 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Size exclusion high performance liquid chromatography analysis of a human monoclonal antibody (mAb) showed the presence of a new species that eluted with a retention time between the dimeric and monomeric species of the antibody. Extensive characterization of this species, referred to as “shoulder,” indicated that it was a mAb containing an extra light chain and had a molecular weight of approximately 175 kDa. The extra light chain was found to be non-covalently associated with the Fab portion of the protein. The relative amount of shoulder (typically 1−3% of the total mAb present) varied with the Chinese hamster ovary cell line producing the mAb and was not influenced by the growth conditions. Our three-step mAb purification platform using protein A, anion exchange, and cation exchange process steps was successful at removing dimer and higher and lower molecular weight species, but not the shoulder impurity. It was found that hydrophobic interaction chromatography could be used in place of cation exchange to exploit the subtle differences in hydrophobicity between monomer and shoulder. We developed an antibody polishing process using Butyl Sepharose HP resin that is capable of removing the majority of high and low molecular weight impurities yielding 99% pure mAb monomer, virtually devoid of the shoulder species, with a step recovery of about 80%.
Introduction
Many biotherapeutics must be delivered to patients in large doses given over long periods of time. To provide the safest and most efficacious drug possible, the protein should be in its purest form. Protein aggregates and certain post-translational modifications have the potential to affect drug performance (potency, pharmacokinetics, pharmacodynamics) and safety (immunogenicity, adverse effects).Citation1-Citation7 As a result, it is imperative that the purity of biotherapeutics is determined using multiple analytic methods so that the levels of unwanted impurities can be closely monitored throughout the cell culture and purification processes.
Size exclusion high-performance liquid chromatography (SE-HPLC) analysis of a human monoclonal antibody (mAb), referred to herein as mAb-X, identified a mAb species that eluted between the monomeric and dimeric species of the antibody (). The presence of similar species has been reported. Researchers at Amgen identified one species as a mAb with oxidation of a heavy chain tryptophan residue by mass spectrometry analysis.Citation8,Citation9 Genentech’s THIOMAB product also included a species that eluted just prior to monomeric mAb during SE-HPLC analysis.Citation10,Citation11 After extensive characterization, the species was identified as a mAb monomer with an additional light chain covalently associated through a disulfide bond formed between a light chain cysteine and an engineered cysteine in either the heavy or light chains of the THIOMAB molecule. Genentech researchers recently reported the thorough characterization of a mAb species containing variants with one and two additional light chains present at 0.2% when produced in Chinese hamster ovary (CHO) cell culture.Citation12 They determined that the relative quantity of this species was related to the redox environment of the cell culture, which implicates disulfide bonding as the mode of binding for the additional light chains.
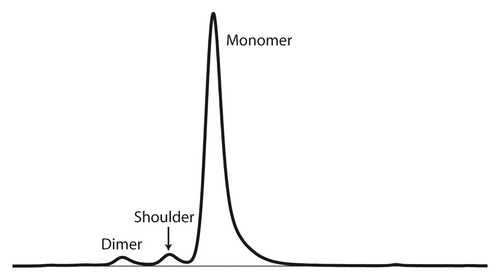
Figure 1. SE-HPLC chromatogram of mAb-X following protein A purification designating peaks for monomer, dimer, and the shoulder species.
In this work, we report the characterization of a mAb size variant using SE-HPLC coupled with multi-angle light scattering (MALS), matrix-assisted laser desorption/ionization-time of flight (MALDI-TOF) mass spectrometry, capillary gel electrophoresis and N-terminal sequencing, and measurement of mAb activity by binding affinity (Octet and ELISA) and neutralization assays. We showed that our mAb species, like Genentech’s, is due to the incorporation of a third light chain; however, it is novel in that the extra light chain is associated through non-covalent interactions with an otherwise properly assembled mAb-X monomer. In addition, we developed a robust and effective purification strategy to remove this unusual mAb species. Using hydrophobic interaction chromatography (HIC), we were able to reduce the levels of the shoulder species from ~3% to < 0.5% with good step recovery.
Results
Initial detection of shoulder
The first indication that an unusual mAb species was present in the mAb-X preparation came from SE-HPLC analysis. The SE-HPLC chromatogram of a protein A-purified mAb-X sample showed about 3% of an impurity that eluted with a retention time between that of dimeric and monomeric mAb-X and appeared as a shoulder on the monomer peak (). The term “shoulder” is used throughout this paper when referring to this mAb species.
Molecular weight determination
The molecular weight (MW) of species present in a mAb-X reference standard and samples enriched in the shoulder or monomer species of mAb-X was determined by SE-HPLC with MALS (). The average MW of the monomer was 152.0 kDa, which is 3.6% above the theoretical MW of 146.7 kDa. The MW of the shoulder was determined to be 175.9 kDa, 23.9 kDa above the measured MW of 152.0 kDa for a monomer. The theoretical MWs do not take glycosylation into account. The difference in measured MW of the shoulder species and the monomer species is very close to the theoretical light chain MW of 23.3 kDa. The finding was consistent with the shoulder species being a mAb with two heavy and three light chains.
Table 1. Molecular weight of mAb-X samples as determined by multiangle light scattering
Shoulder-enriched samples were also analyzed by MALDI-TOF intact mass analysis. A peak was detected in the shoulder sample that had the MW of mAb-X with three light chains (native + 1LC, 172 668 Da). This peak was absent in monomer and reference standard preparations, which contained little or non-detectable amounts of the shoulder species. These observations are consistent with the assignment of shoulder as a mAb monomer with an additional light chain (Fig. S1).
Non-reduced SDS-PAGE
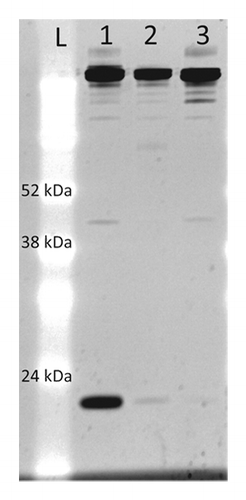
mAb preparations enriched for the shoulder and monomer species were characterized by non-reduced SDS-PAGE. A shoulder-enriched sample (95% monomer) was run on a non-reducing SDS-PAGE gel and compared with a mAb-X reference standard and purified monomer. A very prominent band with a MW of ~24 kDa was evident in the shoulder-enriched sample that was nearly absent in the reference standard and monomer samples (). The MW of this lower band was in good agreement with that of a free light chain (23.9 kDa). The third light chain appeared to dissociate from the mAb under denaturing conditions in SDS, but did not dissociate from the antibody under the high salt and Tween 80 conditions used in the protein A chromatography step for mAb-X purification. This suggests that hydrophobic interactions may be involved in the binding of the third light chain to mAb-X.
Figure 2. Non-reducing SDS-PAGE of mAb-X preparations. Five µg of shoulder-enriched mAb (lane 1), mAb-X reference standard (lane 2), and monomer-enriched (lane 3) were electrophoresed in a 14% polyacrylamide Tris-Glycine SDS gel and stained with SYPRO Ruby.
A gel slice containing the 23 kDa protein from a shoulder-enriched sample was excised from a non-reducing SDS-PAGE gel and digested with trypsin or chymotrypsin. Electrospray ionization mass spectrometry sequencing of the band revealed that it was full length mAb-X light chain (data not shown). We did not detect any cysteine oxidation in these samples. These data rule out a scenario in which the light chain is unable to make an intermolecular disulfide bridge with the heavy chain of the antibody due to either lack of, or oxidation of, the C-terminal cysteine residue.
SDS capillary gel electrophoresis
Reducing SDS-CGE was used to estimate the ratio of heavy chain to light chain. The amount of light chain increased as the purity of the shoulder sample increased (), which suggested that an extra light chain was present. The electropherogram for the reduced monomer- and shoulder-enriched samples () also shows an increase in peak area for the peak associated with the light chain. We calculated the theoretical ratio of heavy chain to light chain mass for a given purity of shoulder assuming that the ratio of subunits is 3LC:2HC (). The experimentally observed peak area ratio of heavy chain to light chain was in agreement with the predicted peak area ratio for shoulder-enriched fractions, indicating that the shoulder was a mAb monomer with a third light chain.
Table 2. Analysis of mAb-X reference standard and monomer- and shoulder-enriched mAb-X preparations by reducing and non-reducing SDS CGE. Values in parenthesis are theoretical. For reducing CGE an assumption of a subunit ratio of 3 light chains (LC) to 2 heavy chains (HC) was used. For non-reducing CGE Equation 1 was used to calculate LC percentages.
Figure 3. Electropherograms for reducing (A) and non-reducing (B) SDS capillary gel electrophoresis experiments. In each electropherogram, a monomer-enriched sample (black) containing 99.5% monomer, 0.5% dimer and a shoulder-enriched sample (red) containing 93.4% shoulder, 1.7% monomer, and 4.9% dimer are overlaid to highlight the differences between the two species.
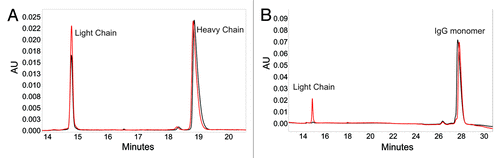
To quantify the amount of the third non-covalently associated light chain present in mAb-X, a non-reduced SDS-CGE experiment was performed. The results from the non-reduced SDS-CGE experiment are presented in . shows an overlay of the monomer-enriched and shoulder-enriched samples, which accentuates the additional light chain present in the shoulder-enriched sample. Six peaks were identified in the mAb-X reference standard, a main peak that corresponded to intact monomer and five minor peaks. Four preparations of mAb with increasing abundance of shoulder species (0, 8, 85, and 93%) were analyzed. As the purity of the shoulder species increased, the amount of 23 kDa protein increased (minor peak 1) with a corresponding loss in the major peak area. Minor peak 1 was also detectable at about 1% in the mAb-X reference standard. The amount of protein in minor peak 1 correlated with the theoretical amount (calculated from Equation 1 in the Materials and Methods section) of the third light chain expected for the mAb-X preparations with differing amounts of shoulder species, shown in parentheses in . These observations are consistent with our assignment of minor peak 1 as the third light chain that was non-covalently associated with mAb-X.
Antigen binding by shoulder species
Association and dissociation rates of shoulder-enriched (> 98%) and monomer-enriched (>99%) preparations for the mAb-X target antigen were measured by Octet (ForteBio). mAb preparations were bound to protein A-coated biosensors that were then incubated in a solution containing the antigen. The kobs and kdissoc experimental rate constants were used to calculate a Kon and KD for both samples. The results are presented in . Analysis of the data determined an affinity constant of 233 nM for a shoulder-enriched preparation, 51 nM for the monomer-enriched preparation, and 75 nM for the mAb-X reference standard. These data also correlated well with ELISA data, in which the affinity of shoulder for antigen was ~20% of the mAb-X reference standard, and results from a cell-based potency assay showing the reduced ability of the shoulder species to neutralize the target antigen (data not shown).
Table 3. Octet affinity analysis of monomer- and shoulder-enriched mAb-X preparations
mAb-X production in CHO cells
The first mAb-X production cell line, cell line 1, produced 0.5−1 g/L with 1−2% shoulder. Because of the need for high titers from cell culture, a second cell line was generated. Cell line 2 produced 3−5 times more mAb than cell line 1 (2.5−3 g/L); however, the amount of shoulder also increased to 2–3%. Our goal was to maximize mAb production and minimize the amount of shoulder using either cell line 1 or 2. Experiments were conducted to investigate the effect of cell culture conditions on shoulder production. We investigated the medium composition, feed solution composition and feed strategy, osmolality, pH, CO2 concentration, and impellor speed to find conditions that resulted in high titers and low shoulder content. CHO cells were cultivated in an in-house, chemically-defined medium with feed supplements, which allowed us to examine their influence on mAb production and the amount of shoulder species. Culture parameters such as maximum cell density, titer, and culture duration were also analyzed.
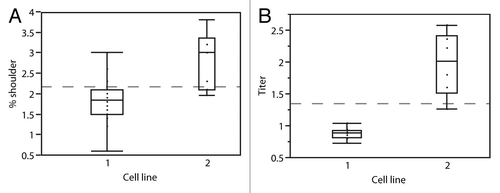
In total, 20 conditions were assessed for cell line 1 and 10 conditions for cell line 2. From the box plot in , the majority of the conditions tested for cell line 1 produced between 1.5 and 2% (median = 1.8%) shoulder and for cell line 2 between 2 and 3.5% (median = 3%). From the box plot in , the majority of the conditions tested resulted in mAb-X production levels of 0.7 and 2 g/L by cell lines 1 and 2, respectively. Within the cell culture conditions examined for the two mAb-X cell lines, there was no set of conditions that maximized mAb production and minimized the amount of shoulder species produced, especially as they relate to the titer () and cell density (data not shown).
Figure 4. (A) Distribution of shoulder species based on culture conditions for the two mAb-X production cell lines. (B) mAb-X production based on culture conditions for the two cell lines. Titer units are grams per liter.
mAb-X purification
Purification of mAb-X and removal of the shoulder species: a platform approach
The current platform for mAb purification was effective in purifying mAb-X monomer from dimer, high and low MW species, and process impurities. Initial purification experiments using harvest material from both cell lines were performed using a three-step purification procedure consisting of a protein A affinity column followed by anion exchange (AEX), then cation exchange (CEX) chromatographies. For cell line 1, the three-step purification process reduced the shoulder species by ~35% from 1.82% to 1.18%. The same purification scheme was followed for mAb-X produced by cell line 2. Shoulder was reduced from 3.21% to 2.84% for an overall reduction of 12%. From these experiments it was clear that this three-step purification process was not sufficient for removing appreciable quantities of the shoulder species.
Optimization of purification platform for shoulder removal
To satisfy stringent regulatory requirements, it is important to manufacture a mAb product that is as homogeneous for the monomer species as possible. As a result, we performed a series of experiments to determine whether optimizing the CEX and AEX steps would aid in the separation of shoulder from monomer. During the characterization of shoulder, it was determined that mAb-X monomer and the shoulder species had slightly different pI’s, 7.8−8.1 and 7.5–7.8, respectively (Fig. S2). We recognized that we could exploit this pI difference on either polishing resin. Our first strategy was to empirically modify the CEX process. We were able to remove some shoulder species through a combination of reducing the protein load density, reducing the elution salt concentration, and fractionating the elution peak. This strategy was sufficient for purification of mAb-X from cell line 1 resulting in final shoulder amounts of ~0.5%; however, purified mAb-X from cell line 2 still contained 2.5–3% shoulder after the modifications to the CEX process. In all cases, there was a significant loss of product. Several other pH and buffer conditions were tested for both polishing steps; however, none of the buffer conditions screened were successful at removing shoulder to acceptable levels. Taken together, the CEX and AEX optimization trials indicate that electrostatic effects, as they relate to charge distribution on the protein, are not strong enough to discriminate between mAb-X shoulder and monomer.
Utilization of hydrophobic interaction chromatography to remove shoulder
Optimization of the process conditions for the three-step purification platform (protein A, AEX, CEX) was not effective in removing appreciable levels of shoulder. As a result, we screened a series of hydrophobic interaction chromatography resins to determine whether shoulder could be discriminated from monomer by hydrophobicity. mAb-X is a particularly hydrophobic protein, so for these experiments we screened six HIC resins of varying hydrophobicities (GE Butyl Sepharose HP, Toyopearl Butyl 650S, GE Phenyl Sepharose Low Sub FF, Toyopearl Phenyl 650S, Toyopearl PPG 600M, and Toyopearl Ether 650S). mAb-X recovered from the Mustang Q purification step was loaded onto each column with 1.2 M ammonium sulfate and eluted using a linear gradient from 1.2–0 M ammonium sulfate in 20 mM citrate, pH 6.
The SE-HPLC analyses of fractions collected during gradient elution for the two resins that showed the best separation of shoulder from monomer are shown in . While all six resins were capable of discriminating between shoulder and monomer to a certain extent, we determined that the more hydrophobic resins resulted in better resolution of monomer and shoulder. We found that monomer binds more tightly to the resin compared with high MW, shoulder, dimer, and low MW species (data not shown).
Figure 5. Species distribution from SE-HPLC chromatograms for linear gradient elution from 1.2M to 0M ammonium sulfate (left to right) on two HIC resins. The blue bars represent the monomer, the green shoulder, the red represents the dimer, the teal represents the high molecular weight species, and the purple represents the low molecular weight mAb fragments.
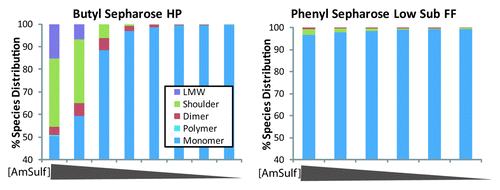
The GE Butyl Sepharose HP resin was the best at discriminating between mAb-X monomer and shoulder, dimer, and high MW species, resulting in product containing 99.7% monomer and < 0.1% shoulder by the end of the gradient. The only other resin tested that was capable of reducing the amount of shoulder to < 1% was GE Phenyl Sepharose Low Sub FF. The Phenyl Sepharose resin also produced very pure monomer (99.2% monomer); however, monomer did not bind as tightly to the resin. A comparison of the first elution fraction from the GE Butyl Sepharose HP resin with the GE Phenyl Sepharose Low Sub FF resin () showed that early fractions on the Butyl Sepharose HP resin were enriched in mAb impurities (51.4% monomer, 30.25% shoulder, 3.75% dimer, 0.6% high MW, and 15% low MW species), whereas the first Phenyl Sepharose Low Sub FF fraction contained 96.5% monomer, 2.8% shoulder, and 0.65% dimer. Because Butyl Sepharose HP produced better resolution of the mAb-X species, we also expected better recoveries from this separation. For these reasons we chose GE Butyl Sepharose HP resin for further evaluation.
Optimization of HIC process
Effect of ethanol on the elution pool volume
Although Butyl Sepharose HP resin was able to effectively separate the shoulder species from mAb-X monomer, elution of the mAb-X monomer from the resin was not efficient using a 20 mM citrate, pH 6.0, buffer. Under these conditions it required approximately 40 column volumes (CVs) of elution buffer to elute all of the mAb from the resin, indicating that mAb-X bound very tightly to the Butyl Sepharose HP resin. Since an elution pool volume of 40 CVs is not practical, ethanol was added to the elution buffer to make the protein elution step more effective. We initially assessed adding 5% ethanol to the elution buffer (20 mM citrate, pH 6.0). Using 5% ethanol in the elution buffer reduced the elution pool from 40 CVs to 20 CVs. The most concentrated elution fraction was quite pure, 98.9% monomer and 1.1% high MW species. Further optimization of the elution step resulted in a final elution buffer formulation of 20 mM citrate, pH 6, 12% ethanol. Under these conditions the mAb-X monomer elutes in approximately 5 CVs with no effect on mAb-X purity (99% monomer, 1% high molecular weight species). mAb recovery was 84%.
DOE study of GE butyl sepharose HP process
Prior to performing the process optimization experiments, a series of screening experiments were performed to determine the process ranges to investigate during the DOE runs. High-throughput process development was used to screen separation conditions because it allows a large number of conditions to be tested in a short period of time while also pushing the failure limits of the process.Citation13,Citation14 We performed HTPD experiments in a 96-well plate format to study the effects of load [NaCl], wash [NaCl] and wash CVs on the recovery and purity of mAb-X from the Butyl Sepharose HP resin. At this stage, we decided to switch the salt in the HIC buffer system from ammonium sulfate to sodium chloride. We saw no difference in the separation as a result of this change (data not shown). These screening experiments provided us with a good interval to design a set of experiments to optimize the GE Butyl Sepharose HP process in columns. We determined that the best range to study load [NaCl] was 0.7–1M, and the range for the wash [NaCl] was 0.2–0.4 M over 4–6 CVs.
A DOE experimental design was used to characterize the mAb-X HIC process. The study used Omnifit columns with a 5 mL resin bed volume. For this experiment, a 24-run response surface design was generated in JMP9. This model included three center point runs. The mAb-X load material for these experiments was pooled from several protein A purifications and was adjusted with 1:2 dilution with a 2× formulation of the appropriate equilibration buffer. mAb-X dimer and shoulder were each present at 3.4% in the load material The input parameters that were tested for this experiment were mAb-X load amount (13.5–27.4 mg/mL resin), load/equilibration [NaCl] (0.7–1 M), flow rate (100–175 cm/h, 2.2–3.8 min residence time), wash [NaCl] (0.2–0.4 M), and wash CVs (4–6 CVs). All buffers were prepared in 10 mM sodium citrate, pH 5.6, which was determined to be the best pH for this process from previous screening experiments. Eluate samples were tested for monomer and shoulder content and recovery by SE-HPLC, residual HCP by ELISA, and residual CHO DNA by qPCR. The contour plots for the raw data for the SE-HPLC and mAb recovery data are shown in .
Figure 6. Contour plots generated from the raw data for percent monomer (top), shoulder (middle), and recovery (bottom) as they relate to wash CVs, load amount and wash [NaCl].
![Figure 6. Contour plots generated from the raw data for percent monomer (top), shoulder (middle), and recovery (bottom) as they relate to wash CVs, load amount and wash [NaCl].](/cms/asset/d3d5efe9-988a-47f4-a623-40b6946b8f7c/kmab_a_10926192_f0006.gif)
The percent monomer, percent shoulder, and percent recovery data were modeled together in JMP9. Since residual HCP and DNA were close to, or below, the detection limit of the assay—and thus acceptable in all instances—for mAb recovered from the GE Butyl Sepharose HP resin under all process conditions, these results were not included in the model. Modeling of all three responses together resulted in a good, statistically significant, model (p < 0.05). Results from the process model indicated that load amount and wash CVs had the greatest effect on % monomer and % recovery, while wash [NaCl] also had an impact on % shoulder (Fig. S3).
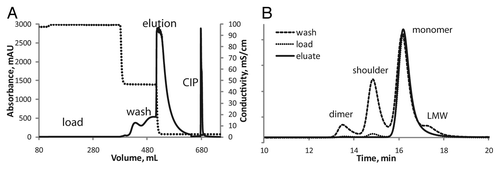
The strong correlation between recovery and purity was expected. In previous experiments, a loss in pure monomer was observed during an extended wash because the resin does not fully retain monomer during this step. shows a representative fast protein liquid chromatography (FPLC) chromatogram of mAb-X purification on a GE Butyl Sepharose HP resin column (A) and the SE-HPLC chromatograms for the load protein, wash, and eluate (B). The A280 does not return to baseline during the wash step even after washing with copious amounts of wash buffer (> 10 CVs). On analysis of the wash fractions, we found that after about six CVs the protein that elutes is predominantly mAb-X monomer (data not shown). Importantly, however, the early wash fractions are enriched in mAb-X shoulder and dimer (, long dashes). In an effort to minimize the loss of mAb-X monomer during the wash step, we halted the wash step at six CVs.
Figure 7. (A) Representative fast protein liquid chromatography chromatogram for the purification of mAb-X on GE Butyl Sepharose HP resin. The A280 is solid and the conductivity is a dashed line. (B) Representative SE-HPLC chromatograms from fractions from the GE Butyl Sepharose HP purification of mAb-X. The three chromatograms are scaled for effect: load (dots), wash fraction (long dashes), and eluate (solid).
The effect of the protein load on purity was not as obvious. We would expect as the amount of protein loaded onto the column increases (increasing the amount of impurities present) the purity of the mAb-X in the eluate would decrease; however, based on our fit model it is apparent that higher load amounts would result in a higher percentage of mAb-X monomer in the eluate. This can be observed in the contour plots from the raw data in (top row, first two panels). Because recovery tracks with percent mAb-X monomer, the fit model supports our observation that higher load amounts result in lower recoveries (e.g., compare data in first and second panels in top and bottom rows).
In addition to load amount and wash CVs, the salt concentration [NaCl] of the wash buffer was determined to have a significant effect on percent shoulder. We have observed that as the [NaCl] of the wash buffer decreases, more mAb-X monomer elutes from the resin during the wash step, resulting in a decrease in protein recovery. When wash [NaCl] is high, less monomer elutes from the resin, but the mAb-X monomer content is quite low. As a result, we must choose wash conditions that will satisfy our specifications for both percent shoulder and percent recovery.
There were also several interaction terms that were significant according to our DOE fit model (Fig. S3). The interactions predicted to have an effect on percent monomer in the eluate were load amount × wash CVs and wash [NaCl] × flow rate. Wash [NaCl] x flow rate was the only interaction term that exhibited an effect on percebt shoulder. The load amount × wash CVs, and load [NaCl] × wash CVs were predicted to play a role in the amount of mAb-X recovered in the eluate. Besides wash [NaCl] × flow rate, the other interaction terms were a combination of the significant main effects, which is to be expected. On closer analysis of the wash [NaCl] x flow rate interaction term, which was significant for percent monomer and percent shoulder, the effect was large at low flow rates, but as the flow rate reached 175 cm/h, the effect was insignificant.
Using the “maximize desirability” function in JMP we obtained the optimal run conditions for the Butyl Sepharose HP purification process. The run conditions were as follows: 25 mg/mL resin loaded in 10 mM citrate, pH 5.6, 1 M NaCl. The resin was washed with 10 mM citrate, pH 5.6, 0.2 M NaCl for 6 CVs. All steps were run at 100 cm/h. We tested these conditions on a 26.5 mL Butyl Sepharose HP column. The purification results were very good resulting in 99.3% monomer and 87.4% recovery by SE-HPLC. All CHO cell residuals were below the detection limit of the assay.
Determining safe operating space
The resulting HIC process was capable of removing the majority of the shoulder species under optimal conditions. The next series of experiments were conducted to understand the robustness of the HIC process. By understanding the process well, ranges for each of the process parameters in which acceptable levels of impurities are achieved can be determined. shows contour plots generated from the fit model for percent monomer and recovery. The shaded areas indicate regions in the experimental space that do not meet our minimum criteria of >98.5% monomer (red), <0.5% shoulder, and >80% recovery (green). The white space represents the safe operating window for the Butyl Sepharose HP process as it relates to wash CVs, wash [NaCl], and load amount. The safe operating ranges for each of the input factors for this purification process are a load amount of 13.5–23.5 mg/mL resin, [NaCl] of load material of 0.7–1.0 M, wash [NaCl] of 0.2–0.3 M over 5–6 CVs, all steps can be run at linear flow rates between 100–175 cm/h.
Figure 8. Design space for the Butyl Sepharose HP process as it relates to wash CVs and load amount (left) and wash [NaCl] and load amount (right). Shaded areas, % monomer in red, % shoulder in blue, and recovery in green, indicate conditions that would result in product < 98.5% monomer, > 0.5% shoulder, and < 80% recovery. The area in white is the region where our product meets, or exceeds, our purity and recovery criteria.
![Figure 8. Design space for the Butyl Sepharose HP process as it relates to wash CVs and load amount (left) and wash [NaCl] and load amount (right). Shaded areas, % monomer in red, % shoulder in blue, and recovery in green, indicate conditions that would result in product < 98.5% monomer, > 0.5% shoulder, and < 80% recovery. The area in white is the region where our product meets, or exceeds, our purity and recovery criteria.](/cms/asset/06ffaf56-ce27-4ef5-95b3-aedbbd1b8299/kmab_a_10926192_f0008.gif)
Five Butyl Sepharose HP experiments were performed to validate our predicted safe operating window. A variety of conditions at the edges of the safe operating window, as well as the optimal conditions, were tested on a 6 mL bed volume (7.6 cm × 0.7854 cm2) column. Using a different column and different mAb-X load material from the DOE runs, the Butyl Sepharose HP purification process was run under optimal conditions and obtained a mAb-X purity of 98.6% monomer and a recovery of 80.7%. Data from the five validation runs are summarized in . The average percent monomer for the five validation runs was 98.2 ± 0.3% and the recovery was 79.7 ± 3.5%. From these experiments, we confirmed that our optimal conditions, and the safe operating ranges, are adequate according to our purity and recovery requirements for this process.
Table 4. Conditions and results for DOE validation runs for the Butyl Sepharose HP column chromatography process
Discussion
We identified a novel mAb species containing a non-covalently associated third light chain that is present at 1% to 3% in crude cell culture supernatants. Two other examples of a mAb containing a third light chain have been reported in the literature. These species are believed to be formed through a disulfide bond either to an engineered cysteine,Citation10,Citation11 or the C-terminal cysteineCitation12 in the mAb light chain, neither is the case for the three light chain species discussed in this paper. We have shown that the third light chain is not associated through disulfide bonding, but rather is non-covalently attached to mAb-X monomer.
Shoulder is produced de novo in cell culture. It is detectable in culture supernatants by SE-HPLC as early as day 4 of growth and does not accumulate during the protein purification process. Accelerated stability studies on mAb-X showed a decrease in the amount of shoulder present over time, while the relative amount of dimer and fragment increased, which implies that shoulder is not produced as a result of mAb instability (data not shown).
After extensive work to characterize the shoulder species, we determined that the third light chain possesses the same sequence as the mAb-X light chain, and thus is not from a host cell IgG. The shoulder species is slightly more acidic than mAb-X monomer and shows ~70% less activity in both ELISA, target binding, and neutralization assays. Evidence from a series of SDS-PAGE gel and SE-HPLC analyses in which LysC, a protease specific for the IgG hinge region, was used to cleave mAb-X into Fc and Fab fragments indicated that the third light chain was associated with the Fab fragment (data not shown). The location of the third light chain is consistent with the potency results indicating that the extra light chain interfers with antigen binding.
Three light chain-containing mAbs may be more common than previously thought. Because we do not currently know the clinical impact of this species, it is important that we have a method to remove this species if it is encountered. Our mAb purification platform, which consists of Protein A → AEX → CEX, was not sufficient to eliminate appreciable amounts of the shoulder species. We determined that a HIC process using Butyl Sepharose HP resin, instead of a CEX chromatography step, was the best option for separating mAb-X monomer from the shoulder impurity. The load, wash and elution conditions for the HIC process were optimized using a design of experiments approach. We now have a robust process capable of purifying mAb-X to >99% monomer and <0.3% shoulder with recoveries greater than 80%.
Materials and Methods
Antibodies
The monoclonal antibody studied is a fully human IgG1 mAb in which the variable region from an IgG3 antibody was grafted onto the constant region of an IgG1 mAb. The antibody was expressed in CHO cells. It is identified as mAb-X in this paper.
Enriching shoulder by SE-HPLC
For purification of the shoulder species, 2 mg portions of mAb-X were loaded onto a ProPak 300 SE-HPLC column (Waters) using a Waters 1525 Binary HPLC Pump. Protein peaks were detected at 280 nm using a Waters 2487 dual wavelength detector. The mobile phase was PBS, pH 6.5, at a flow rate of 0.7 mL/min. The shoulder-containing fraction was collected and kept at 4 °C. Once 25 runs were complete all shoulder fractions were pooled and concentrated by centrifugation through an ultrafiltration membrane (Amicon Ultracel-15, 30 kDa MWCO, Millipore). The concentrated shoulder fraction was again resolved on a ProPak 300 SE-HPLC column. The final shoulder prepared using the SE-HPLC enrichment protocol yielded a sample consisting of 96.5% three-light chain species and 3.5% monomer. Six other batches of shoulder-enriched mAb were prepared in a similar manner; they contained 84, 85, 92, 93, 96.5, and >98% shoulder species. The same procedure was followed to generate purified monomer and dimer species. Purified mAb-X preparation identified as “mAb-X reference standard” that contained 1% shoulder species was also analyzed.
Multiangle light scattering
The MWs of monomer, dimer and shoulder species were determined by SE-HPLC with refractive index and MALS detection, performed at the Keck Core Facility at Yale University. Samples enriched in monomer, dimer or shoulder were resolved on a Waters ProPak 300 SE-HPLC column, using a PBS, pH 6.5, mobile phase with a flow rate of 1.0 mL per min. Eluted peaks were detected with Wyatt OptiLab rEX refractive index and Wyatt DAWN HELEOS multiangle light scattering detectors. MWs were determined by ASTRA software (Wyatt) using a protein concentration determined by the refractive index detector. The dn/dc value for protein was set to 0.175. A Debye fit was used to model the light scattering data.
Sequencing of mAb-X Light chain
The 23.9 kDa band was excised from the SDS-PAGE gel using a clean razor blade, washed with 1% acetic acid, 50% methanol, and electroeluted into PBS buffer. The band was then digested with trypsin or chymotrypsin, and the resulting digested peptide fragments were separated by reverse phase HPLC using a C18 column (Vydac) and increasing gradient of 0.1% TFA/ACN buffer over 40 min. Eluted peptides injected onto a Q-TOF ESI-mass spectrometer (Thermo Scientific) and analyzed by TOF for sequence and oxidative modifications.
Reducing and non-reducing SDS-PAGE and SDS capillary gel electrophoresis (CGE)
SDS CGE was used to analyze the Mab-X reference standard, a preparation enriched for monomer and enriched shoulder preparations of varying purity were diluted to a concentration of 1 mg/mL in SDS sample buffer (100 mM Tris, pH 9, 1% SDS). β-mercaptoethanol (5%) was included in the reducing buffer and sodium iodoacetate (12.5 mM) was included in the non-reducing buffer. A 10 kDa internal migration standard was added to all samples. Samples were denatured by heating at 70°C for 10 min. Species in the denatured samples were separated by electrophoresis though a sieving gel matrix (Beckman Coulter) in a 50 μm I.D. bare-fused silica capillary with 20 cm effective length using a Proteome Lab PA800 CE system. Separation was performed at -15kV for 35 min. Absorbance at 214 nm was monitored during separation. Peak areas were determined using 32 Karat Software (Beckman Coulter).
The theoretical amount of shoulder species present was determined using the following equation:
Where Fs is the fraction of shoulder present in the sample, as determined by SEC-HPLC.
Samples for slab-gel SDS-PAGE were diluted to 0.5 mg/mL in purified water and then diluted in an equal volume of Laemmli SDS sample buffer (BioRad). β-mercaptoethanol (5%) was added for the reducing SDS-PAGE buffers and samples. The samples were denatured by heating to 95 °C for 5 min before loading on a precast 10% NuPAGE bis-Tris gel (Invitrogen, Life Technologies). Separation was performed at a constant 200 V. Proteins in the gels were stained with SYPRO Ruby (Invitrogen, Life Technologies).
Target binding kinetics
Assays to measure kinetics of antibody binding to its target antigen were performed on an Octet QK (ForteBio) system. mAb-X preparations enriched for shoulder (>98%) or monomer (>99%) and the mAb-X reference standard were immobilized onto protein A-coated Octet biosensors, which were then washed in PBS for 300 s. The Octet biosensors were then incubated in a solution containing 1.6 µM target antigen and binding to the antibody-coated biosensors was monitored for 900 s. An association rate constant (kobs) was determined from this incubation step. The biosensors were then incubated in PBS and dissociation of antigen from the antibody was monitored for 1000 s, allowing the dissociation rate constant (kdissoc) to be determined. These kobs and kdissoc experimental values were then used to calculate a kon rate constant and KD affinity constant for shoulder-enriched sample. Experimentally determined rate constants were obtained through partial curve fits with > 0.996 R2 values.
Cell culture conditions for mAb-X production
A suspension-adapted CHO K1 cell line was used in this study. These cells were transfected with the cDNA of the mAb-X molecule using two different proprietary expression vectors. The resulting cell lines (cell lines 1 and 2) were cultivated in shake flasks and in DasGip bioreactors using chemically defined medium and feeds. Cells were seeded at 0.4 million cells/mL and the feed started on day 2–3. Temperature of the cultures was maintained at 36.5 °C, dissolved oxygen was controlled at 40% of air saturation, and the culture pH was maintained at pH 7.0 ± 0.2 using 1 N NaOH or CO2. Fed-batch cultures lasted typically for 14 d and the viability at the end of the culture was higher than 50%. Cell density and viability were determined by CEDEX (Innovatis); glucose, lactate, glutamine, and ammonia were measured using a NOVA analyzer (Bioprofile 100). In addition to feed supplement, glucose was maintained at 4.5 g/L using a 400 g/L stock solution of glucose.
SE-HPLC assay
Various mAb-X species (dimer, shoulder, and monomer) were quantified by SE-HPLC using a Tosoh TSKgel G300SWxl column. The method also quantified high and low MW species if they were present in a sample. An isocratic elution (1.0 mL/min) was performed in PBS, pH 6.5 with 0.02% sodium azide on a Waters 1525 Binary HPLC Pump. Protein peaks were detected at 280 nm using a Waters 2487 dual wavelength detector.
Purification of mAb-X
All Fast Protein Liquid Chromatography (FPLC) experiments were performed on a GE AKTApurifier 100 or a BioRad BioLogic DuoFlow 40 system.
mAb-X was initially purified by a three-step process. The clarified culture was loaded onto a ProSep-vA Ultra (EMD Millipore) column that was equilibrated with 20 mM sodium phosphate, pH 7.5, 150 mM NaCl, 0.5% Tween 80. The column was then washed with 6 column volumes (CVs) of 20 mM sodium phosphate, pH 7.5, 1 M NaCl, 0.5% Tween 80, followed by 6 CVs of 10 mM sodium phosphate, pH 6.0. The mAb was eluted with 20 mM sodium acetate, pH 3.0, and the eluate was then adjusted to pH 5.0 with 2 M Tris.
The protein A eluate was run in flow-through mode on a Mustang Q filter (Pall Corporation). The filter was charged with 1 M NaCl, equilibrated with 20 mM Tris, pH 7.5. The mAb was conditioned for loading by increasing the pH to pH 7.5 with 2 M Tris and then dropping the conductivity to 3.0 mS/cm with MilliQ water. After the mAb was completely loaded, the membrane was washed with five membrane volumes of 20 mM Tris, pH 7.5. Portions of filtrate containing mAb were collected. The membrane was then stripped with 1 M NaCl. The pH of the protein solution was adjusted to pH 5 with 1 M HCl.
Following the Mustang Q step, the mAb was applied to a Fractogel SE HiCap (EMD Millipore) column. The column was equilibrated with 20 mM sodium acetate, pH 5.0. After all of the mAb was loaded, the column was washed with 20 mM sodium citrate, pH 6.0, and the protein was eluted with 20 mM sodium citrate, pH 6.0, 100 mM NaCl. mAb in the eluate was collected in three fractions: rise, main, and tail based on protein absorbance at 280 nm.
HIC resin screen
Six HIC resins, Tosoh’s Toyopearl Butyl 650S, Phenyl 650S, PPG 600M, Ether 650S, and GE’s Butyl Sepharose HP and Phenyl Sepharose Low Sub FF, were evaluated. These experiments were performed on 5 mL columns (Omnifit, 0.7854 cm2 cross section area). The protein load material was adjusted to 1.2 M (NH4)2SO4 by dilution with an equal volume of 2.4 M (NH4)2SO4 in 20 mM citrate, pH 6; the load was mAb-X recovered from the Mustang Q step of the purification process. Toyopearl Butyl 650S and GE Butyl Sepharose HP were loaded at 35 mg/mL resin; Toyopearl PPG 600M was loaded at 22 mg/mL resin; Toyopearl Ether 650S, Toyopearl Phenyl 650S, and GE Phenyl Sepharose Low Sub FF were loaded at 20 mg/mL resin. For all six resins, a linear elution gradient from 1.2 M to 0 M (NH4)2SO4 in 10 mM sodium citrate, pH 6.0, was run over 15 CVs. All steps were run at 100 cm/h. The resins were cleaned with 0.5 N sodium hydroxide and stored in 20% ethanol.
GE butyl sepharose HP elution buffer optimization
A 5 mL Butyl Sepharose HP column (Omnifit, 0.7854 cm2 cross-section area) was used to determine the optimal elution buffer. All steps were run at 100 cm/h. In these experiments, mAb-X recovered from the Mustang Q purification step was adjusted to 1.2 M NaCl with a 1:4 ratio of 4.8 M NaCl in 40 mM sodium citrate, pH 6.0, and loaded onto the column. The column was then washed with 6 CVs of 10 mM sodium citrate, pH 6.0, 0.4 M NaCl. The protein was eluted with 0, 5, 7.5, or 12% ethanol in 10 mM sodium citrate, pH 6.0. The column washes and flow-through were fractionated into 5 mL fractions. All samples were assayed by absorbance at 280 nm for protein concentration and representative fractions were analyzed by SE-HPLC.
High-throughput plate preparation and general procedure
A 96-well filter plate (Pall AcroPrep Advance 96-well, Pall Corporation), with a 1.2 µm Supor NTRL membrane was loaded with 20 µL of GE Butyl Sepharose HP resin. The resin was equilibrated three times with 200 µL of the appropriate equilibration buffer, incubating for 5 min each time. Buffer was removed by centrifugation at 1000 × g for 2 min and the flow through was collected in a Grenier Bio-one 1 mL receiver plate. mAb-X recovered from the Mustang Q step was loaded into the wells; the filter plate was then sealed (top and bottom) and placed on a Bellco Biotechnology Mini Orbital shaker (3-mm radius) at 1300 rpm shaking speed for two hours. The wash, elution, and CIP steps were incubated with shaking for 15 min prior to centrifugation.
HTP Evaluation of load and wash conditions
An HTP screening experiment was run to evaluate load and wash conditions for GE Butyl Sepharose HP resin purification. Load NaCl concentrations ranging from 1 M to 0.5 M (decreasing by 0.1 M changes in NaCl concentration across the plate) in 20 mM citrate, pH 5.6, were examined with different CVs of wash buffer: 5 CV, 6 CV, and 7 CV of 20 mM citrate, pH 5.6, 0.3 M NaCl. The 96-well filter plate was filled with 20 µL of resin per well and protein was loaded at 30 mg/mL resin in 20 mM citrate, pH 5.6, 1 M NaCl. Flow through, wash, and eluate samples were analyzed by SE-HPLC.
The effect of different wash NaCl concentrations on mAb-X purity and recovery was also examined. mAb-X recovered from the Fractogel SE purification step was loaded in 20 mM citrate, pH 5.6, 1 M NaCl at 30 mg/mL resin. The antibody in the load contained 2.54% shoulder species. The wash buffer NaCl concentration ranged from 0.4M to 0.15 M NaCl (decreasing by 0.05 M changes in NaCl concentration) in 20 mM citrate, pH 5.6. The protein eluate was assayed by SE-HPLC.
Design of experiment studies of GE butyl sepharose HP column chromatography
The Custom Designer tool in JMP9 was used to generate a response surface model, I-optimality criterion, yielding 24 column chromatography experiments, including 3 center point runs. These experiments were run on a 5 mL (Omnifit, 0.7854 cm2 cross section area) column. The different equilibration and wash buffers were made in 10 mM sodium citrate, pH 5.6, each containing a range of NaCl concentrations. In all experiments, the mAb was eluted with 6 CVs of 10 mM sodium citrate, pH 5.6, 12% ethanol. The elution peak was fractionated by protein concentration into two fractions: 1 to 3 mg/mL mAb-X (rise) and 3 mg/mL (rise) to 1 mg/mL (tail). Resin CIP used 10 mM sodium hydroxide and the column was stored in 10 mM sodium citrate, pH 6.0, 20% ethanol. The following factors were evaluated: load NaCl concentration (0.7–1 M), load amount (13.5–27.4 mg/mL resin), linear flow rate (100–175 cm/h), wash NaCl concentration (0.2–0.4 M), and wash volumes (4–6 CVs). Percent recovery, monomer and shoulder contents, and residual CHO host cell protein and CHO DNA from the 24 runs were used for the DoE analysis.
Scale-up of optimal conditions for GE butyl sepharose HP column chromatography
The protein load material was mAb-X recovered from the Mustang Q purification step; it was prepared for these studies by making a 1:2 dilution with a 2 × formulation of the equilibration buffer. All steps were run at 100 cm/h. The protein was loaded at 25 mg/mL resin. The column was equilibrated with 10 mM sodium citrate, pH 5.6, with 1 M NaCl and washed with 6 CVs 10 mM sodium citrate, pH 5.6, 0.2 M NaCl. The protein was eluted with 6 CVs of 10 mM sodium citrate, pH 5.6, buffer with 12% ethanol. Load, wash and eluate samples were assayed by SE-HPLC, and for residual CHO HCP and DNA.
Assays for residual CHO host cell protein and DNA
The capacity of GE Butyl Sepharose HP column chromatography to clear CHO HCP and DNA from mAb was measured in the scaled-up chromatography studies. HCP was quantified with an ELISA; DNA with a real-time PCR assay (both as described below). For the residual host cell protein ELISA, 96-well plates were coated overnight at 4°C with a rabbit anti-CHO protein IgG preparation at pH 7.2. The coated plates were incubated with antibody samples and CHO protein reference standards for 60 min at 37°C. The plates were washed and a biotinylated rabbit anti-CHO IgG preparation was added. After incubation for 60 min at 37°C, the plates were washed and a streptavidin-alkaline phosphatase conjugate was added. Unbound conjugate was removed by washing and alkaline phosphatase was detected by incubation with p-nitrophenyl substrate, and measuring absorbance at 405 nm.
The qPCR assay for residual host cell DNA used primers and a fluorescently-labeled probe that are specific for the Alu 49c DNA repeat found in the CHO genome. Assays were performed on an ABI 7300 Real Time PCR system and the data analyzed with SDS v1.4 software. To extract DNA from test samples, proteins were first cleaved using proteinase K in the presence of 3 M urea. Cleavage products were extracted with buffered phenol:chloroform:iosamyl alcohol (25:24:1), then with chloroform. DNA in the aqueous phase was precipitated with ethanol. Ten microliters of DNA sample was added to each 50 µL amplification reaction, which also contained the Taqman Universal PCR Master Mix, forward and reverse primers, and a 5′ fluorescein-3′TAMRA labeled probe. Purified reference DNA was used to calibrate the assay.
| Abbreviations: | ||
| mAb | = | monoclonal antibody |
| SE-HPLC | = | size exclusion HPLC |
| MALS | = | multi-angle light scattering |
| CHO | = | Chinese hamster ovary |
| HIC | = | hydrophobic interaction chromatography |
| SDS | = | sodium dodecyl sulfate |
| PAGE | = | polyacrylamide gel electrophoresis |
| CGE | = | capillary gel electrophoresis |
| MALDI-TOF | = | matrix-assisted laser desorption/ionization-time of flight |
| FPLC | = | fast protein liquid chromatography |
| PBS | = | phosphate-buffered saline |
| HTP | = | high throughput |
| CIP | = | clean in place |
| CV | = | column volume |
| DoE | = | design of experiments |
| HCP | = | host cell protein |
| HC | = | heavy chain |
| LC | = | light chain |
| AEX | = | anion exchange |
| CEX | = | cation exchange |
| HTPD | = | high throughput process development |
Additional material
Download Zip (1.2 MB)Acknowledgments
We would like to thank Chris Kerrigan, Monica Jiang, Lisa Ngu, Stephanie Callahan, Rayna Trietsch, Ying Xu, and Christine Taylor for their contributions to the characterization of shoulder and the development of HIC purification procedure. We would also like to thank Glenn Godwin, Bernie Creswick, and Sophia Kotsalidis for providing the mAb-X starting material. The SE-LS/UV/RI instrumentation was supported by NIH Award Number 1S10RR023748–01. The content of this paper is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Disclosure of Potential Conflicts of Interest
No potential conflict of interest was disclosed.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/mAbs/article/26192
References
- Hermeling S, Crommelin DJ, Schellekens H, Jiskoot W. Structure-immunogenicity relationships of therapeutic proteins. Pharm Res 2004; 21:897 - 903; http://dx.doi.org/10.1023/B:PHAM.0000029275.41323.a6; PMID: 15212151
- Kumar S, Singh SK, Wang X, Rup B, Gill D. Coupling of aggregation and immunogenicity in biotherapeutics: T- and B-cell immune epitopes may contain aggregation-prone regions. Pharm Res 2011; 28:949 - 61; http://dx.doi.org/10.1007/s11095-011-0414-9; PMID: 21437790
- Rosenberg AS. Effects of protein aggregates: an immunologic perspective. AAPS J 2006; 8:E501 - 7; http://dx.doi.org/10.1208/aapsj080359; PMID: 17025268
- Read EK, Park JT, Brorson KA. Industry and regulatory experience of the glycosylation of monoclonal antibodies. Biotechnol Appl Biochem 2011; 58:213 - 9; http://dx.doi.org/10.1002/bab.35; PMID: 21838794
- Raju TS. Terminal sugars of Fc glycans influence antibody effector functions of IgGs. Curr Opin Immunol 2008; 20:471 - 8; http://dx.doi.org/10.1016/j.coi.2008.06.007; PMID: 18606225
- Anthony RM, Ravetch JV. A novel role for the IgG Fc glycan: the anti-inflammatory activity of sialylated IgG Fcs. J Clin Immunol 2010; 30:Suppl 1 S9 - 14; http://dx.doi.org/10.1007/s10875-010-9405-6; PMID: 20480216
- Chung CH, Mirakhur B, Chan E, Le QT, Berlin J, Morse M, Murphy BA, Satinover SM, Hosen J, Mauro D, et al. Cetuximab-induced anaphylaxis and IgE specific for galactose-alpha-1,3-galactose. N Engl J Med 2008; 358:1109 - 17; http://dx.doi.org/10.1056/NEJMoa074943; PMID: 18337601
- Yang J, Wang S, Liu J, Raghani A. Determination of tryptophan oxidation of monoclonal antibody by reversed phase high performance liquid chromatography. J Chromatogr A 2007; 1156:174 - 82; http://dx.doi.org/10.1016/j.chroma.2007.01.140; PMID: 17379231
- Boyd D, Kaschak T, Yan B. HIC resolution of an IgG1 with an oxidized Trp in a complementarity determining region. J Chromatogr B Analyt Technol Biomed Life Sci 2011; 879:955 - 60; http://dx.doi.org/10.1016/j.jchromb.2011.03.006; PMID: 21440514
- Gomez N, Vinson AR, Ouyang J, Nguyen MD, Chen X-N, Sharma VK, Yuk IH. Triple light chain antibodies: factors that influence its formation in cell culture. Biotechnol Bioeng 2010; 105:748 - 60; PMID: 19845001
- Gomez N, Ouyang J, Nguyen MD, Vinson AR, Lin AA, Yuk IH. Effect of temperature, pH, dissolved oxygen, and hydrolysate on the formation of triple light chain antibodies in cell culture. Biotechnol Prog 2010; 26:1438 - 45; http://dx.doi.org/10.1002/btpr.465; PMID: 20568280
- Lu C, Liu D, Liu H, Motchnik P. Characterization of monoclonal antibody size variants containing extra light chains. MAbs 2013; 5:102 - 13; http://dx.doi.org/10.4161/mabs.22965; PMID: 23255003
- Rege K, Pepsin M, Falcon B, Steele L, Heng M. High-throughput process development for recombinant protein purification. Biotechnol Bioeng 2006; 93:618 - 30; http://dx.doi.org/10.1002/bit.20702; PMID: 16369981
- Bhambure R, Kumar K, Rathore AS. High-throughput process development for biopharmaceutical drug substances. Trends Biotechnol 2011; 29:127 - 35; http://dx.doi.org/10.1016/j.tibtech.2010.12.001; PMID: 21255855