Abstract
The Innovative Approaches in Anti-Cancer Monoclonal Antibodies meeting, held on March 20, 2012 in Lyon, was organized by Cancéropôle Lyon Auvergne-Rhône-Alps in partnership with the French competitiveness cluster Lyonbiopôle. CLARA is one of the seven cancer research clusters within France in charge of facilitating Translational Oncology Research by taking into account the objectives of the French National Cancer Plans I and II and, in coordination with the French National Cancer Institute and local authorities (mainly Grand Lyon, Rhône County and Rhône-Alpes Region), to perform economic development of research findings. The contribution of lectures by outstanding speakers as described in this report, the organization of two-round tables: “Antibody treatment in cancer: Unmet needs in solid tumors and hematological malignancies,” and “From chimeric to more than human antibodies,” together with face-to-face meetings, was shared by over 230 participants. The lectures provided an overview of the commercial pipeline of monoclonal antibody (mAb) therapeutics for cancer; discussion of the distinction between biosimilar, biobetter and next generation therapeutic antibodies for cancer; updates on obinutuzumab and the use of mAbs in lymphoma; and discussion of antibody-drug conjugates.
Morning Sessions
Cancéropôle Lyon Auvergne Rhône-Alpes and Innovative Approaches in Anticancer Monoclonal Antibodies
Petrus J. Pauwels and Charles Dumontet
The Cancéropôle Lyon Auvergne-Rhône-Alps (CLARA) is one of the seven cancer research clusters within France in charge of facilitating Translational Oncology Research by taking into account the objectives of the French National Cancer Plans I and II and, in coordination with the French National Cancer Institute and local authorities (mainly Grand Lyon, Rhône County and Rhône-Alpes Region), to perform economic development of research findings.
The CLARA organization went through three major phases. The first phase in 2005 consisted of CLARA’s major contribution to the development of several highly competitive inter-regional platforms that offer researchers a broad spectrum of platform activities, thereby generating the capacity to move easily from the lab bench to clinical testing. CLARA entered the second phase in 2007, by relying on inter-regional strengths as well as key scientific issues. Therefore, CLARA oriented its Oncology Research within three major priority axes: (1) Tumor Escape, Cell Plasticity and Targeted Therapies; (2) Infections and Cancer; and (3) Nanotechnologies, Imaging and Cancer. Each of these axes received a “with honors” evaluation by AERES (French Research and Higher Education Evaluation Agency) in 2011, principally based on the development of applications and their international recognition. This recognition has recently been further awarded in 2011 by three novel structures of excellence in oncology: (1) Lyon Integrated Cancer Research project (LYRIC, one of the first two projects labeled by the French National Cancer Institute, the other one being Institut Curie in Paris); (2) The Laboratory of Excellence selected by the General Investment Committee, the DEVweCAN project, to obtain better knowledge of embryonic mechanisms reactivated during tumor progression; and (3) Carnot Institute: consortium to accelerate innovation and transfer within the field of lymphoma (CALYM).
The third CLARA phase, reinforced in 2011, is a project-driven approach to speed up the development of applications arising from research results. The Cancéropôle CLARA has chosen to actively support Translational Oncology Research, as well as Clinical and Industrial Transfer. Numerous partnerships have been established between academic, clinical and industrial actors within the CLARA Proof of Concept program, which CLARA is co-financing and co-piloting while increasing awareness to transfer research through specific training sessions, detecting opportunities for research and development projects and partnerships, and favoring project maturation with the contribution of appropriate experts.
CLARA’s Proof-of-Concept portfolio contains 30 innovative projects with a global budget of €36 million €25 million have been invested by the industrial partner companies in addition to €11 million obtained from local authorities and the European Regional Development Fund. Today, one product, the mini-robot ViKY for laparoscopic surgery, is approved in the European Union and USA markets; four projects are in clinical development for the indication of sarcoma, melanoma, liver, breast or lung cancer; and 3 projects issued from nanotechnologies have been validated at the preclinical level.
Five major Proof-of-Concept projects use an immunotherapy approach. “IPROMAH: Proof of concept for new monoclonal antibodies for hematological malignancies” and “THERA8: Targeting cell surface protein for the development of therapeutic and diagnostic antibodies for colorectal cancer treatment” are both at the preclinical level. “SYNFRIZZ: Novel antibody therapy against synovial sarcoma” is currently in a Phase 1 first-in-class/first-in-man in clinical study. “GENIUSVAC-MEL4” is a therapeutic vaccine based on dendritic cells for the treatment of melanoma that is preparing its entry into the clinic. An immune-prognosis tool for breast and lung cancer patients under chemotherapy, “LYMPHOS1,” is under development by tracking TCR repertoire distortions to use lymphopenia to prevent deaths due to infections associated with chemotherapy.
Convinced of the importance of sharing knowledge and experiences in Oncology Research, the Cancéropôle CLARA mobilized its network by organizing the 7th Edition of its Scientific Forum on: Innovative Approaches in Anti-Cancer Monoclonal Antibodies on March 20, 2012 in partnership with the French competitiveness cluster Lyonbiopôle. The contribution of lectures by outstanding speakers as described in this report, the organization of two-round tables: “Antibody treatment in cancer: Unmet needs in solid tumors and hematological malignancies,” and “From chimeric to more than human antibodies,” together with face-to-face meetings, was shared by 232 participants who enjoyed the meeting. A second antibody meeting is scheduled to pursue the efforts to develop better antibodies against cancer.
Current Clinical Development of Anticancer mAbs
Janice M. Reichert
Monoclonal antibodies are well-suited as anticancer agents because they can be engineered for a variety of purposes. For example, they can be designed to carry drugs or other cytotoxic agents, bind two different antigens, or have enhanced cytotoxic activity compared with a natural antibody. These non-canonical versions comprise nearly half of the 165 anticancer antibodies currently undergoing evaluation in clinical studies. Although the promise surrounding mAbs as cancer drugs was recognized as early as the 1980s, interest in their development notably increased over the past decade, as evidenced by the rise in the number entering clinical study from ~10 per year in the late 1990s to over 30 in 2011. A total of 12 anticancer mAb products are approved in either the United States (US) or European Union (EU), and two products (nimotuzumab, mogamulizumab) are approved in countries other than the US or EU.Citation1 One mAb, pertuzumab, is undergoing regulatory review in the US and EU, and may be approved in the US by June 2012.
Most approved mAbs are canonical full-length IgG, but these molecules have well-recognized limitations, such as their large size and limited functionality. A number of variations on the canonical IgGs are currently undergoing clinical studies, including antibody-drug conjugates (ADCs), bispecific antibodies, engineered antibodies, and antibody fragments or domains. At least 25 ADCs are currently in clinical studies, with two (trastuzumab emtansine, inotuzumab ozogamicin) in Phase 3 studies.Citation2,Citation3 Trastuzumab emtansine, an IgG1 conjugated to the maytansinoid DM1, targets HER2 and is currently undergoing evaluation in three Phase 3 studies of patients with breast cancer. In the MARIANNE Phase 3 study, the efficacy and safety of the combination of trastuzumab emtansine with pertuzumab is being evaluated. Inotuzumab ozogamicin, an IgG4 conjugated to calicheamicin, targets CD22 and is undergoing evaluation as a combination with rituximab in relapsed/refractory aggressive non-Hodgkin lymphoma patients who are not candidates for intensive high-dose chemotherapy. A study of the mAb as monotherapy in adult patients with relapsed or refractory CD22-positive acute lymphoblastic leukemia is planned by not yet open to patient recruitment as of March 2012.
One bispecific antibody, catumaxomab, is approved in the EU but not the US, and there are at least 10 bispecific antibodies in clinical studies. Of those in the clinical pipeline, blinatumomab has advanced the furthest and has been or is now undergoing evaluation in a total of six clinical studies (one Phase 1, one Phase 1/2, and four Phase 2 studies). Blinatumomab comprises tandem single-chain variable fragments that target CD19 on tumor cells and CD3 on T cells, and thereby acts as a bridge between T cells and tumor cells. The Phase 2 studies are in patients with acute lymphoblastic leukemia.
Engineering of antibodies can be used to tailor functionality. For example, protein engineering in the Fc portion of antibodies can affect the binding to FcRn and extend half-life or glyco-engineering can be done to increase ADCC activity by removing fucose. There are at least 15 antibodies with such engineering in the clinical pipeline. Examples of these are obinutuzumab (GA101), which is a glyco-engineered, anti-CD20 mAb. The mAb most recently approved, mogamulizumab, is a defucosylated anti-CCR4 IgG1 approved in Japan in March 2012.
The antibody fragments, including Fab, single-chain variable fragments and domain antibodies, are commonly modified to increase functionality. At least 16 of these molecules are in clinical study, with half conjugated to cytotoxins or radiolabeled and six (38%) bispecific. The most advanced in the clinic is naptumomab estafenatox (ABR-217620), which is undergoing evaluation in a Phase 2/3 study combined with interferon-α compared with interferon-α alone in patients with advanced renal cell carcinoma.
Overall, the prospects for the development of mAbs as anticancer agents are bright. The increase in the number in clinical studies during the 2000s suggests that the number that will be approved in the next 5–10 y will also increase, given that the average clinical development phase length for anticancer mAbs is ~7.5 y.Citation4 The variety of approaches, e.g., use of ADCs, bispecific antibodies, engineered antibodies and antibody fragments or domains, also holds promise for development of new efficacious treatments.
Biosimilar, Biobetter and Next Generation Therapeutic Antibodies in Oncology
Alain Beck, Liliane Goetsch and Nathalie Corvaïa
Monoclonal antibodies (mAbs) and related products that have been approved in the past decade in oncology show that research laboratories have worked on the diversification and fine tailoring of structures. Following the success of the first-generation blockbusters, second-generation mAbs were recently approved. In addition, many third-generation antibodies and related structures designed to trigger different mechanisms of action simultaneously, such as targeting growth factors, inhibiting angiogenesis and restoring apoptosis, and associated with enhanced or silenced effector functions, e.g., antibody-dependent cell-mediated cytotoxicity (ADCC) or complement-dependent cytotoxicity (CDC) are being investigated in clinical trials. Among the challenges to be faced in the next 10 y are the identification and validation of new targets in oncology, addressing the resistance to current drug treatments and understanding target cross-talk and regulation. In the meantime, efforts must be made to decrease the costs of these biopharmaceuticals, e.g., by optimizing the design of more homogeneous and stable IgGs (OptimAbs). The availability of regulatory pathways to register biosimilar antibodies in Europe and the United States (US) might be another way to decrease healthcare costs and to generalize the use of mAbs, as well as biobetter mAbs.
The research and development of mAbs is a fast-expanding field. In the past 30 y, more than 40 immunoglobulins (IgGs)Citation1 and their derivatives (e.g., Fc fusion proteins)Citation2 have been approved for use in various indications. Nearly half of them have been approved in oncology. Here, we discuss strategies to select tumor-associated antigen targets based on previous clinical or experimental validation or on functional approaches; strategies to optimize the antibody structure and to design related or new structures with additional functions; and challenges to bring more affordable treatments to the most appropriate patient population screened for validated biomarkers.Citation3
Strategies to select the best therapeutic antigen targets. Antigen target selection can be classified in broad terms into two major approaches. The first approach deals with the development of antibodies directed against so-called ‘validated targets’, either because prior antibodies have clearly demonstrated proof of activity in humans or because a vast literature exists on the importance of these targets for the disease mechanism in both in vitro and in vivo pharmacological models. Basically, the strategy consists of developing new generations of antibodies specific for the same antigens, but that target other epitopes (second-generation antibodies) or trigger different mechanisms of action (third-generation antibodies), or antibodies that are even specific for the same epitopes, but with improved properties. This validated approach has a high probability of success, but the scientific field around this class of target proteins is often crowded and the freedom to operate is reduced. By contrast, one can identify novel or less well-studied target proteins that confer particular functions to cells that might be involved in pathogenic disorders. This second ‘functional approach’, in which antibodies are selected based on a functional screen and the targets they bind are then identified using proteomic or cell-based approaches (reverse pharmacology), for example, is associated with greater potential for innovation and intellectual property rights, but more development risk.
Clinically validated targets
‘Blockbuster’ antibodies such as rituximab, trastuzumab, cetuximab and bevacizumab, directed against now highly clinically-validated targets such as CD20, human epidermal growth factor receptor 2 (HER2), epidermal growth factor receptor (EGFR) and vascular endothelial growth factor (VEGF)-A, respectively, are tremendous success stories. Second-generation antibodies directed against these same antigens, but with improved variable domains to decrease immunogenicity or that target distinct epitopes, with higher or lower affinity for their antigens or with different antibody formats (such as panitumumab and ofatumumab) have been developed. In addition, third-generation antibodies targeting different epitopes, triggering other mechanisms of action and often engineered for improved Fc-associated immune functions or half-life, have reached Phase 1 to 3 clinical studies. As an illustration, the third generation CD20-specific antibody obinutuzumab (GA101) is less immunogenic than rituximab, has a different mechanism of action and is glyco-engineered to trigger increased cytotoxicity.
Experimentally validated targets
Diversification and validation of novel targets in oncology is a challenging issue because the causes of malignacies are often multifactorial, redundant and frequently poorly undestood. In addition, patients are becoming resistant to current cancer treatments, leading to the expression of new molecules (potential targets) that drive the tumor growth. Another difficulty in oncology is to determine the best combination of drugs and drug targets, which is not always predictable from pre-clinical studies, as shown by the adverse events (such as skin toxicity, diarrhea and infection) reported for patients with colorectal cancer who were administered both EGFR- and VEGF-A-specific antibodies. Target selection will also require an understanding of cooperative signaling involving, for example, growth factor receptor heterodimers or integrin cross-talk with growth factors.
A source of potential new experimentally-validated targets in oncology is, for example, the abundant literature documenting the important role of tyrosine kinase receptors in malignancies. As an example, insulin-like growth factor 1 receptor (IGF-1R) was proposed more than 20 y ago to be an interesting oncoprotein, but it became possible only recently for patients to benefit from experimental treatments involving IGF-1R-specific antibodies. The development of anti-IGF-1R mAbs is the focus of many pharmaceutical companies, with 12 identified antibodies at the preclinical or clinical stage of development. All these antibodies are specific, high affinity binders for IGF-1R. They block IGF-1R/IGF-1 interaction, inhibit cell proliferation and induce apoptosis by blocking IGF-1R signaling and by internalizing and degrading IGF-1R. Centre d’Immunologie Pierre Fabre (CIPF) used the targeted approach successfully to select IGF-1R as a target. IGF-1R is a tyrosine kinase receptor with 70% homology to the insulin recep¬tor. The target is overexpressed in various tumors where it is mito¬genic, required for maintenance of the transformed phenotype and protects tumor cells from apoptosis. CIPF developed the human¬ized anti-IGF-1R IgG1 dalotuzumab (h7C10 or MK-0646), which was licensed to Merck in 2004 and is currently in Phase 2 studies.
CIPF’s targeted strategy was also used to develop candidates targeting the c-Met receptor, which is a tyrosine kinase recep¬tor, yielding the identification of mAb 224G11 licensed to Abbott in 2010. The ligand of this receptor is hepatocyte growth factor/scatter factor. cMet is overex¬pressed in tumor tissue, where it has anti-apoptotic activity and is involved in transformation/mitogenesis and invasion/metastasis.
Functionally validated targets
A more challenging approach is to select mAbs with a defined biological effect on tumor cells (such as the inhibition of proliferation or apoptosis) and to identify the recognized antigens by proteomics and alternative techniques. These so-called functional approaches (or reverse pharmacology) allow the discovery of novel and original cell surface antigens, but these new targets need extensive and careful clinical validation, which represents a high development risk and involves longer research timelines before entering into the clinic. Using this type of approach, the identification of pathways that control refractoriness or resistance to current therapies should be better investigated to uncover putative targets that might translate into novel and efficient therapies. For example, it has been reported that treatment with EGFR inhibitors leads to c-Met overexpression. Similarly, resistance to HER2-specific antibodies has been reported to be related to IGF-1R overexpression.
CIPF’s functional strategy has been used to identify JAM-A as a potential anti-tumor target. The literature describes JAM-A as a constituent of the tight junctions of epithelium and endothelial cells that has interaction with lymphocyte function-associated antigen 1 and fibroblast growth factor β, and is a receptor for reovirus, but there is very little information on the role of JAM-A in oncology. CIPF has produced an anti-JAM-A mAb, and studies have indicated that the mAb, 6F4, inhibits tumor growth in vivo through inhibition of cell proliferation and downregulation or shedding of the target. The mAb has been humanized and pretoxicological studies in monkeys have demontrated the safety of the humanized mAb.
Strategies to optimize the antibody IgG structure. Detailed knowledge of antibody structure and activity now allows researchers to engineer primary antibodies on a more rational basis. This can yield more homogeneous and stable molecules with additional properties such as increased cytotoxicity or dual targeting, as well as IgG-related structures with additional functions and specificities.
Improving pharmaceutical properties (OptimAbs)
Most approved antibodies are chimeric, humanized or human IgGs with similar constant domains. Numerous studies looking at the structure–function relationships of these antibodies have been published in the past five years with the aim of identifying antibody micro-variants and investigating the impact of these variants on antigen binding, stability, pharmacokinetics and pharmacodynamics. This knowledge is now being used at the lead optimization stages to increase homogeneity and mitigate the chemistry, manufacture and control (CMC) liabilities of pre-clinical antibody candidates by genetic engineering. The removal by mutation of instability or aggregation hot spots in the antibody complementarity-determining regions (CDRs), and the use of hinge-stabilized or aglycosylated IgG4, are just a few examples of antibodies with improved pharmaceutical properties (e.g., decreased heterogeneity) that are currently in development.
To optimize the structure of the next generation of therapeutic antibodies, the CIPF has set up the “OptimAbs” network to transform research antibody leads into optimized clinical candidates in a collaborative project that includes three partners (Promise Advanced Proteomics, CNRS-LSMBO, CNRS-AGIM). To achieve this goal, antibodies/antigens complexes are investigated by emergent mass spectrometry (MS) methods in biological contexts of increasing complexity: in vitro (native MS and crystallography), in sera (quantitative MS), in cells and tumor tissues (Imaging MS). These innovative analytical platforms will allow optimization of the structure of new antibodies targeting original tumor-associated antigens and will facilitate their pharmaceutical development.
Improving antibody functions
The variable domain (Fv) of an antibody is responsible for interactions with antigens and dictates essential properties such as binding affinity and target specificity. The origin of the Fv can be diverse, such as hybridomas, human antibody libraries, rodents with a human antibody repertoire, or primatized/humanized antibodies from various species. Affinity maturation allows the binding affinity of the Fv to be improved or target selectivity to be modulated. The constant domain (Fc) of an antibody is responsible for interactions with immune cells, and the associated properties of the Fc can also be modulated by engineering at several levels, such as altering the glycosylation status to modulate anti- and pro-inflammatory properties; modulation of ADCC by site-directed mutagenesis to modulate binding to Fc receptors; increasing the serum half-life by Fc engineering to increase binding to the neonatal Fc receptor (FcRn), which prevents IgG degradation; and increasing complement activation by isotype chimerism.
Second- and third-generation antibody-drug conjugates
Combining the cytotoxicity of natural or synthetic highly potent anti-neoplastic agents with mAbs conjugated by blood-stable optimized linkers is the key strategy of a new generation of antibody drug conjugates (ADCs). The overall development of ADCs in oncology takes longer and is more complex than for naked antibodies because many more parameters need to be fine-tuned. As a sign of technological advancement, one ADC was approved in 2011 (brentuximab vedotin) in the US and two have reached the late clinical development stages and shown encouraging therapeutic effects against both solid tumors and hematological malignancies (trastuzumab emtansine and inotuzumab ozogamicin).Citation4 In addition, more than 20 ADCs are being investigated in earlier clinical trials, including glembatumumab vedotin and lorvotuzumab mertansine, which are in Phase 2 trials. The US Food and Drug Administration (FDA) and European Medicine Agency (EMA) recently granted orphan drug designation for the latter ADC. Altogether these recent encouraging successes show that the next generation of ADCs has come of age. Renewed promises and hopes emerged, as was the case 10 y ago.Citation5
Strategies to provide more affordable treatments for patients. Antibodies are a successful class of therapeutics, but many treaments remain costly, which may limit their use, particularly when synergistic combinations of IgGs are required.
Biosimilar and biobetter antibodies On November 18, 2010, the EMA released a draft guideline on similar medicinal products containing mAbs, following a workshop organized by the agency in London July 2, 2009.Citation6 The guideline discusses relevant animal model, non-clinical and clinical studies that are recommended to establish the similarity and the safety of a biosimilar compared with an originator mAb approved in the European Union (EU). Legislation establishing an abbreviated approval pathway for biosimilars was signed into law in March 2010 in the US and the FDA is currently working to define the rules for its implementation. Biosimilars, follow-on biologics, biogenerics, biobetters, biosuperiors, second and third generation antibodies are terms used sometimes in a confusing way.
Biosimilar antibodies are “generic” version of “innovator” (or “originator”) antibodies with the same amino acid sequence, but produced from different clones and manufacturing processes.Citation7 As a consequence, biosimilar mAbs may include possible differences in glycosylation and other microvariations such as charge variants that may affect quality, safety and potency. Biosimilars are known as follow-on biologics in the US, but this term is misleading because it could also refer to second- and third-generation antibodies (defined and illustrated below) and should be avoided. In contrast to the low-cost generic versions of small molecules that are off patent, it is currently not possible to produce exact copies of large proteins and glycoproteins such as antibodies owing to their structural complexity, and so the term biogeneric should also be avoided. Nevertheless, tremendous progress has been made in bioproduction and analytical sciences,Citation8 and it is now possible to produce proteins and glycoproteins that are highly similar to reference products. The EMA has pioneered the regulatory framework for approval of these products. Since 2005, EMA has initiated regulatory pathways for biosimilar products, resulting to date in marketing authorization in Europe for 14 recombinant drugs encompassing 3 product classes (human growth hormone, granulocyte colony-stimulating factor and erythropoietins). Specific guidelines are also available in Europe for biosimilar insulin, interferon and low molecular weight heparins.
Bio-better antibodies are antibodies that target the same validated epitope as a marketed antibody, but have been engineered to have improved properties, e.g., optimized glycosylation profiles to enhance effector functions or an engineered Fc domain to increase the serum half-life. Such “Me better” antibodies with controlled and optimized glycosylation have been obtained in glyco-engineered CHO cells or yeast strains, e.g., copies of rituximab and trastuzumab amino acid sequences with afucosylated glycoforms that result in a 40- to 100-fold increase in ADCC, or with increased plasmatic half-life, e.g., copies of rituximab, trastuzumab, bevacizumab that have a mutation of two or three amino acids in the Fc domain resulting in extended pharmacokinetics. In these cases, the cost of treatment is expected to decrease because of the lower cost of the products, less frequent administration regimens or lower dosages.
Cetuximab, a chimeric mouse-human IgG1 targeting EGFR, is approved for use in the EU and US as a treatment for colorectal cancer and squamous cell carcinoma of the head and neck. A high prevalence of hypersensitivity reactions to cetuximab has been reported in some areas of the US. Among 76 cetuximab-treated subjects, 25 had a hypersensitivity reaction to the drug. The IgE antibodies thought to be responsible for the reaction were shown to be specific for an oligosaccharide, galactose-α-1,3-galactose, that is present on the Fab portion of the cetuximab heavy chain when the molecule is produced in the murine SP2/0 cell line used for commercial manufacturing, but not in the CHO cells used as control. The mechanism underlying a hypersensitivity reaction to cetuximab involves pre-existing IgE antibodies that target an oligosaccharide present on the recombinant molecule produced in the SP2/0 cell line. These results have implications for evaluating risks associated with antibody-based therapeutics, and for understanding the relevance of IgE antibodies specific for post-translational modifications of natural and recombinant molecules. The second N-glycosylation site in the Fab portion on heavy chain Asn88 of cetuximab is of prime importance. For the marketed version of cetuximab produced in SP2/0 cells, 21 different glycoforms were identified with ~30% capped by at least one α-1,3-galactose residue, 12% capped by a N-glycolylneuraminic acid (NGNA) residue, and traces of oligomannose. Importantly, both α-1,3-galactose and NGNA were found only in the Fab moieties, rather than the Fc fragment for which only typical IgG G0F, G1F and G2F glycoforms were identified. In cases of cetuximab-induced anaphylaxis, pre-existing IgEs specific for this α-1,3-galactose epitope were detected in patients treated with cetuximab. Using a solid phase immunoassay, these IgEs were found to bind to SP2/0-produced cetuximab and F(ab’)2 fragment, but not to the Fc fragment. Interestingly, no IgE immunoreactivity was found against a CHO-produced version of cetuximab (CHO-C225). A bio-better version of cetuximab produced in CHO cells is currently in development in China.
Engineering Antibodies to Improve Their Anti-Tumoral Efficacy: 1. GA101, A Type II Glycoengineered CD20 Antibody; 2. CrossMab Approach for the Generation of Human Bispecific Bivalent Antibodies
Christian Klein
Various approaches to antibody engineering are applied within Roche Pharma Research and Early Development (pRED) to generate differentiated cancer therapies. The introduction of rituximab (Mabthera®, Rituxan®) in combination with chemotherapy for the treatment of non-Hodgkin lymphoma (NHL) and B cell chronic lymphocytic leukemia (B-CLL) has revolutionized the treatment of the diseases. The exact mechanism of action of rituximab remains unconfirmed, but like other therapeutic antibodies is thought to involve a combination of complement dependent cytotoxicity (CDC), apoptosis or direct cell death induction and antibody dependent cellular cytotoxicity (ADCC). Obinutuzumab (GA101), a novel humanized and glycoengineered Type II CD20 monoclonal antibody,Citation1 was designed to differentiate in key features from the type I CD20 antibody rituximab as current standard of care for the therapy of NHL. GA101 has two distinct features: (1) Enhanced direct cell death induction with a concomitant reduction of CDC by virtue of being a type II antibody. The Type II character of GA101 is most likely related to the particular epitope recognized on the large extracellular loop of CD20 by GA101 as elucidated by X-ray crystallography and may have to do with the particular geometry of CD20 recognition; and (2) Enhanced ADCC as a consequence of application of the GlycoMab technology that results in enhanced affinity for the FcgRIIIa receptor on immune effector cells such as NK cells and macrophages/monocytes.Citation1-Citation5 Enhanced ADCC may be an important mechanism, in particular in the 80–85% of patients who are carriers of the low-affinity FcγRIIIa allele.
Recent data have indicated that cell death induced by the type II CD20 antibody GA101, as well as HLA-DR and other lymphocyte antibodies, is related to homotypic adhesion and represents a novel type of actin dependent and lysosome-induced cell death mediated through a novel reactive oxygen species-dependent pathway.Citation3,Citation6,Citation7 In line with these findings, confocal time lapsed microscopy demonstrated that cell death induction is independent of mechanical disruption. Comparison of GA101 with rituximab, as well as the Type I CD20 antibody ofatumumab (Arzerra®) that was recently approved for the treatment of patients with B-CLL refractory to fludarabine and alemtuzumab, in various in vitro assays and in vivo xenograft experiments show difference in the antibodies. For example, in vivo the properties of GA101 result in superior anti-tumoral efficacy of obinutuzumab compared with rituximab and ofatumumab (both Type 1 mAbs). Obinutzumab has been studied in several Phase 1 and Phase 2 clinical studies in NHL and B-CLL patients,Citation8,Citation9 and is currently in pivotal clinical studies for the treatment of B-CLL, indolent NHL and diffuse large B cell lymphoma in direct comparison with rituximab based regimens. These head-to-head clinical studies will be essential to demonstrate whether the findings from preclinical and Phase 1/2 clinical studies translate into a clinical benefit in large randomized clinical studies.
During recent years, bispecific antibodies have experienced a renaissance and many investigators and companies are now working on the development of these agents; most of them in the preclinical stage. A novel technology is being developed at Roche pRED for the generation of bispecific bivalent IgG antibodies, also known as CrossMab technology.Citation10 The key challenge in the production of bispecific bivalent IgG antibodies comes from the requirement to express four different antibody chains (two heavy and light chains each) in one expression cell line to generate one bispecific bivalent antibody. The isolation of the desired bispecific antibody out of the mixture of the various antibodies formed during this process made the manufacturing of bispecific bivalent IgG antibodies almost impossible for many years.
Different approaches to overcome this so-called chain association problem have been described, e.g., the knob-into-holes technology to enforce correct heavy chain association.Citation11 However, this approach still did not overcome the issue of random light chain association and only recently Schaefer and colleaguesCitation10 proposed and described a novel and generic approach to enforce correct light chain association by domain exchange within the Fab domain of one half of the bispecific antibody in combination with the knob-into-holes technology. Basically, according to this approach the whole Fab region, the VH-VL or the CH1-CL domains in one arm of the antibody are exchanged by domain crossover between the heavy and light chain Fab domains. This approach enforces correct light chain association and reduces mispairing by a minimal change in immunoglobulin domain arrangement.
As expected from theoretical considerations the CrossMabCH1-CL with the crossover of only the CH1-CL domains showed the best side product profile and was selected to generate a bispecific bivalent antibody targeting vascular endothelical growth factor A (VEGF-A) and Angiopoietin-2 based on bevacizumab and LC06 as parental antibodies with the goal to block these two angiogenic factors simultaneously.Citation10 The Ang-2-VEGF CrossMabCH1-CL showed potent tumor growth inhibition and anti-angiogenic activity in xenograft tumor models at least as good as the combination of the two monotherapies and resulted in a complete shutdown of angiogenesis in a VEGF-induced cornea pocket assay. Moreover, it could be produced in CHO cells with volumetric productivities in the range of several grams per liter, which is similar to standard IgG processes, with thermodynamic and long-term stability comparable to conventional IgG antibodies and with IgG-like pharmacokinetic properties in non-human primates.
2012 Update on Monoclonal Antibodies in Lymphoma
Bertrand Coiffier
The introduction of monoclonal antibodies (mAbs) in the therapy of lymphomas was a major improvement in the treatment of these patients. The first anticancer mAb registered was rituximab (Rituxan®), a chimeric anti-CD20 antibody, that demonstrated activity in relapsing follicular lymphoma in a Phase 1 study.Citation1 Rituximab was registered for the treatment of follicular lymphoma, then diffuse large B cell lymphoma in combination with chemotherapy, and chronic lymphocytic leukemia (CLL).Citation2-Citation4 Rituximab has secondarily demonstrated efficacy in other B cell lymphomas, as well as in autoimmune cytopenias, and B cell-driven diseases like rheumatoid arthritis.Citation5-Citation7 Because of this efficacy, other mAbs targeting the CD20 antigen or other lymphoid antigens were developed for the treatment of lymphomas. The benefits of such therapies, particularly focusing on the pitfalls of drug development for mAb in lymphoma are summarized here.
Lymphoma will serve as an example of what can be done with mAbs, but other cancers or diseases might have been chosen because the use of mAbs developed quickly after the demonstration of its possibility in lymphomas.
The different monoclonal antibodies. Several types of mAb exist, including mouse anti-human antibodies, chimeric, humanized and fully human antibodies (regarding the percentage of persisting mouse protein in the final molecule). Rituximab is a chimeric mouse/human molecule with ~20% of the original mouse Fab fraction persisting. This was not really associated with clinical reaction or development of anti-rituximab antibodies in the setting of lymphoma where some immunodeficiency exists related to the lymphoma or the previous therapies, but the presence of non-human sequence may cause difficulties when rituximab is used as a treatment in autoimmune diseases.Citation8 For the moment, there has been no difference in mAbs anti-lymphoma activity for molecules with varying percentages of mouse protein in the molecules.
Two subgroups of mAb exist regarding their mechanism of action, the naked antibodies where the antibody have a direct immunologic effect or trigger intracellular direct effects on homeostasis of lymphoma cells and the conjugated antibodies where the antibody acts as a transporter to bring to the cell a toxic product, isotope, toxin or drug. In this latter case, after the antibody has targeted the antigen, it must be internalized to produce its effects.Citation9
The number of antigens at the surface of normal or pathologic lymphocytes that can be targeted is quite important, with more than 30 antigens having been described so far. The most commonly targeted are CD19, CD20, CD22, and CD30, but this is not limited and any new mAb against a previously untargeted antigen may bring a new site to attention.Citation10
Mechanisms of action. The mechanisms of action of rituximab are described here as an example, and the mechanisms of actions for other mAb are described later.
The mechanisms of action of rituximab include complement-dependent cytotoxicity (CDC), antibody-dependent cell-mediated cytotoxicity (ADCC) and other Fcγ receptor-mediated mechanisms, and induction of apoptosis ().Citation11 It is difficult to evaluate the contribution of each of these mechanisms when treating a patient because all of them have been described in vitro on cell lines, fresh lymphoma cells, or animals. Moreover, these mechanisms are probably different according to the tumor type.Citation11 Binding of type I anti-CD20 antibodies such as rituximab to CD20 induces rapid translocation of the complex to lipid rafts, signal transduction zones that enables the co-localization of receptors and signal effectors. This is not observed in type II antibodies such as obinutuzumab. This activity can be modulated by patient-related parameters such as gender, polymorphism of FCGR3A, genetic heterogeneity of CD11b, or natural killer cell effectors ().Citation11
Figure 1. Mechanism of rituximab action, resistance, and causes of variability of response. (1) FCGR3A-158FV genotype affects the affinity of rituximab for human IgG1. (2) Rituximab binds more strongly to homozygous FcγRIIIa-158V effector cells than to FcγRIIIa-158F carrier effector cells, resulting in significantly different response rates to rituximab. (3) Polymorphism located in the C3-interacting site of CR3/CD11b has been shown to be associated with PFS in patients with follicular lymphoma treated with rituximab alone. (4) NK cell target recognition depends mainly on the surveillance of HLA class I molecules by B-cells interacting with KIRs on NK cells modulating ADCC and CR3-ADCC. (5) Apoptosis as an effector mechanism of rituximab has been shown to be modulated, resulting in in vitro resistance to both rituximab and chemotherapy. (6) CD20 expression influences CDC induced by rituximab. (7) A rare cause of rituximab resistance may be mutations of the CD20 gene, resulting in reduced CD20 expression or affinity for rituximab. Antitumor effects of rituximab are dependent on translocation of antibody-bound receptor to lipid rafts in the plasma membrane. All changes in the cholesterol content of the plasma membrane can thus affect the lipid-raft–dependent activity of rituximab. Reprinted with permission from Cartron G et al. Clin Cancer Res 2011; 17:19–30.
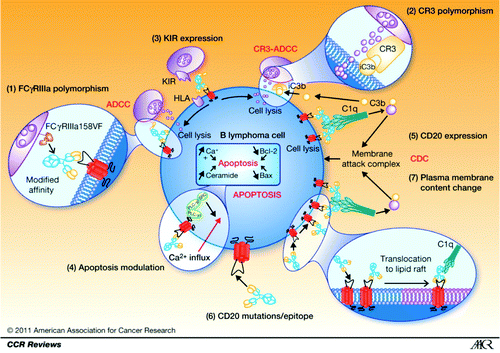
The importance of these parameters can change from one antibody to another, and according to the targeted antigen. Pharmacokinetics or pharmacodynamics can be different. For example, veltuzumab, a humanized anti-CD20 antibody, needs smaller dose for activity and thus can be injected subcutaneously.Citation12 Ofatumumab, another fully humanized anti-CD20 mAb, targets a different epitope than rituximab.Citation13
In case of conjugated antibodies, the antibody serves as a transporter of the active product to the pathologic cells and it does not need to have activity by itself. Active products can be a radionuclide (131I or 90Y), a toxin (immunotoxins), or a small drug (antibody-drug conjugate or ADC).Citation9 The three parts of the drug are important: the specificity of the antibody, the linker, and the active drug molecule. The linker is particularly important for immunotoxins or antibody-drug conjugates because it must be sufficiently stable not to release the active molecule during infusion and in plasma, but sufficiently labile to release it once phagocyted by the tumor cell.
Rituximab. The benefit of adding rituximab to chemotherapy has been demonstrated in several randomized studies for follicular lymphoma, diffuse large B cell lymphoma, and CLL.Citation3,Citation4,Citation14 This benefit has also been demonstrated in maintenance for follicular lymphoma and mantle cell lymphoma.Citation15,Citation16 For the less frequent B cell lymphomas, Phase 2 studies have shown that a benefit probably exists, but it has not been fully demonstrated for the moment. This benefit was gained without an increase in toxicity. In these randomized studies, the benefit is ~15–20% for progression-free survival and ~10–20% for overall survival.
Because of these results, rituximab plus chemotherapy is the standard regimen to treat patients with B cell malignancies in first line. Chemotherapy regimen changes according to the lymphoma subtypes, but R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone) is certainly the most used because of its very good safety profile. These studies have set a very high standard even if some progress is needed for the most aggressive patients.
Other anti-CD20 mAb. Besides the biosimilars of rituximab, which are not yet registered in Europe or the United States, there are several new molecules that target CD20. Veltuzumab and ocralizumab are humanized; ofatumumab targets different epitopes of CD20; obinutuzumab and ublituximab are engineered mAb.Citation10 None of these mAbs have yet been tested in comparison to rituximab, so their potential benefit over rituximab is not known. They have demonstrated some advantages over rituximab in in vitro tests. Ofatumumab might have a better efficacy in CLL patients.Citation17 Obinutuzumab has a greater in vitro activity, particularly for ADCC and induction of direct cell death.Citation18,Citation19 Randomized studies are ongoing for these drugs.
Other anti-B cell antibodies. A number of companies are developing mAbs that target other antigens on B cells., including MEDI-551 (MedImmune), an anti-CD19 mAb;Citation20 and epratuzumab (Immunomedics), an anti-CD22 mAb;Citation21 dacetuzumab (Seattle Genetics), an anti-CD40 mAb.Citation22 However, these mAbs are in Phase 1 or Phase 2 and their potential has not been fully demonstrated yet.
Alemtuzumab (Campath®) targets CD52, a pan-lymphocyte antigen that is present on B and T cells. It is registered for CLL patients, but has demonstrated low activity in lymphoma, mainly in cutaneous T cell lymphomas.Citation23-Citation25
mAb targeting T cells. Several mAb have been developed for T cell lymphoma, but none has yet succeeded in demonstrating good efficacy, and some were associated with severe toxicity. Siplizumab targets CD2 antigen and was associated with severe opportunistic infections.Citation26 Two companies have developed an anti-CD30 mAb without success.Citation27,Citation28 Zanolimumab was associated with some responses at high-dose, but was licensed to different companies every 2–3 y and was not well developed.Citation29,Citation30 Mogamulizumab (POTELIGEO®) was recently approved in Japan as a treatment for patients with relapsed or refractory CCR4-positive T cell leukemia-lymphoma, but only study results from its early development have been published.Citation31 Alemtuzumab has also been used in T cell lymphoma with some responses in CTCL.Citation32
This list shows clearly that generally anti-T cell mAbs have not been well-developed, or they had no real anti-lymphoma activity, and a number of antigens on T cells have not proven to be good targets.
Conjugated antibodies. As said previously, three groups of conjugated mAb exist: radionuclides, immunotoxins, and ADC. The two radionuclides, 131I-tositumomab (Bexxar®) and 90Y-ibritumomab tiuxetan (Zevalin®) have not really succeeded in finding their place in the treatment of B cell lymphomas. This is mostly secondary to a poor development plan without randomized study.Citation33 Immunotoxins and ADC use an antibody to target an antigen and bring the toxin or the drug into the cell, as discussed by Beverly Teicher in the final presentation summary of this meeting report.
Conclusion. MAbs have created a breakthrough in the treatment of several cancers, particularly B cell lymphomas. More progresses will be reached with the refinement of technology and in vitro engineering for naked or conjugated antibodies. Conjugated antibodies will probably allow what was still only a dream not so long ago: targeting only pathological cells with anti-cancer drugs.
Afternoon Sessions
The afternoon session started with two concurrent Round Tables, followed by the plenary talk featuring Dr. Beverly Teicher from the US National Cancer Institute. The topic of one Round Table was “Antibody Treatment in Cancer: Unmet Needs in Solid Tumors and Hematological Malignancies,” with moderator: Charles Dumontet and the panelists were Oliver Tredan (Léon Bérard Cancer Center), Stéphane Dalle (South Lyon Hospital, CRCL, UCBL) and Jérôme Honnorat (CRNL,Inserm/CNRS). The other Round Table was dedicated to discussion on “From Chimeric to more than Human Antibodies,” with moderator Christian Klein (Roche Glycart AG) and Chairpersons Marie-Paule Lefranc (IMGT, Montpellier) and Liliane Goetsch (CIPF, Saint Julien en Genevois). The panelists were Hervé Watier (Labex MabImprove/GDR ACCITH), Majid Mehtali (Vivalis), Claudine Vermot-Desroches, IDD Biotech), Jérôme Tiollier (Innate Pharma).
Antibody Drug Conjugates
Beverly A. Teicher
Antibody conjugates are a diverse class of therapeutics consisting of a cytotoxic agent linked covalently to an antibody or antibody fragment directed toward a specific cell surface target expressed by tumor cells. The main approaches using antibodies to target cytotoxic agents to malignant cells: antibody-protein toxin (or antibody fragment-protein toxin fusion) conjugates, antibody-chelated radionuclide conjugates, antibody-small-molecule drug conjugates (ADCs). Although ADCs are highly selective, they are quite inefficient in delivering drugs to tumor and thus require very potent cytotoxins as the drug component.Citation1,Citation2 The drugs used on most ADCs in clinical trials are dolastatin 10 analog, monomethylauristatin E or F, maystansine analogs, calicheamicin, or duocarmycin derivatives. The targets for these potent drugs are either tubulin or DNA.Citation2,Citation3 At least 25 ADCs are currently in clinical trials. Brentuximab vedotin (SGN-35) was recently approved by the US Food and Drug Administration as a treatment of Hodgkin lymphoma and anaplastic large cell lymphoma.Citation4 The next ADC likely to enter regulatory review is trastuzumab emtansine, which is a treatment of HER2+ breast cancer.Citation5 Preclinical pharmacokinetic and pharmacodynamic assays indicate that there are several drug metabolites present in xenograft tumor tissue that reflect lysosomal degradation of the ADC and that the role of the cell surface target in vivo can be proven by negating the ADC therapeutic activity by administration of excess naked antibody.Citation6 Several antibody formats including bispecific antibodies, minibodies, Fabs, diabodies, scFv fragments are under investigation as methods for delivery of radionuclides for imaging and for radiation therapy.Citation7 Finally, antibody fusion proteins continue to be explored. Overcoming immunogenicity of the fusion proteins remains an important goal. The anti-CD22 recombinant immunotoxin moxetumomab pasudotox, which includes the protein toxin PE38, is currently in clinical study. However, research is continuing to develop even less immunogenic variants of PE38 that retain potent cytotoxicity.Citation8
Identifying and validating cell surface proteins as suitable for ADC targets requires rigorous attention to tumor and normal tissue expression of the target protein.Citation1,Citation2 The protein endosialin, which is expression by sarcoma, illustrates the steps needed to determine whether an ADC targeting endosialin may be worthwhile pursuing.Citation9,Citation10 Endosialin is primarily expressed during embryo development and is found in rare cells in some adult tissues. As a beginning, antibodies were raised to endosialin. The expression of endosialin was determine by flow cytometry in about 45 human sarcoma lines and several types of normal cells in culture. About 50% of the sarcoma lines expressed endosialin, some expressed very high levels of the protein on the cell surface. When an antibody to endosialin bound to the target protein on the cell surface, the antigen-antibody complex was internalized into the cell. In formalin-fixed paraffin-embedded human clinical specimens, about 100 carcinomas and 100 sarcomas, endosialin was found in the vasculature (pericytes) of about 33% of the carcinomas, but was found in the vasculature of 78% and 51% of the malignant cells of the sarcomas, as well as about 25% of the tumor-associated fibroblasts.Citation9,Citation10 In frozen human clinical specimens, 93% of carcinomas had detectable endosialin in the vasculature. In sarcomas, 100% of the specimens had endosialin in the vasculature, 50% in the malignant cells and 100% in the tumor associated fibroblasts. The expression of endosialin in the sarcomas occurred across all sarcoma subtypes tested and across all age groups of patients. A model ADC was tested and found to be cytotoxic only to sarcoma cells that expressed endosialin in cell culture. When grown as subcutaneous xenograft tumors in nude mice, sarcoma cell lines retained expression of endosialin; however, the intensity of expression varied from line to line and was not always identical to expression in culture. Metastatic sarcoma produced in nude mice by injection the sarcoma cell intravenously demonstrated that metastatic disease also retained expression of endosialin. An efficacy study with an anti-endosialin drug conjugate results in highly promising results with 5 of 9 mice in the ADC treated group tumor free at 100 d while the control group had to be removed from the study due to tumor size by day 18.
Varied and interesting ADCs directed toward targets on liquid and solid tumors are in clinical trials and more are being evaluated in preclinical studies. The next hurdle is demonstration of clinical activity worthy of regulatory approval. ADCs are chemotherapeutics that will be used in combination treatment regimens, the time may have come for this technology to become an important contributor to improving treatment for cancer patients.
References
- Reichert JM. Marketed therapeutic antibodies compendium. MAbs 2012; 4:413 - 5; http://dx.doi.org/10.4161/mabs.19931; PMID: 22531442
- Reichert JM. Which are the antibodies to watch in 2012?. MAbs 2012; 4:1 - 3; http://dx.doi.org/10.4161/mabs.4.1.18719; PMID: 22327425
- Reichert JM. Antibody-based therapeutics to watch in 2011. MAbs 2011; 3:76 - 99; http://dx.doi.org/10.4161/mabs.3.1.13895; PMID: 21051951
- Reichert JM. Metrics for antibody therapeutics development. MAbs 2010; 2:695 - 700; http://dx.doi.org/10.4161/mabs.2.6.13603; PMID: 20930555
References
- Reichert JM. Marketed therapeutic antibodies compendium. MAbs 2012; 4:413 - 5; http://dx.doi.org/10.4161/mabs.19931; PMID: 22531442
- Beck A, Reichert JM. Therapeutic Fc-fusion proteins and peptides as successful alternatives to antibodies. MAbs 2011; 3:415 - 6; http://dx.doi.org/10.4161/mabs.3.5.17334; PMID: 21785279
- Beck A, Wurch T, Bailly C, Corvaia N. Strategies and challenges for the next generation of therapeutic antibodies. Nat Rev Immunol 2010; 10:345 - 52; http://dx.doi.org/10.1038/nri2747; PMID: 20414207
- Beck A, Senter P, Chari R. World Antibody Drug Conjugate Summit Europe: February 21-23, 2011; Frankfurt, Germany. MAbs 2011; 3:331 - 7; http://dx.doi.org/10.4161/mabs.3.4.16612; PMID: 21691144
- Beck A, Haeuw JF, Wurch T, Goetsch L, Bailly C, Corvaïa N. The next generation of antibody-drug conjugates comes of age. Discov Med 2010; 10:329 - 39; PMID: 21034674
- Reichert JM, Beck A, Iyer H. European Medicines Agency workshop on biosimilar monoclonal antibodies: July 2, 2009, London, UK. MAbs 2009; 1:394 - 416; http://dx.doi.org/10.4161/mabs.1.5.9630; PMID: 20065643
- Beck A. Biosimilar, biobetter and next generation therapeutic antibodies. MAbs 2011; 3:107 - 10; http://dx.doi.org/10.4161/mabs.3.2.14785; PMID: 21285536
- Beck A, -Cianférani S, Van Dorsselaer A. Biosimilar, biobetter and next generation antibody characterization by mass spectrometry. Anal Chem 2012; In press http://dx.doi.org/10.1021/ac3002885; PMID: 22510259
References
- Mössner E, Brünker P, Moser S, Püntener U, Schmidt C, Herter S, et al. Increasing the efficacy of CD20 antibody therapy through the engineering of a new type II anti-CD20 antibody with enhanced direct and immune effector cell-mediated B-cell cytotoxicity. Blood 2010; 115:4393 - 402; http://dx.doi.org/10.1182/blood-2009-06-225979; PMID: 20194898
- Niederfellner G, Lammens A, Mundigl O, Georges GJ, Schaefer W, Schwaiger M, et al. Epitope characterization and crystal structure of GA101 provide insights into the molecular basis for type I/II distinction of CD20 antibodies. Blood 2011; 118:358 - 67; http://dx.doi.org/10.1182/blood-2010-09-305847; PMID: 21444918
- Alduaij W, Ivanov A, Honeychurch J, Cheadle EJ, Potluri S, Lim SH, et al. Novel type II anti-CD20 monoclonal antibody (GA101) evokes homotypic adhesion and actin-dependent, lysosome-mediated cell death in B-cell malignancies. Blood 2011; 117:4519 - 29; http://dx.doi.org/10.1182/blood-2010-07-296913; PMID: 21378274
- Ferrara C, Grau S, Jäger C, Sondermann P, Brünker P, Waldhauer I, et al. Unique carbohydrate-carbohydrate interactions are required for high affinity binding between FcgammaRIII and antibodies lacking core fucose. Proc Natl Acad Sci U S A 2011; 108:12669 - 74; http://dx.doi.org/10.1073/pnas.1108455108; PMID: 21768335
- Dalle S, Reslan L, Besseyre de Horts T, Herveau S, Herting F, Plesa A, et al. Preclinical studies on the mechanism of action and the anti-lymphoma activity of the novel anti-CD20 antibody GA101. Mol Cancer Ther 2011; 10:178 - 85; http://dx.doi.org/10.1158/1535-7163.MCT-10-0385; PMID: 21220500
- Honeychurch J, Alduaij W, Azizyan M, Cheadle EJ, Pelicano H, Ivanov A, et al. Antibody-induced nonapoptotic cell death in human lymphoma and leukemia cells is mediated through a novel reactive oxygen species-dependent pathway. Blood 2012; 119:3523 - 33; http://dx.doi.org/10.1182/blood-2011-12-395541; PMID: 22354003
- Jak M, van Bochove GG, Reits EA, Kallemeijn WW, Tromp JM, Umana P, et al. CD40 stimulation sensitizes CLL cells to lysosomal cell death induction by type II anti-CD20 mAb GA101. Blood 2011; 118:5178 - 88; http://dx.doi.org/10.1182/blood-2011-01-331702; PMID: 21948297
- Sehn LH, Assouline SE, Stewart DA, Mangel J, Gascoyne RD, Fine G, et al. A phase 1 study of obinutuzumab induction followed by 2 years of maintenance in patients with relapsed CD20-positive B-cell malignancies. Blood 2012; 119:5118 - 25; http://dx.doi.org/10.1182/blood-2012-02-408773; PMID: 22438256
- Salles G, Morschhauser F, Lamy T, Milpied NJ, Thieblemont C, Tilly H, et al. Phase 1 study results of the type II glycoengineered humanized anti-CD20 monoclonal antibody obinutuzumab (GA101) in B-cell lymphoma patients. Blood 2012; 119:5126 - 32; http://dx.doi.org/10.1182/blood-2012-01-404368; PMID: 22431570
- Schaefer W, Regula JT, Bähner M, Schanzer J, Croasdale R, Dürr H, et al. Immunoglobulin domain crossover as a generic approach for the production of bispecific IgG antibodies. Proc Natl Acad Sci U S A 2011; 108:11187 - 92; http://dx.doi.org/10.1073/pnas.1019002108; PMID: 21690412
- Merchant AM, Zhu Z, Yuan JQ, Goddard A, Adams CW, Presta LG, et al. An efficient route to human bispecific IgG. Nat Biotechnol 1998; 16:677 - 81; http://dx.doi.org/10.1038/nbt0798-677; PMID: 9661204
References
- Maloney DG, Liles TM, Czerwinski DK, Waldichuk C, Rosenberg J, Grillo-Lopez A, et al. Phase I clinical trial using escalating single-dose infusion of chimeric anti-CD20 monoclonal antibody (IDEC-C2B8) in patients with recurrent B-cell lymphoma. Blood 1994; 84:2457 - 66; PMID: 7522629
- McLaughlin P, Grillo-López AJ, Link BK, Levy R, Czuczman MS, Williams ME, et al. Rituximab chimeric anti-CD20 monoclonal antibody therapy for relapsed indolent lymphoma: half of patients respond to a four-dose treatment program. J Clin Oncol 1998; 16:2825 - 33; PMID: 9704735
- Coiffier B, Lepage E, Briere J, Herbrecht R, Tilly H, Bouabdallah R, et al. CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large-B-cell lymphoma. N Engl J Med 2002; 346:235 - 42; http://dx.doi.org/10.1056/NEJMoa011795; PMID: 11807147
- Hallek M, Fischer K, Fingerle-Rowson G, Fink AM, Busch R, Mayer J, et al, International Group of Investigators, German Chronic Lymphocytic Leukaemia Study Group. Addition of rituximab to fludarabine and cyclophosphamide in patients with chronic lymphocytic leukaemia: a randomised, open-label, phase 3 trial. Lancet 2010; 376:1164 - 74; http://dx.doi.org/10.1016/S0140-6736(10)61381-5; PMID: 20888994
- Schulz H, Bohlius JF, Trelle S, Skoetz N, Reiser M, Kober T, et al. Immunochemotherapy with rituximab and overall survival in patients with indolent or mantle cell lymphoma: a systematic review and meta-analysis. J Natl Cancer Inst 2007; 99:706 - 14; http://dx.doi.org/10.1093/jnci/djk152; PMID: 17470738
- Zaja F, Iacona I, Masolini P, Russo D, Sperotto A, Prosdocimo S, et al. B-cell depletion with rituximab as treatment for immune hemolytic anemia and chronic thrombocytopenia. Haematologica 2002; 87:189 - 95; PMID: 11836170
- Dörner T, Radbruch A, Burmester GR. B-cell-directed therapies for autoimmune disease. Nat Rev Rheumatol 2009; 5:433 - 41; http://dx.doi.org/10.1038/nrrheum.2009.141; PMID: 19581902
- Thurlings RM, Teng O, Vos K, Gerlag DM, Aarden L, Stapel SO, et al. Clinical response, pharmacokinetics, development of human anti-chimaeric antibodies, and synovial tissue response to rituximab treatment in patients with rheumatoid arthritis. Ann Rheum Dis 2010; 69:409 - 12; http://dx.doi.org/10.1136/ard.2009.109041; PMID: 19596693
- Teicher BA, Chari RV. Antibody conjugate therapeutics: challenges and potential. Clin Cancer Res 2011; 17:6389 - 97; http://dx.doi.org/10.1158/1078-0432.CCR-11-1417; PMID: 22003066
- Derby L, Czuczman MS. Update on novel monoclonal antibodies and immunoconjugates for the treatment of lymphoproliferative disorders. Future Oncol 2011; 7:963 - 79; http://dx.doi.org/10.2217/fon.11.79; PMID: 21823892
- Cartron G, Trappe RU, Solal-Céligny P, Hallek M. Interindividual variability of response to rituximab: from biological origins to individualized therapies. Clin Cancer Res 2011; 17:19 - 30; http://dx.doi.org/10.1158/1078-0432.CCR-10-1292; PMID: 21208903
- Negrea GO, Elstrom R, Allen SL, Rai KR, Abbasi RM, Farber CM, et al. Subcutaneous injections of low-dose veltuzumab (humanized anti-CD20 antibody) are safe and active in patients with indolent non-Hodgkin’s lymphoma. Haematologica 2011; 96:567 - 73; http://dx.doi.org/10.3324/haematol.2010.037390; PMID: 21173095
- Teeling JL, Mackus WJ, Wiegman LJ, van den Brakel JH, Beers SA, French RR, et al. The biological activity of human CD20 monoclonal antibodies is linked to unique epitopes on CD20. J Immunol 2006; 177:362 - 71; PMID: 16785532
- Salles G, Mounier N, de Guibert S, Morschhauser F, Doyen C, Rossi JF, et al. Rituximab combined with chemotherapy and interferon in follicular lymphoma patients: results of the GELA-GOELAMS FL2000 study. Blood 2008; 112:4824 - 31; http://dx.doi.org/10.1182/blood-2008-04-153189; PMID: 18799723
- Salles G, Seymour JF, Offner F, López-Guillermo A, Belada D, Xerri L, et al. Rituximab maintenance for 2 years in patients with high tumour burden follicular lymphoma responding to rituximab plus chemotherapy (PRIMA): a phase 3, randomised controlled trial. Lancet 2011; 377:42 - 51; http://dx.doi.org/10.1016/S0140-6736(10)62175-7; PMID: 21176949
- Kluin-Nelemans H, Hoster E, Hermine O, et al. Induction and maintenance therapy in elderly patients with mantle cell lymphoma. Double-randomized trial by the European Mantle Cell Lymphoma Network. N Engl J Med 2012; In press
- Nightingale G. Ofatumumab: a novel anti-CD20 monoclonal antibody for treatment of refractory chronic lymphocytic leukemia. Ann Pharmacother 2011; 45:1248 - 55; http://dx.doi.org/10.1345/aph.1P780; PMID: 21896924
- Mössner E, Brünker P, Moser S, Püntener U, Schmidt C, Herter S, et al. Increasing the efficacy of CD20 antibody therapy through the engineering of a new type II anti-CD20 antibody with enhanced direct and immune effector cell-mediated B-cell cytotoxicity. Blood 2010; 115:4393 - 402; http://dx.doi.org/10.1182/blood-2009-06-225979; PMID: 20194898
- Patz M, Isaeva P, Forcob N, Müller B, Frenzel LP, Wendtner CM, et al. Comparison of the in vitro effects of the anti-CD20 antibodies rituximab and GA101 on chronic lymphocytic leukaemia cells. Br J Haematol 2011; 152:295 - 306; http://dx.doi.org/10.1111/j.1365-2141.2010.08428.x; PMID: 21155758
- Ward E, Mittereder N, Kuta E, Sims GP, Bowen MA, Dall’Acqua W, et al. A glycoengineered anti-CD19 antibody with potent antibody-dependent cellular cytotoxicity activity in vitro and lymphoma growth inhibition in vivo. Br J Haematol 2011; 155:426 - 37; http://dx.doi.org/10.1111/j.1365-2141.2011.08857.x; PMID: 21902688
- Leonard JP, Goldenberg DM. Preclinical and clinical evaluation of epratuzumab (anti-CD22 IgG) in B-cell malignancies. Oncogene 2007; 26:3704 - 13; http://dx.doi.org/10.1038/sj.onc.1210370; PMID: 17530024
- Advani R, Forero-Torres A, Furman RR, Rosenblatt JD, Younes A, Ren H, et al. Phase I study of the humanized anti-CD40 monoclonal antibody dacetuzumab in refractory or recurrent non-Hodgkin’s lymphoma. J Clin Oncol 2009; 27:4371 - 7; http://dx.doi.org/10.1200/JCO.2008.21.3017; PMID: 19636010
- Kluin-Nelemans HC, van Marwijk Kooy M, Lugtenburg PJ, van Putten WL, Luten M, Oudejans J, et al. Intensified alemtuzumab-CHOP therapy for peripheral T-cell lymphoma. Ann Oncol 2011; 22:1595 - 600; http://dx.doi.org/10.1093/annonc/mdq635; PMID: 21212158
- Enblad G, Hagberg H, Erlanson M, Lundin J, MacDonald AP, Repp R, et al. A pilot study of alemtuzumab (anti-CD52 monoclonal antibody) therapy for patients with relapsed or chemotherapy-refractory peripheral T-cell lymphomas. Blood 2004; 103:2920 - 4; http://dx.doi.org/10.1182/blood-2003-10-3389; PMID: 15070664
- Lundin J, Hagberg H, Repp R, Cavallin-Ståhl E, Fredén S, Juliusson G, et al. Phase 2 study of alemtuzumab (anti-CD52 monoclonal antibody) in patients with advanced mycosis fungoides/Sezary syndrome. Blood 2003; 101:4267 - 72; http://dx.doi.org/10.1182/blood-2002-09-2802; PMID: 12543862
- O’Mahony D, Morris JC, Stetler-Stevenson M, Matthews H, Brown MR, Fleisher T, et al. EBV-related lymphoproliferative disease complicating therapy with the anti-CD2 monoclonal antibody, siplizumab, in patients with T-cell malignancies. Clin Cancer Res 2009; 15:2514 - 22; http://dx.doi.org/10.1158/1078-0432.CCR-08-1254; PMID: 19293260
- Ansell SM, Horwitz SM, Engert A, Khan KD, Lin T, Strair R, et al. Phase I/II study of an anti-CD30 monoclonal antibody (MDX-060) in Hodgkin’s lymphoma and anaplastic large-cell lymphoma. J Clin Oncol 2007; 25:2764 - 9; http://dx.doi.org/10.1200/JCO.2006.07.8972; PMID: 17515574
- Forero-Torres A, Leonard JP, Younes A, Rosenblatt JD, Brice P, Bartlett NL, et al. A Phase II study of SGN-30 (anti-CD30 mAb) in Hodgkin lymphoma or systemic anaplastic large cell lymphoma. Br J Haematol 2009; 146:171 - 9; http://dx.doi.org/10.1111/j.1365-2141.2009.07740.x; PMID: 19466965
- d’Amore F, Radford J, Relander T, Jerkeman M, Tilly H, Osterborg A, et al. Phase II trial of zanolimumab (HuMax-CD4) in relapsed or refractory non-cutaneous peripheral T cell lymphoma. Br J Haematol 2010; 150:565 - 73; http://dx.doi.org/10.1111/j.1365-2141.2010.08298.x; PMID: 20629661
- Kim YH, Duvic M, Obitz E, Gniadecki R, Iversen L, Osterborg A, et al. Clinical efficacy of zanolimumab (HuMax-CD4): two phase 2 studies in refractory cutaneous T-cell lymphoma. Blood 2007; 109:4655 - 62; http://dx.doi.org/10.1182/blood-2006-12-062877; PMID: 17311990
- Ishida T, Joh T, Uike N, Yamamoto K, Utsunomiya A, Yoshida S, et al. Defucosylated anti-CCR4 monoclonal antibody (KW-0761) for relapsed adult T-cell leukemia-lymphoma: a multicenter phase II study. J Clin Oncol 2012; 30:837 - 42; http://dx.doi.org/10.1200/JCO.2011.37.3472; PMID: 22312108
- Dearden C. The role of alemtuzumab in the management of T-cell malignancies. Semin Oncol 2006; 33:Suppl 5 S44 - 52; http://dx.doi.org/10.1053/j.seminoncol.2006.01.029; PMID: 16720203
- Chamarthy MR, Williams SC, Moadel RM. Radioimmunotherapy of non-Hodgkin’s lymphoma: from the ‘magic bullets’ to ‘radioactive magic bullets’. Yale J Biol Med 2011; 84:391 - 407; PMID: 22180677
References
- Teicher BA, Chari RV. Antibody conjugate therapeutics: challenges and potential. Clin Cancer Res 2011; 17:6389 - 97; http://dx.doi.org/10.1158/1078-0432.CCR-11-1417; PMID: 22003066
- Teicher BA. Antibody-drug conjugate targets. Curr Cancer Drug Targets 2009; 9:982 - 1004; http://dx.doi.org/10.2174/156800909790192365; PMID: 20025606
- Ricart AD. Antibody-drug conjugates of calicheamicin derivative: gemtuzumab ozogamicin and inotuzumab ozogamicin. Clin Cancer Res 2011; 17:6417 - 27; http://dx.doi.org/10.1158/1078-0432.CCR-11-0486; PMID: 22003069
- Katz J, Janik JE, Younes A. Brentuximab Vedotin (SGN-35). Clin Cancer Res 2011; 17:6428 - 36; http://dx.doi.org/10.1158/1078-0432.CCR-11-0488; PMID: 22003070
- LoRusso PM, Weiss D, Guardino E, Girish S, Sliwkowski MX. Trastuzumab emtansine: a unique antibody-drug conjugate in development for human epidermal growth factor receptor 2-positive cancer. Clin Cancer Res 2011; 17:6437 - 47; http://dx.doi.org/10.1158/1078-0432.CCR-11-0762; PMID: 22003071
- Blanc V, Bousseau A, Caron A, Carrez C, Lutz RJ, Lambert JM. SAR3419: an anti-CD19-Maytansinoid Immunoconjugate for the treatment of B-cell malignancies. Clin Cancer Res 2011; 17:6448 - 58; http://dx.doi.org/10.1158/1078-0432.CCR-11-0485; PMID: 22003072
- Steiner M, Neri D. Antibody-radionuclide conjugates for cancer therapy: historical considerations and new trends. Clin Cancer Res 2011; 17:6406 - 16; http://dx.doi.org/10.1158/1078-0432.CCR-11-0483; PMID: 22003068
- Kreitman RJ, Pastan I. Antibody fusion proteins: anti-CD22 recombinant immunotoxin moxetumomab pasudotox. Clin Cancer Res 2011; 17:6398 - 405; http://dx.doi.org/10.1158/1078-0432.CCR-11-0487; PMID: 22003067
- Rouleau C, Smale R, Fu Y-S, Hui G, Wang F, Hutto E, et al. Endosialin is expressed in high grade and advanced sarcomas: evidence from clinical specimens and preclinical modeling. Int J Oncol 2011; 39:73 - 89; PMID: 21537839
- Rouleau C, Curiel M, Weber W, Smale R, Kurtzberg L, Mascarello J, et al. Comprehensive assessment of endosialin protein expression in human cancer: therapeutic implications for synovial sarcoma, fibrosarcoma, malignant fibrous histiocytoma and osteosarcoma. Clin Cancer Res 2008; 14:7223 - 36; http://dx.doi.org/10.1158/1078-0432.CCR-08-0499; PMID: 19010839