Abstract
Antibody-based therapeutics are of great value for the treatment of human diseases. In addition to functional activity, affinity or physico-chemical properties, antibody specificity is considered to be one of the most crucial attributes for safety and efficacy. Consequently, appropriate studies are required before entering clinical trials.
High content protein arrays are widely applied to assess antibody specificity, but this commercial solution can only be applied to final therapeutic antibody candidates because such arrays are expensive and their throughput is limited. A flexible, high-throughput and economical assay that allows specificity testing of IgG or Fab molecules during early discovery is described here. The 384-well microtiter plate assay contains a comprehensive panel of 32 test proteins and uses electrochemiluminescence as readout.
The Protein Panel Profiling (3P) was used to analyze marketed therapeutic antibodies that all showed highly specific binding profiles. Subsequently, 3P was applied to antibody candidates from early discovery and the results compared well with those obtained with a commercially available high content protein chip. Our results suggest that 3P can be applied as an additional filter for lead selection, allowing the identification of favorable antibody candidates in early discovery and thereby increasing the speed and possibility of success in drug development.
Introduction
As protein therapeutics, monoclonal antibodies (mAbs) offer enormous potential for the treatment of a large variety of human diseases.Citation1-Citation3 In the development and engineering of antibodies, an understanding of the underlying antibody–antigen association is of major importance. An antibody interacts with its target (antigen) through a number of non-covalent interactions, including hydrogen bonds, electrostatic and hydrophobic interactions and van der Waals forces. Antibody specificity is defined as the ability of an antibody to react with a certain antigenic determinant and not with a different one. There is, however, no general rule concerning the requirements for a specific antibody-antigen interaction.Citation4 Non-specific (also known as cross-reactive and polyreactive) binding behavior can result from multiple mechanisms. For instance, hydrophobic interactions, derived either from the paratope itself or the adoption of a non-native conformation, are reported to contribute to non-specific binding. In general, antibodies that recognize many different antigens (polyreactivity) have a lower affinity for each of the antigens than antibodies that recognize only one (mono-reactive) or a limited number of antigens. It is assumed that flexibility in the antigen binding pocket contributes to polyreactivity.Citation4 Mohan and coworkers analyzed the association of three different murine anti-lysozyme mAbs with their target and described how specific and cross-reactive interactions differ thermodynamically; they concluded that “antibody specificity was dominated by enthalpic contributions whereas antibody cross-reactivity was mediated by configurational entropy.”Citation5
It is generally accepted that therapeutic antibodies must be antigen-specific for successful drug development. Hoetzel and coworkers recently showed that off-target binding in ELISA correlated with an increased risk for fast clearance of IgG molecules. They used coated baculovirus particles representing a complex mixture of antigens to detect unspecific binding of antibodies.Citation6 The faster clearance they observed is most probably caused by unspecific binding of the antibody in vivo, which increases the volume of distribution (VD) of the drug in the body and therefore decreases the effective serum concentration levels. Additionally, off-target binding could affect the pharmacodynamic and safety profile of the antibody by leading to unfavorable effects. For instance, an antibody that binds other proteins in addition to its target may exhibit unwanted side effects such as killing of healthy cells, e.g., by antibody-dependent cell-mediated cytotoxicity, or the release of highly toxic drug payloads of an antibody drug conjugate. Furthermore, prior to clinical trials with mAbs, the immunological properties of the antibody should be described in detail, including its antigenic specificity and any unintentional reactivity toward human tissues distinct from the intended target. Such cross-reactivity studies are routinely performed by immunohistochemical procedures using a range of human tissues. For the above reasons, the early assessment of specificity during the discovery phase of antibodies is of high importance because it can be used to improve the quality of candidates selected for clinical development.
Fluorescence-activated cell sorting (FACS) and cell-based potency assays using different cell lines or immunohistochemistry (IHC) on different biological tissues are typically used to assess antibody specificity, but these methods are time consuming, laborious and the evidence concerning product specificity is limited. As a consequence, protein microarray-based methods are evolving as alternative approaches for specificity profiling. A well-established commercial example is ProtoArray®, which consists of more than 9,000 human proteins. The proteins are expressed as N-terminal GST fusion proteins using a Baculovirus expression system, affinity purified, and then spotted under non-denaturing conditions on ultra-thin nitrocellulose-coated glass slides. ProtoArray® is based on the yeast protein microarray technology developed by Zhu and coworkers.Citation7 In addition to the identification of protein–protein interactions, this protein array can be used for profiling of antibody specificity.Citation8 An alternative protein microarray, the UNIchip® family (Protagen) contains different sets of proteins such as membrane, intracellular and extracellular proteins. The Unichip® array consists of 384 pre-defined, His-tag purified recombinant human proteins, immobilized on a nitrocellulose-coated glass slide.Citation9 UNIchip® allows assessment of an antibody’s ability to bind its target and hundreds of other proteins in parallel using a fluorescent readout. Typically, different candidate antibodies are tested and compared for their so-called off-target activities (OTA).Citation10,Citation11
Phage display selection of large antibody repertoires routinely results in a high number of hits recognizing the target of interest.Citation12,Citation13 Thus, the identification of a lead candidate with the greatest possibility of success in clinical development requires extensive screening, as well as functionalCitation14 and physico-chemical characterization.Citation15 Testing for binding specificity is typically done at multiple stages of the discovery process. During ELISA-based primary and secondary screenings, a limited set of negative control proteins is usually applied.Citation16 Cell-based screenings are typically performed in parallel with a suited negative control cell line, if available. In later phases of discovery, the biological activity, affinity, epitopes,Citation17,Citation18 manufacturing and pharmaceutical development aspectsCitation19,Citation20 are evaluated. Extensive characterization of specificity on large protein panels, as described above, is expensive and time consuming and is reserved for the final few lead candidates, if at all. Especially for difficult targets, e.g., aggregated or hydrophobic antigens, early antibody specificity screening might further improve the lead candidate selection. Thus, there is substantial need for a method that allows high-throughput specificity testing that can be practicably used at an earlier stage of discovery. For this reason, the Protein Panel Profiling (3P) method was developed for antibody screening on a comprehensive set of test proteins. The automated 384-well assay utilizes electrochemiluminescence (ECL) technology, which is characterized by high sensitivity, having several logs of dynamic range and a high throughput.Citation21 The 3P assay was characterized using marketed therapeutic antibodies and antibodies from MorphoSys’s proprietary discovery pipeline. It was found to serve as a valuable selection filter that can be applied during early antibody discovery.
Results
Rationale for selection of the protein panel for assay development
The motivation for development of the 3P assay was the need of an efficient tool for the early assessment of antibody specificity during discovery. Requirements for the method were high throughput at low consumables costs and the suitability for automation. Furthermore, a flexible system in terms of tailored composition of proteins included in the panel was highly desired. The decision to run the assay on a microtiter plate-based ECL platform (MSD technology), was due to its high sensitivity, broad dynamic range and high throughput, which results from the possibility to use 384-well plates, and the very short detection time per plate.Citation21 Additionally, the MSD platform uses microtiter plates with a carbon surface that enables gentle immobilization of proteins and cells.Citation22 Thus, proteins are more likely to be displayed in a native conformation on the carbon compared with plastic surfaces or coated glass slides. To strike a balance between throughput, costs and the number of proteins tested, two 384-well plates are used for one assay, with 30 different proteins (plus two blanks) and up to 10 antibody samples in total at two different concentrations (for assay layout see ). To confirm the addition and identity of the sample (i.e., Fab or IgG molecule), several controls were included: Protein A, two different anti-human Fab antibodies and one anti-human Fc antibody (see Materials). For further control and maximum flexibility, three positions on the plates were reserved, e.g., for the coating of targets and counter targets. For referencing purposes (see Data evaluation) lysozyme as target of reference antibody “MOR ref. mAb” was included. The selection of the remaining 22 proteins for the panel was based on theoretical considerations and results from previous experiments. For instance, during assay development, a number of different human Fc receptors were tested in the protein panel. However, these showed too high background with the detection antibodies due to the cross-reactivity of mouse or goat antibodies with human Fc receptors (data not shown). Therefore, they were excluded from the current protein panel. To compose a predictive panel, abundant proteins found in human blood were chosen and purchased as serum purified versions. Since these often contain trace amounts of human IgG, proteins from human and bovine serum were tested. If possible the human variant was selected (e.g., serum albumin, hemoglobin), if cross reactivity with the anti-human detection antibody was observed, the bovine ortholog was used (e.g., fibrinogen, transferrin). As representatives for soluble cytokines and cell surface receptors, six recombinant protein targets (marked as cytokine 1, 2, 3 and cell surface receptor 1, 2, 3) were chosen. Furthermore, to assess interactions based on electrostatic forces, a panel of eight proteins from a broad pI range, ranging from 2.8 (pepsinogen) to 10.7 (cytochrome c) was included. Finally, phosphorylated and glycosylated ovalbumin was added, as well as milk powder containing 80% of phosphorylated β casein and the highly glycosylated RNase B (see Materials and Methods for a complete list of selected proteins).
Figure 1. Flow scheme (A) and microtiter plate layout (B) of the 3P assay. A shows flow scheme of the 3P assay. Incubation times are given in parentheses. B shows the microtiter plate layout of the 3P assay. Proteins as indicated in the figure are immobilized in rows (wells 1–24) on two 384-well MSD plates. Rows B and P on plate 1 and row O on plate 2 are variable positions, e.g., for antibody target and counter targets. Rows J–O on plate 1 contain cytokines and cell surface receptors. Samples are dispensed in columns on both 384-well plates: Columns 1 and 2 are used for two blank controls or one blank control and a system suitability test (SST), respectively. Up to 10 samples to be analyzed for binding specificity are dispensed to columns 3–22 at each 100 nM and 10 nM concentration, respectively. Columns 23 and 24 are used for the reference monoclonal antibody (MOR ref. mAb, anti lysozyme) at 100 nM and 10 nM, respectively.

During assay development, the 3P assay was optimized in terms of the following parameters: optimal coating concentration (concentrations ranging from 0.05 to 3.0 µg/ml were tested), selection of the most appropriate detection antibody (three different antibodies tested), optimal concentrations for antibody samples (ranging between 1 and 100 nM), and different buffers and washing conditions (data not shown).
3P analysis of marketed and HuCAL® derived antibodies
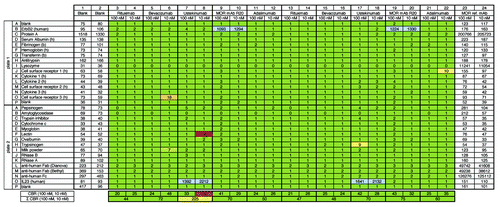
As a starting point, the binding behaviors of four marketed therapeutic IgGs (rituximab and bevacizumab, which are approved for cancer; ustekinumab and adalimumab, which are approved for immunological indications) were analyzed using the 3P assay (). In addition, an IgG molecule composed of a human IgG1 with constant regions of HuCAL® IgG with VL and VH domains of trastuzumab was included. As controls, human ErbB2 and human IL23 were included in the protein panel on “variable positions” (see for plate layout). Specific binding of the mAb R20 (anti-ErbB2) and ustekinumab (anti-IL12 / IL23) to their targets was clearly visible (, specific target interaction shown as blue shaded wells, row B on plate 1, row O on plate 2). To assess intra-assay reproducibility, all samples were analyzed in duplicates in one assay (left and right part). No unspecific binding of any of the tested marketed therapeutic IgGs to coated proteins was visible (green shaded wells). Only four out of the 320 wells showed slightly increased binding over the reference antibody (MOR ref. mAb, one red shaded and three yellow wells). These were regarded as false positives because binding was observed only at one concentration (either at 100 or 10 nM) and not in replicates. The false positives were not significant for the overall result. In summary, as expected, none of the tested marketed IgGs and the mAb R20 showed undesirable binding properties in the 3P analysis.
Figure 2.3P analysis of four marketed therapeutic monoclonal IgG molecules and one IgG with trastuzumab VH and VL regions and HuCAL® constant regions (MOR mAb R20). Raw ECL signals for background binding of the ECL-labeled detection antibody to coated proteins (as indicated on the left) are shown in columns 1 and 2 (blank). Raw ECL signals for binding of reference antibody (MOR ref. mAb) at 100 nM and 10 nM are shown in columns 23 and 24, respectively. Commercial therapeutic and tool antibodies rituximab, bevacizumab, ustekinumab, MOR mAb R20 (see Materials and Methods and Results), and adalimumab were tested at 100 nM and 10 nM in duplicates (see left and right part of figure). Numbers in columns 3–22 indicate x-fold binding (rounded numbers) of analyzed sample to respective coated protein over binding of the MOR ref. mAb to the same coated protein at the same concentration analyzed, e.g., 1,093 (see plate 1, well B9) indicates that ECL signal for binding of 100 nM MOR mAb R20 to coated human ErbB2 (trastuzumab target) was 1,093-fold increased over binding of 100 nM MOR ref. mAb to coated human ErbB2. As expected, ustekinumab bound to its target IL23 (see plate 2, wells O7, 8, 17, 18). A binding ratio of 0 always indicates that the sample binds weaker to the immobilized protein than the MOR ref. mAb. Colored shadings in columns 3–22 visualize degree of x-fold binding over reference antibody: green represents a binding ratio of up to 5; yellow is up to 10; red is above 50. Blue shading indicates binding to target proteins. Note, only the target proteins for ustekinumab and MOR mAb R20 (IL23 and ErbB2) were included here. The cumulative binding ratio (CBR) for each concentration, 100 nM and 10 nM, respectively, and the sum of these CBRs are given at the two bottom lines. Colored shadings of sum of CBRs is as follows: up to 150 green (i.e., overall no/low degree of unspecific binding); up to 250 yellow (overall moderate degree of unspecific binding); above 400 red (overall high degree of unspecific binding). Note that the anti-human Fab detection antibody (goat) shows cross-reactivity to coated Protein A (plate 1, wells C1, 2); however, since the binding signals for IgG molecules to Protein A are much larger (plate 1, wells C23, 24) and this coated protein serves as a positive control, the observed cross-reactivity was regarded to be acceptable. The same is true for binding of the detection antibody to coated anti-human antibodies (plate 2, rows L, M, N). As expected, the MOR ref. mAb strongly interacts with its target lysozyme (plate 1, wells I23, 24).
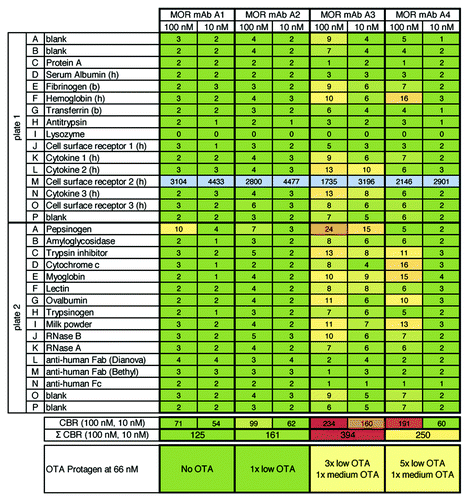
Next, the correlation between the 3P assay and commercially-available high density protein arrays for antibody profiling was investigated (). Four HuCAL® derived IgG molecules, A1–A4, were compared in 3P and UNIchip® AV-VAR-EP Premium analysis. The IgG molecules were tested at 66 nM (UNIchip®) and 10 and 100 nM (3P analysis). IgG molecules MOR mAb A1 and A2 showed low off-target activity OTA using UNIchip®, which is in excellent agreement with the 3P results were no/low unspecific binding was observed. For MOR mAb A4, 3P analysis yielded moderate unspecific binding, which is in good correlation with moderate OTA (6 hits, 5–29% OTA) using UNIchip®. Only for MOR mAb A3 the agreement was less striking: high unspecific binding was observed with 3P whereas moderate OTA (4 hits, 5–20% OTA) were found with the Protagen UNIchip®. Overall, these results were assessed to be in good qualitative agreement. These data underline that 3P, although representing a smaller panel of 32 different proteins and controls, might deliver compatible results to established state-of-the-art technologies for antibody profiling.
Figure 3.3P analysis of four HuCAL® derived IgG molecules (targeted against a cell surface receptor: plate 1, row M) and correlation with data from Protagen using the UNIchip®. Figure shows results from 3P analysis of four HuCAL® derived IgG molecules that were previously analyzed by Protagen using the UNIchip® on a fee-for-service basis. Results shown as OTA from Protagen’s analysis are indicated below the 3P results. In 3P, as expected, strong binding to the target (cell surface receptor 2) was observed for all antibodies. For two IgGs, MOR mAb A1 and A2, no unspecific binding was observed (see Σ CBR = 125 and 161, green shaded). The degree of unspecific binding of MOR mAb A3 and mAB A4 was ranked with moderate to high (Σ CBR = 394, light red shaded) and moderate (Σ CBR = 250, yellow shaded), respectively. Overall, the 3P and Protagen UNIchip® results are in good qualitative agreement.
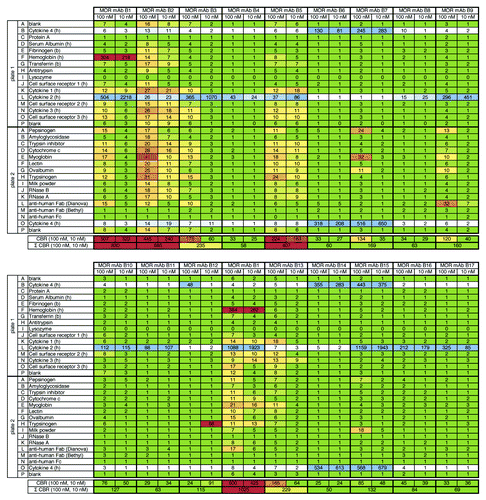
To gain more insight into how 3P analysis works in a real life setting during the drug discovery phase, a number of IgG molecules that were selected either for binding to its target cytokine (cytokine 2) or its closest homologous protein (cytokine 4) were tested. A total of 17 IgG molecules identified using phage display and previously selected by primary screening in ELISA or FACS were subjected to 3P analysis; the results are shown in . As previously observed in ELISA, IgGs recognized cytokine 2 (row L on plate 1, e. g., MOR mAb B9) or cytokine 4 (rows B on plate 1 and O on plate 2, e. g., MOR mAB B7). As was seen before, MOR mAb B15 was confirmed to be cross-reactive, i.e., the IgG strongly bound to its target and its closest homolog. In total, 12 out of 17 IgG molecules showed no or low unspecific binding, two molecules were ranked with moderate unspecific binding and three molecules were ranked with high unspecific binding. To get an impression about inter-assay reproducibility, antibody MOR mAb B1 was analyzed in duplicates in two independent experiments (, upper and lower part). Cumulative binding ratios from both experiments were comparable. This IgG was ranked with a high degree of unspecific binding. Although not yet understood, reproducible, strong binding of this IgG to human hemoglobin was found. This finding was not observable for the other IgG molecules analyzed here. This data set further supports the capability of the 3P assay for ranking and classification of molecules according to their binding characteristics in terms of cross-reactivity to related proteins and overall specificity.
Figure 4.3P analysis of 17 HuCAL® derived IgG molecules (targeted against a cytokine). Figure shows results from 3P analysis of 17 different HuCAL® derived IgG molecules that had been generated against cytokine 2 and tested during discovery phase. As expected, IgGs recognized their target (cytokine 2, row L on plate 1, e.g., MOR mAb B9) or its closest homolog, cytokine 4 (rows B on plate 1 and O on plate 2, e.g., MOR mAb B7). As previously observed in FACS and ELISA, MOR mAb B15 was found to be cross-reactive to both cytokine 2 and cytokine 4. A total of 12 IgG molecules were ranked with no/low degree of unspecific binding, two showed a moderate and three molecules showed a high degree of unspecific binding. The MOR mAb B1 had been analyzed as a replicate in an independent experiment (Σ CBR = 830 and 1,025, respectively) to assess the inter-assay reproducibility.
So far, a total of 298 Fab and IgG molecules were analyzed in 3P. From all these molecules, the percentage distribution for each degree of unspecific binding (no/low, moderate or high) was calculated. At both 100 nM and 10 nM, the majority of binding molecules exhibited no/low unspecific binding in 3P (63% and 83%, respectively). A relatively high degree of unspecific binding was observed for 11% of molecules at 10 nM and 25% at 100 nM. This finding confirmed the assumption that the tendency for unspecific binding increased with the analyte concentration. In this context, it must be noted that 3P analysis was mainly performed in projects with difficult targets such as aggregated, hydrophobic or heterogeneous protein preparations, or surface proteins presented at low copy numbers on cells. Independent of the antibody source (in vitro or in vivo), these targets often bear the risk of generating antibodies with undesired properties (authors’ unpublished observations).
Discussion
Target selectivity is considered a critical factor for safety and efficacy assessments of therapeutic antibodies and should be carefully evaluated. Arrays with large panels of immobilized proteins are tools that are widely used for that purpose. It is proposed that antibodies showing clean profiles on such protein arrays will possess a minimized risk of undesired off-target activity. In addition, there is evidence that protein arrays may be predictive for immunohistochemical outcome.Citation11 Due to the relatively high costs of advanced protein arrays, however, this method can only be applied to a very limited number of lead candidates. Therefore, the approach bears the adherent risk that unfavorable specificity profiles may be discovered during late stage discovery or during non-clinical and clinical development. In the present work, a microtiter plate-based specificity profiling assay (3P assay) for early discovery is described. We demonstrated that the 3P assay produces results qualitatively comparable to a state-of-the-art high density protein array and concluded that use of 3P analysis as a pre-selection tool in early phase discovery could markedly enhance the chance of “clean” lead candidates in a final protein array verification.
The effective use of the 3P assay in early discovery is manageable due to a relatively high throughput (ten samples per assay), attractive consumables costs and use of a liquid handling platform for the majority of pipetting steps. Moreover, the microtiter plate format facilitates a customized design of the protein panel, i.e., inclusion of the target protein, homologous forms of pre-clinically relevant species, counter-targets or cell derived vesicles. These obvious advantages, however, go along with a rather restricted number of only 32 proteins and controls used for profiling. Using such a limited panel in comparison to high content protein arrays might raise the question if observed heterogeneous binding profiles are rather due to stickiness caused by hydrophobicity than by real polyreactivity as described by Notkins.Citation4 To get additional evidence about the nature of the observed unspecific binding, a number of antibodies were subjected to forced thermal stress, which should lead to display of hydrophobic patches of the partially unfolded proteins. Interestingly, the specificity profile before and after forced stress application did not significantly change at the 10 and 100 nM concentrations tested (see Fig. S1). Moreover, there was no obvious correlation between 3P assay results and data from physico-chemical methods like hydrophobic interaction chromatography or high performance-size exclusion chromatography (HP-SEC). For instance, the amount of aggregates was below 10% as detected by HP-SEC (data not shown), indicating that observed unspecific binding is not simply caused by aggregation. Thus, the 3P assay may not be considered a redundant method for identification of hydrophobicity or aggregation tendency of antibodies, but rather a complementary test that addresses more complex binding properties. This observation appears to be in contrast to those of Scholz and LükingCitation23 who recently suggested stability-indicating properties of UNIchip® for adalimumab; although it should be noted that this study was performed with a rather high mAb concentration (~17 μM). Moreover, the proteins immobilized on the array are mostly denatured. Under such conditions, it is not unexpected that increased hydrophobic interactions contribute to the protein interaction profile of the stressed samples. A rather gentle and native immobilization on a carbon surface, as applied in the 3P assay, might deliver different results with stressed samples, but additional experiments will be required for final assessment.
Selection of antibody candidates based on their specificity should be well balanced. During primary or secondary screenings, specificity criteria should not be too stringent, which reduces the risk of de-selecting candidates with promising functional properties. In subsequent recovery steps, a more stringent threshold should be applied to ensure the selection of candidates with favorable target selectivity. To fulfill this requirement, an appropriate data evaluation strategy was found to be crucial. A typical approach is normalization by setting the binding signal for a cognate antigen to 100%, as, for instance, performed for Protagen’s UNIchip®.Citation10 Especially in early development stages prior to affinity optimization, however, signals of antibodies to their coated targets strongly vary (see for binding of different IgG molecules to their target, cytokine 2). As an alternative approach, normalization by a reference antibody was performed in this study. Signals of the antibody sample on a certain protein were divided by the respective signals of the reference molecule. For referencing, MOR ref. mAb (anti-lysozyme) was used; this mAb had been extensively characterized in a broad spectrum of in vitro assays and is routinely used as negative control in in vitro, in vivo and immunohistology studies. Accordingly, this antibody showed very low overall background in the 3P assay and a high stability toward thermal stress (see Fig. S1). Furthermore, for evaluation, the cumulative binding ratio (CBR) was then calculated, and CBRs at 100 nM and 10 nM sample concentration were summarized. Antibody concentrations in the nM range as used in 3P are actually found in human serum during therapeutic treatments (e.g., up to 500 μg/ml serum, single digit μM range) in oncology indications.Citation24 From different evaluation modes tested, the approach described above was considered the most meaningful one, although it requires an advanced read-out system with very low intrinsic background noise and a high dynamic range. This requirement could be achieved by using ECL, a technology which is well-established in biotechnological and pharmaceutical industry, e.g., for immunogenicity screeningCitation25 or highly sensitive functional assays.Citation26 Thus, the 3P assay can be easily integrated into existing platforms and can serve as additional tool to close the potentially critical gap in specificity profiling during early antibody discovery.
Verification that in vitro specificity profiling will translate into beneficial pharmacological performance in vivo is still needed, but we are confident that selection tools like the 3P assay are valuable approaches to increase the possibility of success in clinical development.
Materials and Methods
Materials
Antigens
The following proteins were purchased from Sigma-Aldrich, origin/sources and catalog numbers are given in parentheses: Protein A (Staphylococcus aureus, P6031), serum albumin (human serum, A3782), serum albumin (bovine serum, A7906), fibrinogen (bovine plasma, F8630), hemoglobin (human, H7379), apo-transferrin (bovine, T1428), α1-antitrypsin (human plasma, A6150), lysozyme (chicken egg white, L6876), pepsinogen (porcine stomach, P4656), amyloglycosidase (Aspergillus niger, 10115), trypsin inhibitor [Glycine max (soybean), T9003], cytochrome c (bovine heart, C3131), myoglobin (equine heart, M1882), lectin (Lens culinaris, L9267), ovalbumin (chicken egg white, A5503), trypsinogen (bovine pancreas, T1143), RNase A (bovine pancreas, R5503) and RNase B (bovine pancreas, R7884). Low fat milk powder (80% β-Casein) was from Saliter.
Human cytokines and cell surface proteins were purchased (one protein) or produced at MorphoSys (six proteins). Human ErbB2 was from R&D Systems (1129-ER-050); human IL23 (p19/p40) was from eBioscience (34-8239-82).
Stock solutions from all proteins were prepared at a concentration of 1.0 mg/ml in PBS, aliquoted into PCR stripes (Thermo Scientific) and stored at −20°C.
Antibodies
Commercial therapeutic antibodies rituximab (Rituxan®), bevacizumab (Avastin®), adalimumab (Humira®) and ustekinumab (Stelara®) were purchased from pharmacy.
Goat anti-human IgG F(ab')2 fragment (109-005-097) and IgG Fcγ fragment (109-005-098) specific antibodies were purchased from Dianova; goat anti-human IgG Fab fragment antibody (A80-114A) was from Biomol. Goat anti-human IgG Fab Fragment antibody was labeled with ECL-sulfo-tag N-hydroxysuccinimide (NHS)-ester from MSD according to the manufacturer’s protocol at a concentration of 1.0 mg/ml.
Antibody mAb R20, targeting EGFR2 (ErbB2; HER-2/neu), was constructed by insertion of DNA encoding VH and VL regions [as published in the trastuzumab (Herceptin®) patent] into pMORPH-4 expression vector using HuCAL®-specific restriction enzyme cleavage sites.
The reference antibody (MOR ref. mAb, IgG1 format) was selected from the HuCAL GOLD® library by using lysozyme as a target. Other applied antibodies were obtained from the HuCAL PLATINUM® library by phage display technologyCitation13 via selection on the respective targets.
All HuCAL® derived antibodies were transiently produced in IgG1 format by using the human cell line HKB11. Antibodies were affinity purified via Protein A, eluted with glycine buffer at low pH, and the buffer was exchanged to PBS.
Reagents and buffers
PBS (Dulbecco’s Phosphate Buffered Saline, without CaCl2 and MgCl2) was purchased from Gibco; Tween 20 was from Merck Millipore and Read buffer T with surfactant was from MSD.
Protein Panel Profiling using an ECL-based assay
Proteins were coated on two 384-well MSD plates. Stock solutions (see Materials) were thawed at RT, diluted to a final concentration of 1.0 μg/ml in PBS, and 30 μl of coating solution were added per well. Plates were incubated at 4°C over night (see for plate layout). The coating solution was discarded and plates were blocked with 50 μl 3% (w/v) BSA in PBS for one h at RT on a microtiter plate shaker (~500 rpm) followed by three washing steps with 50 μl washing buffer [PBS-T: PBS with 0.05% (v/v) Tween 20].
Antibody samples were diluted to 100 nM and 10 nM in assay buffer [PBS with 0.5% (w/v) BSA, 0.05% (v/v) Tween 20]. As controls, the MOR ref. mAb (, columns 23, 24) and assay buffer (columns 1, 2) were used. Samples and controls were added at 30 μl/well and incubated for 3 h at RT on a microtiter plate shaker (see for assay flow scheme).
The plates were washed three times with 50 μl washing buffer and 30 μl detection antibody (ECL-labeled anti-human Fab, diluted 1:2,000 in assay buffer) were added per well and incubated for 1 h on a microtiter plate shaker (~500 rpm). After washing, 35 μl/well 1× Read buffer T with surfactant was added. ECL signals were detected immediately using an MSD SECTOR® Imager 6000 instrument.
All pipetting and dispensing steps were performed using the liquid handling station epMotion® 5075 (Eppendorf).
Data evaluation
Data evaluation was done with Microsoft Excel 2010. Raw ECL signals from the measurement were transferred to a customized Excel evaluation template. The raw ECL signals in columns 1 (blank, secondary ECL-labeled detection antibody only), 2 [second blank or System Suitability Test (SST)], 23 (signal of 100 nM MOR ref. mAb) and 24 (10 nM MOR ref. mAb) were kept as uncorrected ECL signals and reported as those in the evaluation sheet. For all wells from columns 3–22 (samples 1–10), the following calculation was performed: Each ECL signal of a sample at a concentration of “x” nM for binding to protein “y” was divided by the ECL signal of MOR ref. mAb at a concentration of “x” nM for binding to protein “y,” i.e., each binding signal was normalized to binding of the “MOR ref. mAb” (adapted from Lueking et al.Citation11).
These binding ratios (BRs) were reported in the evaluation sheet and ranked using a color scheme. Specific antigen (i.e., target) binding was shown in graded color scale from white to blue, where blue was representing a BR of ≥ 50. For all other proteins, BRs up to 5 were considered as noncritical and were shown in green. BRs from 5 to 10 represent a low to moderate unspecific binding and were shown with a graded color scale from green to yellow. Higher BRs (from 10 to 50 and above) were shaded red and represent significant unspecific binding to the respective protein.
For ranking of an antibody, the BRs of all proteins (25 in total including BSA blanks) with the exception of the targets and control proteins, i.e., Protein A, anti-human Fab (Dianova), anti-human Fab (Bethyl) and anti-human Fc were summed up individually for 100 nM and 10 nM sample concentration. This calculation procedure yielded the cumulative binding ratio, CBR. A CBR up to 75 (i.e., an average BR of 3 for each protein) is depicted in green, a CBR up to 125 in yellow and a CBR above 200 in red. For an overall ranking, the sum of CBR at 100 nM and 10 nM was calculated and the same color scheme was used as before, but with doubled thresholds: up to 150, green; up to 250, yellow; above 400, red. For a final evaluation, the values were reviewed thoroughly. If single outliers would have changed the overall ranking of the antibody, as shown in for ustekinumab (plate 2, well F8), the coloring scheme was adapted manually to exclude single false positive values from the evaluation.
| Abbreviations: | ||
| 3P | = | Protein Panel Profiling |
| BSA | = | bovine serum albumin |
| ECL | = | electrochemiluminescence |
| ELISA | = | enzyme-linked immunosorbent assay |
| GFP | = | green fluorescent protein |
| HuCAL® | = | human combinatorial antibody library |
| mAb | = | monoclonal antibody |
| MSD | = | Meso Scale Discovery |
| OTA | = | off-target activity |
| PBS | = | phosphate buffered saline |
| RT | = | room temperature |
| SST | = | system suitability test |
Additional material
Download Zip (113 KB)Acknowledgments
The authors thank Jürgen Klattig, Swen Zierow, Francis Dodeller and the Protein Production Group for helpful discussions and provision of antibodies and Stefan Härtle, Josef Prassler and Paul Wiegel for helpful comments and reviewing of the manuscript.
Disclosure of Potential Conflicts of Interest
All authors are employees of MorphoSys AG. HuCAL®, HuCAL GOLD® and HuCAL PLATINUM® are registered trademarks of MorphoSys AG.
References
- Zhou H, Zhang YL, Lu G, Ji H, Rodi CP. Recombinant antibody libraries and selection technologies. N Biotechnol 2011; 28:448 - 52; http://dx.doi.org/10.1016/j.nbt.2011.03.013; PMID: 21477669
- Reichert JM. Which are the antibodies to watch in 2012?. MAbs 2012; 4:1 - 3; http://dx.doi.org/10.4161/mabs.4.1.18719; PMID: 22327425
- Reichert JM. Marketed therapeutic antibodies compendium. MAbs 2012; 4:413 - 5; http://dx.doi.org/10.4161/mabs.19931; PMID: 22531442
- Notkins AL. Polyreactivity of antibody molecules. Trends Immunol 2004; 25:174 - 9; http://dx.doi.org/10.1016/j.it.2004.02.004; PMID: 15039043
- Mohan S, Kourentzi K, Schick KA, Uehara C, Lipschultz CA, Acchione M, et al. Association energetics of cross-reactive and specific antibodies. Biochemistry 2009; 48:1390 - 8; http://dx.doi.org/10.1021/bi801901d; PMID: 19166328
- Hötzel I, Theil FP, Bernstein LJ, Prabhu S, Deng R, Quintana L, et al. A strategy for risk mitigation of antibodies with fast clearance. MAbs 2012; 4:753 - 60; http://dx.doi.org/10.4161/mabs.22189
- Zhu H, Bilgin M, Bangham R, Hall D, Casamayor A, Bertone P, et al. Global analysis of protein activities using proteome chips. Science 2001; 293:2101 - 5; http://dx.doi.org/10.1126/science.1062191; PMID: 11474067
- Diehnelt CW, Shah M, Gupta N, Belcher PE, Greving MP, Stafford P, et al. Discovery of high-affinity protein binding ligands--backwards. PLoS One 2010; 5:e10728; http://dx.doi.org/10.1371/journal.pone.0010728; PMID: 20502719
- Lueking A, Possling A, Huber O, Beveridge A, Horn M, Eickhoff H, et al. A nonredundant human protein chip for antibody screening and serum profiling. Mol Cell Proteomics 2003; 2:1342 - 9; http://dx.doi.org/10.1074/mcp.T300001-MCP200; PMID: 14517340
- Feyen O, Lueking A, Kowald A, Stephan C, Meyer HE, Göbel U, et al. Off-target activity of TNF-alpha inhibitors characterized by protein biochips. Anal Bioanal Chem 2008; 391:1713 - 20; http://dx.doi.org/10.1007/s00216-008-1938-7; PMID: 18344017
- Lueking A, Beator J, Patz E, Müllner S, Mehes G, Amersdorfer P. Determination and validation of off-target activities of anti-CD44 variant 6 antibodies using protein biochips and tissue microarrays. [Pi-Pv.] Biotechniques 2008; 45; http://dx.doi.org/10.2144/000112898; PMID: 18855764
- Ponsel D, Neugebauer J, Ladetzki-Baehs K, Tissot K. High affinity, developability and functional size: the holy grail of combinatorial antibody library generation. Molecules 2011; 16:3675 - 700; http://dx.doi.org/10.3390/molecules16053675; PMID: 21540796
- Prassler J, Thiel S, Pracht C, Polzer A, Peters S, Bauer M, et al. HuCAL PLATINUM, a synthetic Fab library optimized for sequence diversity and superior performance in mammalian expression systems. J Mol Biol 2011; 413:261 - 78; http://dx.doi.org/10.1016/j.jmb.2011.08.012; PMID: 21856311
- Steidl S, Ratsch O, Brocks B, Dürr M, Thomassen-Wolf E. In vitro affinity maturation of human GM-CSF antibodies by targeted CDR-diversification. Mol Immunol 2008; 46:135 - 44; http://dx.doi.org/10.1016/j.molimm.2008.07.013; PMID: 18722015
- Ewert S, Honegger A, Plückthun A. Structure-based improvement of the biophysical properties of immunoglobulin VH domains with a generalizable approach. Biochemistry 2003; 42:1517 - 28; http://dx.doi.org/10.1021/bi026448p; PMID: 12578364
- Brocks B, Garin-Chesa P, Behrle E, Park JE, Rettig WJ, Pfizenmaier K, et al. Species-crossreactive scFv against the tumor stroma marker “fibroblast activation protein” selected by phage display from an immunized FAP-/- knock-out mouse. Mol Med 2001; 7:461 - 9; PMID: 11683371
- Abdiche YN, Malashock DS, Pons J. Probing the binding mechanism and affinity of tanezumab, a recombinant humanized anti-NGF monoclonal antibody, using a repertoire of biosensors. Protein Sci 2008; 17:1326 - 35; http://dx.doi.org/10.1110/ps.035402.108; PMID: 18505735
- Abdiche YN, Lindquist KC, Stone DM, Rajpal A, Pons J. Label-free epitope binning assays of monoclonal antibodies enable the identification of antigen heterogeneity. J Immunol Methods 2012; 382:101 - 16; http://dx.doi.org/10.1016/j.jim.2012.05.010; PMID: 22609372
- Lauer TM, Agrawal NJ, Chennamsetty N, Egodage K, Helk B, Trout BL. Developability index: a rapid in silico tool for the screening of antibody aggregation propensity. J Pharm Sci 2012; 101:102 - 15; http://dx.doi.org/10.1002/jps.22758; PMID: 21935950
- Spencer S, Bethea D, Raju TS, Giles-Komar J, Feng Y. Solubility evaluation of murine hybridoma antibodies. MAbs 2012; 4:319 - 25; http://dx.doi.org/10.4161/mabs.19869; PMID: 22531448
- Rhyne PW, Wong OT, Zhang YJ, Weiner RS. Electrochemiluminescence in bioanalysis. Bioanalysis 2009; 1:919 - 35; http://dx.doi.org/10.4155/bio.09.80; PMID: 21083063
- Lu Y, Wong WL, Meng YG. A high throughput electrochemiluminescent cell-binding assay for therapeutic anti-CD20 antibody selection. J Immunol Methods 2006; 314:74 - 9; http://dx.doi.org/10.1016/j.jim.2006.05.011; PMID: 16814318
- Scholz M, Lüking A. A protein-stabilizing technology for enhanced antibody stability and antibody-binding profiles in a microchip array. Biotechnol J 2012; 7:1002 - 7; http://dx.doi.org/10.1002/biot.201100494; PMID: 22422601
- Berinstein NL, Grillo-López AJ, White CA, Bence-Bruckler I, Maloney D, Czuczman M, et al. Association of serum Rituximab (IDEC-C2B8) concentration and anti-tumor response in the treatment of recurrent low-grade or follicular non-Hodgkin’s lymphoma. Ann Oncol 1998; 9:995 - 1001; http://dx.doi.org/10.1023/A:1008416911099; PMID: 9818074
- Loyet KM, Deng R, Liang WC, Wu Y, Lowman HB, DeForge LE. Technology comparisons for anti-therapeutic antibody and neutralizing antibody assays in the context of an anti-TNF pharmacokinetic study. J Immunol Methods 2009; 345:17 - 28; http://dx.doi.org/10.1016/j.jim.2009.03.012; PMID: 19345224
- Dabitao D, Margolick JB, Lopez J, Bream JH. Multiplex measurement of proinflammatory cytokines in human serum: comparison of the Meso Scale Discovery electrochemiluminescence assay and the Cytometric Bead Array. J Immunol Methods 2011; 372:71 - 7; http://dx.doi.org/10.1016/j.jim.2011.06.033; PMID: 21781970