnol 2007; 25:107 - 16; http://dx.doi.org/10.1038/nbt1269; PMID: 17173035
- Farady CJ, Sellers BD, Jacobson MP, Craik CS. Improving the species cross-reactivity of an antibody using computational design. Bioorg Med Chem Lett 2009; 19:3744 - 7; http://dx.doi.org/10.1016/j.bmcl.2009.05.005; PMID: 19477127
- Demarest SJ, Glaser SM. Antibody therapeutics, antibody engineering, and the merits of protein stability. Curr Opin Drug Discov Devel 2008; 11:675 - 87; PMID: 18729019
- Honegger A. Engineering antibodies for stability and efficient folding. Handb exp Pharmacol 2008; 181:47-68.
- Kügler M, Stein C, Schwenkert M, Saul D, Vockentanz L, Huber T, et al. Stabilization and humanization of a single-chain Fv antibody fragment specific for human lymphocyte antigen CD19 by designed point mutations and CDR-grafting onto a human framework. Protein Eng Des Sel 2009; 22:135 - 47; http://dx.doi.org/10.1093/protein/gzn079; PMID: 19188138
- Nieba L, Honegger A, Krebber C, Plückthun A. Disrupting the hydrophobic patches at the antibody variable/constant domain interface: improved in vivo folding and physical characterization of an engineered scFv fragment. Protein Eng 1997; 10:435 - 44; http://dx.doi.org/10.1093/protein/10.4.435; PMID: 9194169
- Ewert S, Honegger A, Plückthun A. Stability improvement of antibodies for extracellular and intracellular applications: CDR grafting to stable frameworks and structure-based framework engineering. Methods 2004; 34:184 - 99; http://dx.doi.org/10.1016/j.ymeth.2004.04.007; PMID: 15312672
- Ewert S, Honegger A, Plückthun A. Structure-based improvement of the biophysical properties of immunoglobulin VH domains with a generalizable approach. Biochemistry 2003; 42:1517 - 28; http://dx.doi.org/10.1021/bi026448p; PMID: 12578364
- Honegger A, Malebranche AD, Röthlisberger D, Plückthun A. The influence of the framework core residues on the biophysical properties of immunoglobulin heavy chain variable domains. Protein Eng Des Sel 2009; 22:121 - 34; http://dx.doi.org/10.1093/protein/gzn077; PMID: 19136675
- Wörn A, Auf der Maur A, Escher D, Honegger A, Barberis A, Plückthun A. Correlation between in vitro stability and in vivo performance of anti-GCN4 intrabodies as cytoplasmic inhibitors. J Biol Chem 2000; 275:2795 - 803; http://dx.doi.org/10.1074/jbc.275.4.2795; PMID: 10644744
- Jung S, Honegger A, Plückthun A. Selection for improved protein stability by phage display. J Mol Biol 1999; 294:163 - 80; http://dx.doi.org/10.1006/jmbi.1999.3196; PMID: 10556036
- Brockmann EC, Cooper M, Strömsten N, Vehniäinen M, Saviranta P. Selecting for antibody scFv fragments with improved stability using phage display with denaturation under reducing conditions. J Immunol Methods 2005; 296:159 - 70; http://dx.doi.org/10.1016/j.jim.2004.11.008; PMID: 15680160
- Miller BR, Demarest SJ, Lugovskoy A, Huang F, Wu X, Snyder WB, et al. Stability engineering of scFvs for the development of bispecific and multivalent antibodies. Protein Eng Des Sel 2010; 23:549 - 57; http://dx.doi.org/10.1093/protein/gzq028; PMID: 20457695
- Perchiacca JM, Bhattacharya M, Tessier PM. Mutational analysis of domain antibodies reveals aggregation hotspots within and near the complementarity determining regions. Proteins 2011; 79:2637 - 47; http://dx.doi.org/10.1002/prot.23085; PMID: 21732420
- Chennamsetty N, Helk B, Voynov V, Kayser V, Trout BL. Aggregation-prone motifs in human immunoglobulin G. J Mol Biol 2009; 391:404 - 13; http://dx.doi.org/10.1016/j.jmb.2009.06.028; PMID: 19527731
- Igawa T, Tsunoda H, Tachibana T, Maeda A, Mimoto F, Moriyama C, et al. Reduced elimination of IgG antibodies by engineering the variable region. Protein Eng Des Sel 2010; 23:385 - 92; http://dx.doi.org/10.1093/protein/gzq009; PMID: 20159773
- Igawa T, Ishii S, Tachibana T, Maeda A, Higuchi Y, Shimaoka S, et al. Antibody recycling by engineered pH-dependent antigen binding improves the duration of antigen neutralization. Nat Biotechnol 2010; 28:1203 - 7; http://dx.doi.org/10.1038/nbt.1691; PMID: 20953198
- Adachi M, Kurihara Y, Nojima H, Takeda-Shitaka M, Kamiya K, Umeyama H. Interaction between the antigen and antibody is controlled by the constant domains: normal mode dynamics of the HEL-HyHEL-10 complex. Protein Sci 2003; 12:2125 - 31; http://dx.doi.org/10.1110/ps.03100803; PMID: 14500870
- Röthlisberger D, Honegger A, Plückthun A. Domain interactions in the Fab fragment: a comparative evaluation of the single-chain Fv and Fab format engineered with variable domains of different stability. J Mol Biol 2005; 347:773 - 89; http://dx.doi.org/10.1016/j.jmb.2005.01.053; PMID: 15769469
- Qu Z, Sharkey RM, Hansen HJ, Goldenberg DM, Leung S. Structure determination of N-linked oligosaccharides engineered at the CH1 domain of humanized LL2. Glycobiology 1997; 7:803 - 9; http://dx.doi.org/10.1093/glycob/7.6.803; PMID: 9376682
- Qu Z, Sharkey RM, Hansen HJ, Shih LB, Govindan SV, Shen J, et al. Carbohydrates engineered at antibody constant domains can be used for site-specific conjugation of drugs and chelates. J Immunol Methods 1998; 213:131 - 44; http://dx.doi.org/10.1016/S0022-1759(97)00192-0; PMID: 9692846
- Teerinen T, Valjakka J, Rouvinen J, Takkinen K. Structure-based stability engineering of the mouse IgG1 Fab fragment by modifying constant domains. J Mol Biol 2006; 361:687 - 97; http://dx.doi.org/10.1016/j.jmb.2006.06.073; PMID: 16876195
- Daëron M. Fc receptor biology. Annu Rev Immunol 1997; 15:203 - 34; http://dx.doi.org/10.1146/annurev.immunol.15.1.203; PMID: 9143687
- Ravetch JV, Bolland S. IgG Fc receptors. Annu Rev Immunol 2001; 19:275 - 90; http://dx.doi.org/10.1146/annurev.immunol.19.1.275; PMID: 11244038
- Bruhns P, Iannascoli B, England P, Mancardi DA, Fernandez N, Jorieux S, et al. Specificity and affinity of human Fcgamma receptors and their polymorphic variants for human IgG subclasses. Blood 2009; 113:3716 - 25; http://dx.doi.org/10.1182/blood-2008-09-179754; PMID: 19018092
- Basta M. Ambivalent effect of immunoglobulins on the complement system: activation versus inhibition. Mol Immunol 2008; 45:4073 - 9; http://dx.doi.org/10.1016/j.molimm.2008.07.012; PMID: 18706699
- Liu XY, Pop LM, Vitetta ES. Engineering therapeutic monoclonal antibodies. Immunol Rev 2008; 222:9 - 27; http://dx.doi.org/10.1111/j.1600-065X.2008.00601.x; PMID: 18363992
- Strohl WR. Optimization of Fc-mediated effector functions of monoclonal antibodies. Curr Opin Biotechnol 2009; 20:685 - 91; http://dx.doi.org/10.1016/j.copbio.2009.10.011; PMID: 19896358
- Jeong KJ, Jang SH, Velmurugan N. Recombinant antibodies: engineering and production in yeast and bacterial hosts. Biotechnol J 2011; 6:16 - 27; http://dx.doi.org/10.1002/biot.201000381; PMID: 21170983
- Sondermann P, Huber R, Oosthuizen V, Jacob U. The 3.2-A crystal structure of the human IgG1 Fc fragment-Fc gammaRIII complex. Nature 2000; 406:267 - 73; http://dx.doi.org/10.1038/35018508; PMID: 10917521
- Shields RL, Namenuk AK, Hong K, Meng YG, Rae J, Briggs J, et al. High resolution mapping of the binding site on human IgG1 for Fc γ RI, Fc γ RII, Fc γ RIII, and FcRn and design of IgG1 variants with improved binding to the Fc γ R. J Biol Chem 2001; 276:6591 - 604; http://dx.doi.org/10.1074/jbc.M009483200; PMID: 11096108
- Lazar GA, Dang W, Karki S, Vafa O, Peng JS, Hyun L, et al. Engineered antibody Fc variants with enhanced effector function. Proc Natl Acad Sci U S A 2006; 103:4005 - 10; http://dx.doi.org/10.1073/pnas.0508123103; PMID: 16537476
- Sibéril S, Ménez R, Jorieux S, de Romeuf C, Bourel D, Fridman WH, et al. Effect of zinc on human IgG1 and its FcγR interactions. Immunol Lett 2012; 143:60 - 9; http://dx.doi.org/10.1016/j.imlet.2012.02.002; PMID: 22553781
- Lobo ED, Hansen RJ, Balthasar JP. Antibody pharmacokinetics and pharmacodynamics. J Pharm Sci 2004; 93:2645 - 68; http://dx.doi.org/10.1002/jps.20178; PMID: 15389672
- Martin WL, West AP Jr., Gan L, Bjorkman PJ. Crystal structure at 2.8 A of an FcRn/heterodimeric Fc complex: mechanism of pH-dependent binding. Mol Cell 2001; 7:867 - 77; http://dx.doi.org/10.1016/S1097-2765(01)00230-1; PMID: 11336709
- Petkova SB, Akilesh S, Sproule TJ, Christianson GJ, Al Khabbaz H, Brown AC, et al. Enhanced half-life of genetically engineered human IgG1 antibodies in a humanized FcRn mouse model: potential application in humorally mediated autoimmune disease. Int Immunol 2006; 18:1759 - 69; http://dx.doi.org/10.1093/intimm/dxl110; PMID: 17077181
- Dall’Acqua WF, Kiener PA, Wu H. Properties of human IgG1s engineered for enhanced binding to the neonatal Fc receptor (FcRn). J Biol Chem 2006; 281:23514 - 24; http://dx.doi.org/10.1074/jbc.M604292200; PMID: 16793771
- Oganesyan V, Damschroder MM, Woods RM, Cook KE, Wu H, Dall’acqua WF. Structural characterization of a human Fc fragment engineered for extended serum half-life. Mol Immunol 2009; 46:1750 - 5; http://dx.doi.org/10.1016/j.molimm.2009.01.026; PMID: 19250681
- Pop LM, Liu X, Ghetie V, Vitetta ES. The generation of immunotoxins using chimeric anti-CD22 antibodies containing mutations which alter their serum half-life. Int Immunopharmacol 2005; 5:1279 - 90; http://dx.doi.org/10.1016/j.intimp.2005.03.013; PMID: 15914332
- Kenanova V, Olafsen T, Crow DM, Sundaresan G, Subbarayan M, Carter NH, et al. Tailoring the pharmacokinetics and positron emission tomography imaging properties of anti-carcinoembryonic antigen single-chain Fv-Fc antibody fragments. Cancer Res 2005; 65:622 - 31; PMID: 15695407
- Datta-Mannan A, Witcher DR, Tang Y, Watkins J, Wroblewski VJ, Wroblewski VJ. Monoclonal antibody clearance. Impact of modulating the interaction of IgG with the neonatal Fc receptor. J Biol Chem 2007; 282:1709 - 17; http://dx.doi.org/10.1074/jbc.M607161200; PMID: 17135257
- Sazinsky SL, Ott RG, Silver NW, Tidor B, Ravetch JV, Wittrup KD. Aglycosylated immunoglobulin G1 variants productively engage activating Fc receptors. Proc Natl Acad Sci U S A 2008; 105:20167 - 72; http://dx.doi.org/10.1073/pnas.0809257105; PMID: 19074274
- Jung ST, Reddy ST, Kang TH, Borrok MJ, Sandlie I, Tucker PW, et al. Aglycosylated IgG variants expressed in bacteria that selectively bind FcγRI potentiate tumour cell killing by monocyte-dendritic cells. Proc Natl Acad Sci USA •••; 201:604 - 9
- Jung ST, Kang TH, Kelton W, Georgiou G. Bypassing glycosylation: engineering aglycosylated full-length IgG antibodies for human therapy. Curr Opin Biotechnol 2011; 22:1 - 10; http://dx.doi.org/10.1016/j.copbio.2011.03.002; PMID: 21190838
- Makino T, Skretas G, Kang TH, Georgiou G. Comprehensive engineering of Escherichia coli for enhanced expression of IgG antibodies. Metab Eng 2011; 13:241 - 51; http://dx.doi.org/10.1016/j.ymben.2010.11.002; PMID: 21130896
- Traxlmayr MW, Faissner M, Stadlmayr G, Hasenhindl C, Antes B, Rüker F, et al. Directed evolution of stabilized IgG1-Fc scaffolds by application of strong heat shock to libraries displayed on yeast. Biochim Biophys Acta 2012; 1824:542-9.
- Wozniak-Knopp G, Stadlmann J, Rüker F. Stabilisation of the Fc fragment of human IgG1 by engineered intradomain disulfide bonds. PLoS One 2012; 7:e30083; http://dx.doi.org/10.1371/journal.pone.0030083; PMID: 22272277
- Wozniak-Knopp G, Bartl S, Bauer A, Mostageer M, Woisetschläger M, Antes B, et al. Introducing antigen-binding sites in structural loops of immunoglobulin constant domains: Fc fragments with engineered HER2/neu-binding sites and antibody properties. Protein Eng Des Sel 2010; 23:289 - 97; http://dx.doi.org/10.1093/protein/gzq005; PMID: 20150180
- Traxlmayr MW, Wozniak-Knopp G, Antes B, Stadlmayr G, Rüker F, Obinger C. Integrin binding human antibody constant domains--probing the C-terminal structural loops for grafting the RGD motif. J Biotechnol 2011; 155:193 - 202; http://dx.doi.org/10.1016/j.jbiotec.2011.06.042; PMID: 21771617
- Nelson AL, Reichert JM. Development trends for therapeutic antibody fragments. Nat Biotechnol 2009; 27:331 - 7; http://dx.doi.org/10.1038/nbt0409-331; PMID: 19352366
- Holt LJ, Herring C, Jespers LS, Woolven BP, Tomlinson IM. Domain antibodies: proteins for therapy. Trends Biotechnol 2003; 21:484 - 90; http://dx.doi.org/10.1016/j.tibtech.2003.08.007; PMID: 14573361
- Holliger P, Hudson PJ. Engineered antibody fragments and the rise of single domains. Nat Biotechnol 2005; 23:1126 - 36; http://dx.doi.org/10.1038/nbt1142; PMID: 16151406
- Weiner LM, Carter P. Tunable antibodies. Nat Biotechnol 2005; 23:556 - 7; http://dx.doi.org/10.1038/nbt0505-556; PMID: 15877072
- Romer T, Leonhardt H, Rothbauer U. Engineering antibodies and proteins for molecular in vivo imaging. Curr Opin Biotechnol 2011; 22:882 - 7; http://dx.doi.org/10.1016/j.copbio.2011.06.007; PMID: 21708456
- Vaneycken I, D’huyvetter M, Hernot S, De Vos J, Xavier C, Devoogdt N, et al. Immuno-imaging using nanobodies. Curr Opin Biotechnol 2011; 22:1 - 5; http://dx.doi.org/10.1016/j.copbio.2011.06.009; PMID: 21190838
- Olafsen T, Wu AM. Antibody vectors for imaging. Semin Nucl Med 2010; 40:167 - 81; http://dx.doi.org/10.1053/j.semnuclmed.2009.12.005; PMID: 20350626
- Wu AM, Senter PD. Arming antibodies: prospects and challenges for immunoconjugates. Nat Biotechnol 2005; 23:1137 - 46; http://dx.doi.org/10.1038/nbt1141; PMID: 16151407
- Beck A, Wagner-Rousset E, Wurch T, Corvaia N. [Therapeutic antibodies and related products: choosing the right structure for success]. Med Sci (Paris) 2009; 25:1024 - 32; http://dx.doi.org/10.1051/medsci/200925121024; PMID: 20035674