Abstract
Effective characterization of protein-based therapeutic candidates such as monoclonal antibodies (mAbs) is important to facilitate their successful progression from early discovery and development stages to marketing approval. One challenge relevant to biopharmaceutical development is, understanding how the stability of a protein is affected by the presence of an attached oligosaccharide, termed a glycan. To explore the utility of molecular dynamics simulations as a complementary technique to currently available experimental methods, the Fc fragment was employed as a model system to improve our understanding of protein stabilization by glycan attachment. Long molecular dynamics simulations were performed on three Fc glycoform variants modeled using the crystal structure of a human IgG1 mAb. Two of these three glycoform variants have their glycan carbohydrates partially or completely removed. Structural differences among the glycoform variants during simulations suggest that glycan truncation and/or removal can cause quaternary structural deformation of the Fc as a result of the loss or disruption of a significant number of inter-glycan contacts that are not formed in the human IgG1 crystal structure, but do form during simulations described here. Glycan truncation/removal can also increase the tertiary structural deformation of CH2 domains, demonstrating the importance of specific carbohydrates toward stabilizing individual CH2 domains. At elevated temperatures, glycan truncation can also differentially affect structural deformation in locations (Helix-1 and Helix-2) that are far from the oligosaccharide attachment point. Deformation of these helices, which form part of the FcRn, could affect binding if these regions are unable to refold after temperature normalization. During elevated temperature simulations of the deglycosylated variant, CH2 domains collapsed onto CH3 domains. Observations from these glycan truncation/removal simulations have improved our understanding on how glycan composition can affect mAb stability.
Introduction
Proteins-based therapeutics are an important class of biopharmaceuticals that have substantially increased the number of treatable diseases.Citation1,Citation2 Among the biopharmaceuticals currently in development, monoclonal antibodies (mAbs) make up the largest portion. Antibodies perform multiple functions—each antibody is composed of two Fab regions capable of binding antigen and one Fc region, which modulates antibody clearance and activation of antigen destruction through effector function pathways such as antibody-dependent cell-mediated cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC).Citation3 The Fc region consists of four structural domains, two CH2 and two CH3 domains (). In antibodies of the IgG class, each CH2 domain in the Fc region has a complex biantennary oligosaccharide, termed a glycan (), attached at Asn297 (in Kabat numbering schemeCitation4). Due to the presence of different terminal sugars, the composition of these glycans can be one of several forms G2F, G0F, and G2FS2 (). Micro-heterogeneity in Fc glycan composition varies with species and expression systems used to produce candidate mAbs during research and commercial manufacturing.Citation5,Citation6 Glycan composition is also known to influence the activation of specific effector function pathways differentially.Citation7,Citation8 For example, truncating the G2F glycoform by degalactosylation to G0F can decrease CDC activation without affecting ADCC activation.Citation9,Citation10 Effector function activity can also be modulated by defucosylation, which has been shown to increase ADCC activity.Citation11 These discoveries have inspired the development of different engineering approaches to enhance/optimize antibody effector function activity by altering the Fc-bound glycan profile or by exchanging amino acids in the protein backbone.Citation12,Citation13 Glycan modification techniques such as these have led to the first market-approved glycoengineered monoclonal antibody,Citation14 and may be useful in optimizing effector function activities of therapeutic Fc-fusion proteins as well.Citation15
Figure 1. Fc structure (A) indicating the locations of structural domains, two attached oligosaccharides colored cyan, and the position of Gly237 residues used to calculate the CH2-CH2 distance. For chain H, the CH2 domain is red and the CH3 domain is orange. For chain K, the CH2 domain is green and the CH3 domain is blue. The hinge, which was present in all glycoform variant simulations, has been left out of this representation for the sake of clarity. CH2 domain schematic (B) with β-strands labeled and residue indices for strands in Kabat numbering scheme. Two phenylalanine residues 241 and 243, important for CH2 domain stability, are located on β-strand A. The oligosaccharide attachment residue, Asn297, is located in loop C’E.
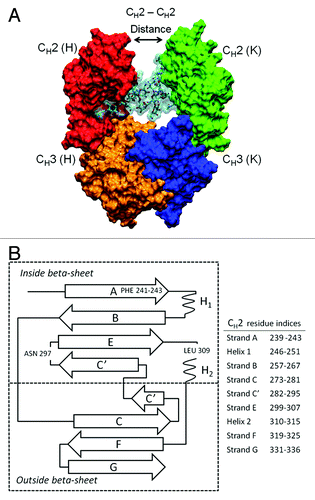
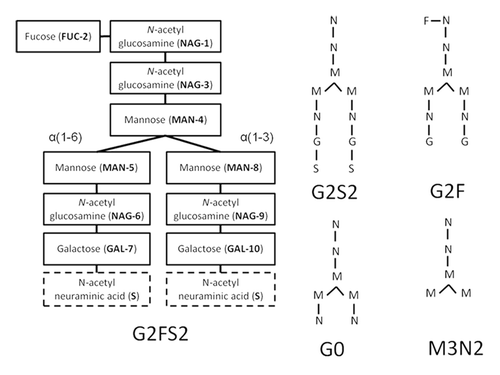
Figure 2. Structure, composition, and branches are labeled for oligosaccharides of G2FS2. The glycan has three terminal carbohydrates: FUC-2, and two N-acetyl neuraminic acids or sialylic acids, labeled S. Desialylating G2FS2 produces G2F. Alternate glycan compositions and structures are shown using a simplified notation. Note that MN2, not shown, can be formed by removing MAN5 and MAN8 from M3N2.
From a biopharmaceutical development perspective, truncating or removing glycans attached to therapeutic mAb candidates can also have undesirable consequences such as reduced Fc thermostability and increased aggregation. Consequently, it is important to ensure a consistent glycan profile across manufacturing batches or to match the glycan profile of an innovator product when developing a biosimilar version.Citation16 Therefore, much effort has been devoted to the development of rapid high through-put methods for glycan profile determination.Citation17,Citation18 In a set of experiments demonstrating the effect glycan composition can have on Fc effector function activity, the thermostability of CH2 domains decreased as more carbohydrates were removed from the glycan.Citation19 In a separate study by Zheng et al., full deglycosylation was shown to expand the Fc hydrodynamic radius, increasing its susceptibility to proteolytic cleavage by papain.Citation20 Other studies have shown that partial and full deglycosylation can also increase the propensity of IgG1 Fc fragments to aggregate.Citation21 Therefore, understanding the mechanism that underlies the stability differences of fully and partially deglycosylated IgG Fc fragments is important to the development of therapeutic mAbs.Citation22
Effective characterization of protein-based therapeutics such as mAbs is important to facilitate their translation from discovery to commercially-available drugs. Achieving this goal partly depends on the sensitivity of analytical tools to provide chemical/physical information. Due to the size and complexity of full-length mAbs, however, characterization of their higher order structure with currently available experimental methodologies is challenging. The tools available for characterization, including differential scanning calorimetry, analytical ultracentrifugation, circular dichroism, isothermal calorimetry, can provide a global view of mAb structure/stability, but cannot determine specific protein sequence or structural sites that can affect experimental observations. On the other hand, X-ray crystallography can provide detailed structural information, but structures are relatively static and do not provide dynamic information beyond crystallographic B-factors.Citation23 In principle, protein solution dynamics can be determined using nuclear magnetic resonance (NMR), but intact and IgG fragments are too large for routine NMR 3-D structural analyses. Moreover, NMR experiments require samples with highly concentrated protein, which can distort antibody structure and lead to aggregation.Citation24 Therefore, it is important for biopharmaceutical development to seek new analytical tools to detect protein structural changes with a high degree of sensitivity.
Atomistic and coarse-grained molecular dynamics (MD) simulations can play an important role in fulfilling this need.Citation25-Citation29 MD can assess both the global and local conformational behavior of mAbs in solution under a variety of different conditions. Our recent analyses of a murine IgG2a using molecular dynamics provided new insights into mAb dynamics by comparing structural change differences upon thermal stress and deglycosylation.Citation30 Studies such as these and others illustrate how MD can be an important addition to the set of tools available for characterization of protein structural changes that may occur during the shipping and storage of biopharmaceuticals.Citation31,Citation32
In this work, explicit water simulations were performed on the following Fc glycoform variants DEGLYCO, M3N2, and G2F, which were modeled using the crystal structure of a human mAb, PDB entry 1HZH (; ). Each system was simulated at 300 K and 375 K for 200 ns in two independent production runs of 100 ns each. Total simulation time was 400 ns per Fc glycoform variant, and the overall total simulated time for this study was 1.2 μs. Simulations were evaluated for differences in domain ensemble averages, FcR (I-III) binding site closure, local structure perturbations, and sugar dynamics/conformational preferences. Clustering glycan-glycan conformations in simulation trajectories revealed a preferred glycan-glycan conformation that is significantly different than the one in the crystal structure (1HZH). Moreover, the preferred glycan-glycan conformation has several novel inter-glycan contacts that are not formed in the crystal structure (1HZH). Consistent with the above observations, structural differences among the glycoform variants during simulations suggest that glycan truncation/removal can cause quaternary structural deformation as a result of the loss or disruption of a significant number of these inter-glycan contacts. Glycan truncation can also increase the structural deformation of CH2 domains, demonstrating the importance of specific carbohydrates in stabilizing CH2 domains. In elevated temperature simulations, glycan truncation appears to affect structure deformation differentially in locations (Helix-1 residues 246–251 and Helix-2 residues 310–315) that are far from the oligosaccharide attachment point (Asn 297). These helices form part of the FcRn binding site and their preservation is important in maintaining pharmacokinetic and pharmacodynamic properties of therapeutic mAb candidates. Observations from these glycan truncation simulations have improved our understanding on how glycan composition can impact pharmaceutical stability of protein therapeutics and demonstrated the usefulness of molecular dynamics simulations in biopharmaceutical development studies.
Table 1. Simulations performed in this study
Results
Individual domain ensemble averages
The Fc structure is described in , and presents the location of carbohydrates in glycan structure and glycan composition for different glycoform variants. Previous studies have shown that glycan truncation affects Fc thermostability of CH2 domains while CH3 domain thermostability remains relatively unchanged.Citation19,Citation30,Citation33 In experiments by Mimura et al. using Wid, an IgG1 extracted from myeloma serum, CH3 domain melting temperatures were virtually unchanged by the removal of any glycan carbohydrate from the heterogeneous mixture (NATIVE) of Wid glycoform variants, e.g., G2S2, G2, G1, G0 (CH3 domain Tm(s) range from 82.2 °C to 81.5 °C for NATIVE and DEGLYCO).Citation19 On the other hand, the removal of sialic acids, galactoses, and NAG6 and NAG9 from NATIVE by sequential enzymatic reactions to produce M3N2 lowered the CH2 melting temperature by ~4 degrees (CH2 Tm for NATIVE is 71.4 °C and for M3N2 is 67.7 °C).Citation19 Desialylation and degalactosylation, however, did not affect CH2 domain thermostability to the same degree (CH2 Tm for G0 is 71.5 °C).Citation19 Because NAG9 does not form interactions with CH2 domains, the loss of CH2 domain thermostability upon the removal of NAG6 (see above for M3N2) can be attributed to key CH2 domain stabilizing interactions formed between NAG6 and Phe243 (Kabat) of β-strand A.Citation34,Citation35 No further destabilization of CH2 domains was observed upon removal of MAN5 or MAN8 from M3N2 to produce MN2 (NAG1–NAG2–MAN4), (CH2 Tm for MN2 is 67.3 °C).Citation19 However, full deglycosylation did lower the CH2 melting temperature below that of MN2 (CH2 Tm for DEGLCYO is 65.9 °C).Citation19 This provides supporting evidence for the importance of interactions between MAN4 and Phe241.Citation34,Citation35 The significance of the MAN4-Phe241 and NAG6-Phe243 interactions in stabilizing CH2 domains was also supported by the work of Voynov et al. who showed that mutations, F241S and F243S, significantly lowered CH2 domain melting temperatures for fully glycosylated glycoforms (G0, G1, and G2).Citation28
Here, individual domain ensemble averages are reported to understand the stabilizing effect that full length glycans have on CH2 and CH3 thermostability. Simulation evaluations began by measuring the degree of structural change using root mean square deviation (RMSD), radius of gyration (Rg), and hydrophobic accessible surface area (HpASA) for glycoform variants during equilibrated simulations. For each of these global measures, the magnitudes of structural change were then compared with the thermodynamic melting temperature differences among glycoform variants or other available experimental data. To calculate domain ensemble averages for all glycoform variants, the last 90 ns of both trajectories at the same temperature were used. Salient observations are described below.
At 300 K, the average RMSDs for CH2 domains of different chains (H and K) for Fc glycoforms were similar (RMSDs for CH2 domains of H/K chains at 300 K for DEGLYCO, 2.15/2.31 Å; M3N2, 2.12/2.13 Å; G2F, 1.94/2.14 Å), (). At 375K, average RMSDs for CH2 domains of different chains were again similar (RMSDs for H/K chains at 375 K for DEGLYCO, 2.89/3.0 Å; M3N2, 2.91/2.84 Å; G2F, 2.43/2.45 Å), (). The same trends held for CH3 domains of different chains at both 300 K and 375 K (). The average RMSDs for CH2 domains were also greater than those for CH3 domains for all glycoform variants at both temperatures. Furthermore, among CH2 domains, there was an increasing trend for average RMSD when comparing glycoform variants G2F, M3N2, and DEGLYCO at both temperatures (RMSDs for CH2 domains of H/K chains at 300 K for G2F, 1.94/2.14 Å; M3N2, 2.12/2.13 Å; DEGLYCO, 2.15/2.31 Å). Comparing the standard error for each mean, it can be seen that average RMSDs for CH2 domains among glycoform variants at 375 K are sufficiently differentiated (). At 375 K, the difference in average CH2 domain RMSD between K chains of DEGLYCO (3.0 Å) and M3N2 (2.84 Å), (3.0 Å - 2.84 Å = 0.16 Å), is 5% of the average CH2 RMSD for M3N2. Between M3N2 and G2F (2.45 Å), the corresponding percentage is 16%. The increasing trend for CH2 domain average RMSDs of glycoform variants was consistent with the decreasing trend in CH2 domain thermostability (CH2 Tm for NATIVE, 71.4 °C; M3N2, 67.7 °C; DEGLYCO, 65.9 °CCitation19). Thus, these simulations have qualitatively recapitulated the CH2 domain thermodynamic stability trend among glycoform variants.
Table 2. Ensemble averages, standard deviations, and standard error of the mean
Full deglycosylation is known to expand the Fc hydrodynamic radius, thereby, increasing its susceptibility to proteolytic cleavage by papain.Citation20 To assess if the size or hydrophobic surface area among individual structural domains of equilibrated Fc glycoform variants was different, the Rg and HpASA were compared at both temperatures. At 300 K, the average Rg for CH2 domains was similar among all glycoform variants (average Rg range for CH2 domains of chains H and K was 14.81–14.92 Å), (). For CH3 domains, average Rg values at 300 K were again similar among all glycoform variants (average Rg range for CH3 domains of chains H and K was 14.35–14.39 Å), (); however, in comparing the average Rg values for CH2 and CH3 domains, the range for CH3 domains was slightly smaller than the range for CH2 domains. Furthermore, standard deviations in Rg among CH3 domains did not overlap with standard deviations in Rg for CH2 domains. Similar trends in average Rg values also held for simulations performed at 375 K. The lack of differences in average Rg values among glycoform variants suggests that glycan truncation/removal does not affect the average Rg of individual structural domains. Note that Rg values were calculated for individual CH2 and CH3 domains and these values do not represent the overall quaternary structure of the Fc in different glycoform variants.
At 300 K, the average HpASA for individual structural domains was larger for CH2 domains (average HpASA range for chains H/K 3396.6–3631.8 Å2) than for CH3 domains (average HpASA range for chains H/K 2346.3–2374.3 Å2) across all glycoforms. This was expected because the average HpASA was calculated in the absence of the oligosaccharide and there are two hydrophobic residues (Phe241, Phe243) that become solvent-exposed when ignoring the glycan. In comparing the average HpASA for CH2 domains across glycoform variants at 300 K, values were approximately the same given that the range of HpASAs for CH2 domains among glycoform variant was ~125 Å2, which is a small percentage of the total average CH2 domain HpASA (~3500 Å2). HpASA standard deviations for CH2 domains of glycoform variants were even smaller than the range (). At 375 K, the average HpASA for CH2 domain (chain K) of DEGLYCO is less than it was at 300 K (3509.5 Å2, 300 K; 3170.6 Å2, 375 K). This was due to the collapse of Fc quaternary structure at elevated temperature (Fc ensemble averages are described in the next section). All other domains of both chains (H and K) for glycoforms variants had similar average HpASA values between 300 K and 375 K simulations (± 100 Å2). The lack of differences in average HpASA values among glycoform variants suggests that glycan truncation/removal does not impact the average HpASA of individual structural domains.
Overall, analyses of Fc individual structural domains during simulations indicate that tertiary structure changes in CH2 and CH3 domains were relatively small among different glycoform variants at both 300 K and 375 K (). Furthermore, CH3 domain tertiary structure changes upon glycan truncation/removal were smaller than those in CH2 domains. This is expected because glycosylation sites are located within CH2 domains. Moreover, the destabilization of CH2 domains upon glycan truncation (G2F vs. M3N2) and deglycosylation (G2F vs. DEGLYCO) was consistent with thermodynamic measurements summarized at the beginning of this section. Qualitative recapitulation of experiment results provides confidence in subsequent simulation analyses.
Fc quaternary structure ensemble averages
In the study by Mimura et al., it was also established that glycan truncation lowers Fc activity similar to thermostability (percent activity retention for NATIVE 100%, G0 93%, M3N2 85%, MN2 58%, DEGLCYO 3%Citation19). To understand how glycan truncation lowered Fc activity, the structures of glycoform variants were determined using crystallography.Citation35 Evaluations of these structures discovered that the distance between CH2 domains (CH2-CH2 distance) became increasingly shorter as more of the glycan was truncated. For example, the CH2-CH2 distance in the crystal structure of MN2 is 4.7 Å shorter than it is in the structure for G2F.Citation35 Full deglycosylation shortened the CH2-CH2 distance even further. Based on these observations, it was concluded that progressive glycan truncation reduces Fc activity by shortening the distance between CH2 domains, appearing to close the FcR (I-III) binding site and decrease FcR binding affinity. To investigate the effect glycan truncation has on overall Fc quaternary structure/activity during simulations, Fc average quaternary structure changes were determined at both temperatures. FcR binding site closure was monitored by computing the distance between Gly237 residues of β-strand A (see Methods).
At 300 K, the CH2-CH2 distance did not change appreciably in G2F and M3N2 simulations (). For DEGLYCO, CH2 domains moved closer together such that the average CH2-CH2 distance was ~4 Å less than the average distance for G2F (). The mutual approach of CH2 domains in deglycosylated Fc simulations is consistent with observations from crystallographic studies (described above). At an elevated temperature (375 K), the CH2-CH2 distance actually increased for DEGLYCO over time. The increase was not due to domain separation as disulfide bonds in the hinge prevent this from happening. Instead, CH2 domains collapsed onto CH3 domains by twisting relative to one another, extending the distance between Gly237 residues (). For DEGLCYO at 375 K, the extent of CH2 domain collapse onto CH3 domains can also be seen by comparing average HpASA values for both chains. At 300 K, the average HpASAs for CH2 and CH3 domains of chain K were 3509.5 Å2 and 2367.2 Å2, respectively. Collapse of CH2 domains onto CH3 domains of chain K decreased the average HpASA for both domains in 375 K simulations (CH2, 3170.6 Å2; CH3, 23041.1 Å2). In M3N2 simulations at 375 K, CH2 domains partially collapsed onto CH3 domains, but the CH2-CH2 distance was restored to the initial simulation distance of 15 Å after 40 ns. CH2 domains began collapsing again after 70 ns suggesting that the collapse of CH2 domains in M3N2 simulations at 375 K could be a transient event. In G2F simulations at 375 K, the CH2-CH2 distance remained relatively constant and similar to the initial simulation distance (15 Å).
Figure 3. The average CH2-CH2 distance during simulations at (A) 300 K and (B) 375 K is plotted. Distances are averaged over a 5 ns window and plotted in the window center. Even though glycoform variant starting structures are the same, initial CH2-CH2 distances for the three glycoform variants are slightly different because the first data points reported are at 2.5 ns due to averaging. Figure insets (α) and (β) represent DEGLYCO structures along the trajectory. At 300 K, simulations began with DEGLYCO in an open conformation. As DEGLYCO simulations progressed, the CH2-CH2 distance decreased and the FcR binding site closed. At 375 K, the CH2-CH2 distance increased as CH2 domains collapsed onto CH3 domains. The FcR binding site was also closed in the collapsed structure.
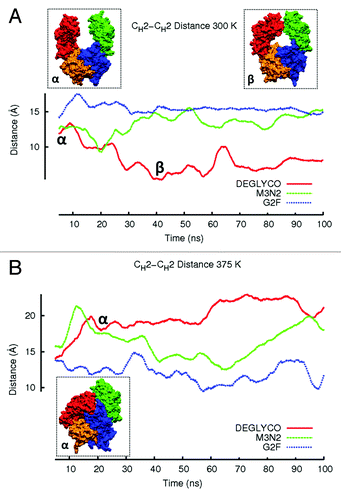
RMSD ensemble averages for the complete Fc fragment () were similar to the changes in CH2-CH2 distance during simulations ( and ). At 300 K, the structure of DEGLYCO deformed by 4.65 Å compared with the crystal structure, and M3N2 and G2F deformed by 4.25Å and 3.74 Å, respectively. The same increasing trend of average RMSD among glycoform variants was even more apparent at 375 K (DEGLYCO, 8.09 Å; M3N2, 6.97 Å; G2F, 5.60 Å). The Fc ensemble average RMSD for DEGLYCO was 2.5 Å greater than the average RMSD for G2F in elevated temperature simulations (). The greater Fc quaternary structural deformations in elevated temperature simulations compared with 300 K simulations was likely due to the collapse of CH2 domains onto CH3 domains. By comparing Fc average RMSD differences among glycoform variants to their CH2-CH2 distance changes during simulations, it appears that these two descriptors of structural change are inter-related, i.e., the movements of the individual CH2 domains with respect to each other were at least partially responsible for Fc quaternary structure changes.
The Fc average radius of gyration (Rg) during DEGLYCO simulations was slightly larger than it was during M3N2 and G2F simulations at 300 K (DEGLYCO, 26.68 Å; M3N2, 26.48 Å; G2F, 26.30 Å), (). This observation is unexpected because CH2-CH2 approach would seemingly decrease the overall volume of the quaternary structure (see above), but it is in agreement with experimental observations of a slight increase in hydrodynamic radius and susceptibility to papain cleavage upon deglycosylation.Citation20 In simulations at 375 K, the Fc average Rg for DEGLYCO was less than it was for G2F and M3N2 due to the collapse of CH2 domains onto CH3 domains (DEGLYCO, 24.12 Å; M3N2, 25.75 Å, G2F, 25.55 Å), (). The collapse of CH2 domains onto CH3 domains in DEGLCYO simulations also affected the Fc average HpASA, which was calculated in the absence of the glycan for all glycoform variants (). While the Fc average HpASA was approximately the same for all glycoform variants at 300 K (DEGLYCO, 14034.9 Å2; M3N2, 14125.8 Å2; G2F, 14015.6 Å2), at 375 K, the average HpASA decreased due to the reduction in overall accessible surface area, resulting from CH2 domain collapse (DELGYCO, 13185.5 Å2; M3N2, 14081.0 Å2; G2F, 14126.8 Å2, see ). Overall, the observations from Fc and structural domain ensemble averages indicate that glycan truncation and deglycosylation can affect both CH2 domain tertiary structure and Fc quaternary structure. The differences in quaternary structure deformation among glycoform variants were, however, significantly larger than differences in tertiary structure deformation. This suggests that individual CH2 domains move with respect of each other and with respect to CH3 domains. Similar observations have also been made in earlier simulations of a full length murine IgG2a mAb.Citation30 The consequence of these inter-domain movements is that the effector function activity can become impaired.
Local structure deformations upon glycan truncation/removal
Glycan truncation/removal can also perturb residues in loops that constitute the FcR binding site such as BC, FG, and C'E loops (). Crystal structures of G2F, G0F, and M3N2F suggest that removal of NAG6 perturbs the C’E loop.Citation35 Variants with an attached NAG6 carbohydrate (e.g., G0F) do not show differences in C'E loop perturbations when compared with the crystal structure of G2F. NMR studies have supported these findings and added that glycan truncation can also perturb the lower hinge region as well as the FG loop after full deglycosylation.Citation33 To monitor the local structure changes upon glycan truncation, average residue-wise RMSD values were computed. Since CH3 domains were mostly unaffected by glycan truncation (), this section focuses on the local structure changes in CH2 domains only. The average residue-wise RMSD values are reported for β-strands and connecting loops by averaging between CH2 domains of heavy chains, H and K (see Materials and Methods). The average residue-wise RMSD values were compared for similarities and differences at 300 K and 375 K among glycoform variants to identify local structure that is susceptible to glycan truncation and removal.
At 300 K, local structure perturbations of CH2 domains were confined to loops while β-strands remained relatively intact (). The most perturbed loops and β-strands in simulations of DEGLYCO at 300 K were Helix-1 (2.05 Å), loop BC (3.0 Å), β-strand C (2.0 Å), β-strand C' (2.0 Å), loop C’E (3.0 Å), Helix-2 (2.5 Å), and loop FG (2.5 Å), (). In G2F simulations at 300 K, the most perturbed local structures were loops BC (3.0 Å) and EF (2.7 Å). Comparing average residue-wise RMSDs among glycoform variants, the regions of local structure with similar perturbations were in Helix-1, loop BC, β-strand C, β-stand C’, and Helix-2 (see values above). Local regions with perturbation differences among glycoform variants were in residues that constitute loop C’E, which also contains the glycan attachment site Asn297. Compared with the crystal structure, the maximum residue-wise RMSD in the C’E loop was 1.5 Å for G2F, 2.0 Å for M3N2, and 3.0 Å for DEGLYCO. Perturbations in loop FG were also different among glycoform variants (DEGLYCO, 2.5 Å; M3N2, 2.6 Å; G2F, 2.2 Å); however, at an elevated temperature (375 K), the perturbation differences in loop FG among variants did not exist (see below). Therefore, local structure deformation differences among glycoform variants were mostly localized to loop C'E loop. Furthermore, the localization of local structure perturbation differences to loop C’E suggests that differences in CH2 domain ensemble average RMSDs among glycoform variants () could be a result of loop C’E deformation differences.
Figure 4. Residue-wise RMSDs for all residues of CH2 domains calculated in simulations at (A) 300 K and (B) 375 K. Residue-wise RMSD values for each glycoform variant were averaged between the two independent simulation runs at each temperature and then averaged over a 5 ns window. Local structure including β-strands and loops are illustrated with black bars.
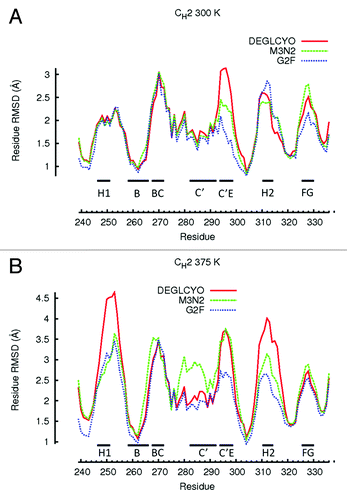
At 375 K, local structure perturbations in CH2 domains were greater than at 300 K, and glycan truncation generally increased the extent of perturbation for local structure regions (). Comparing local structure perturbations between 300 K and 375 K, the local regions most affected by an increase in temperature were Helix-1 and Helix-2. At 375 K, both these helices deformed 2.0 Å more than they did at 300 K, indicating that these regions were highly susceptible to deformation at elevated temperatures. For DEGLCYO, the local regions most perturbed from the crystal structure were in Helix-1 (4.5 Å), loop BC (3.5 Å), β-strand C (3.0 Å), β-strand C' (3.0 Å), loop C’E (3.5 Å), Helix-2 (4.0 Å), and loop FG (2.5 Å). Comparing average residue-wise RMSDs among glycoform variants at 375 K, the regions of local structure with similar perturbations were in loops BC and FG loop, and all β-strands (). Only β-strand C’ differed among glycoform variants as it was heavily perturbed in one of the M3N2 simulations at 375 K. Local regions with differences in perturbation among glycoform variants were in Helix-1 (DEGLYCO, 4.0 Å; M3N2, 3.0 Å; G2F, 2.5 Å), loop AB (DEGLYCO, 4.5 Å; M3N2 and G2F, 3.0 Å), Helix-2 (DEGLYCO, 4.0 Å; M3N2, 3.0 Å; G2F, 2.5 Å), and loop C’E (DEGLYCO and M3N2, 3.5 Å; G2F, 2.5Å). Based on these differences, the extent of perturbation for these local structure regions generally increased due to glycan truncation/removal. Taken together, observations from 300 K and 375 K simulations suggest that the amount of local structure perturbation within CH2 domains depended both on the simulation temperature (300 K vs. 375 K) and the extent of glycan truncation. Helix-1 (residues 246–251) and Helix-2 (residues 310–315), which form part of the FcRn binding site, are located far from the glycan attachment site (Asn 297). Deformation of these helices could affect FcRn binding if these regions are unable to refold after temperature normalization. FcRn binding is an important determinant of pharmacokinetic and pharmacodynamic properties of therapeutic mAb candidates.Citation36
Glycan dynamics
Enzymatic sialylation of galactosylated glycan termini was shown to be anti-inflammatory in mice.Citation37-Citation39 From a glycoengineering standpoint, enzymatic sialylation of normally galactosylated IgGs has the potential to improve therapy for patients suffering from inflammatory diseases such as rheumatoid arthritis. While a recent report suggests that sialylation produces an anti-inflammatory effect by biasing the FcR binding site toward the “closed” conformation,Citation40 it remains unclear how galactosylated termini are accessible for sialylation given their location in the cavity formed by CH2 domains. Crystal structures of galactosylated Fc fragments show that glycan termini are normally buried between CH2 domains. In early solution NMR measurements of Fc glycoforms, the α1–6 branch was completely immobilized to the surface of CH2 domains while the α1–3 branch appeared to be more dynamic ().Citation41,Citation42 Immobilization would seemingly prevent the α1–6 branch from enzymatic sialylation or interactions with receptors. A more recent NMR study indicates that the termini of both glycan branches are highly dynamic.Citation43,Citation44 Here, we investigated the dynamics of both glycan branches at 300 K and 375 K using Root Mean Square Fluctuation (RMSF), (). RMSF is a measure of how much a carbohydrate fluctuates about its average position. It is, therefore, an appropriate measure of glycan dynamic behavior. RMSF values for each carbohydrate were averaged between H and K glycans and across trajectories (see Methods).
Table 3. Glycan contacts for G2F and M3N2 at 300 K
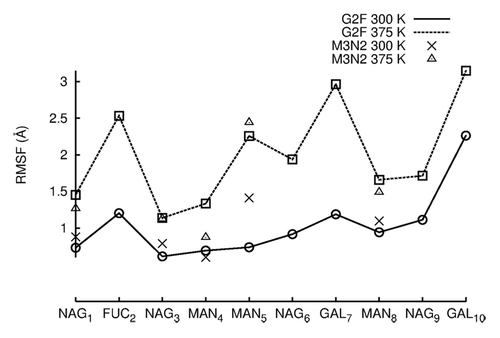
At 300 K, the most mobile carbohydrates in G2F as measured by average RMSF were the branch termini GAL7 (1.0 Å), GAL10 (2.0 Å), and FUC2 (1.0 Å), ( and ). In comparing carbohydrates of the α1–6 and α1–3 branches, NAG9 and MAN8 (α1–3) fluctuated more than NAG6 and MAN5 (α1–6). Therefore, the α1–6 branch was less mobile than the α1–3 branch in 300 K simulations, agreeing with earlier NMR studies. Moreover, upon visual inspection of simulation trajectories, it was found that the α1–6 branch never lost all protein-glycan interactions as was proposed to explain α1–6 sialylation.Citation43,Citation44 The retention of contacts between α1–6 carbohydrates and CH2 domains can also be seen in . The NAG6-F243 contact and all GAL7 contacts were retained during simulations ~100% of the time (NAG6, GAL7 are located on the α1–6). It is possible, however, that 100 ns of simulation time may not be enough to see GAL7 lose its contacts, despite a reported α1–6 branch correlation time of 37 ns.Citation44 Average RMSF values between M3N2 and G2F were similar except at MAN5 (α1–6), (). The truncated glycan, M3N2, had similar carbohydrate mobilities to G2F in the more mobile α1–3, but the α1–6 branch was more mobile in M3N2 than in G2F due to the loss of glycan-protein interactions. Thus, GAL7 and NAG6 glycan-protein interactions are important in restricting α1–6 branch carbohydrate dynamics. As in 300 K simulations, the most mobile carbohydrates at 375 K were the branch termini GAL7 (3.0 Å), GAL10 (3.0 Å), and FUC2 (2.5 Å). A comparison G2F carbohydrate mobilities in 300 K and 375 K simulations shows that there was an average 1.5 Å increase in average RMSF among carbohydrate moieties. At 375 K, MAN5 and NAG6 (α1–6) were more mobile than MAN8 and NAG9 (α1–3), (). Upon visual inspection of G2F simulation trajectories at 375 K, GAL7 did not leave the protein surface, but the greater mobility of the α1–6 branch than that of the α1–3 branch, at an elevated temperature, does suggest that this branch could expose its termini for sialylation over time.
Figure 5. RMSF values are plotted for individual carbohydrates of G2F and M3N2. RMSF values were averaged between the two independent simulation runs at each temperature. RMSF values for data points simulated at 300 K are connected by lines. RMSF values for simulations preformed at 375 K are not connected by lines due to the absence of terminal GAL and NAG carbohydrates.
Carbohydrate conformation/contacts
The crystal structure complex of FcRIII bound to the Fc fragment has revealed the existence of a single contact between oligosaccharides and FcRIII.Citation45 Because FcRIII activation is dependent on Fc glycan compositionCitation19 (see section Fc quaternary structure ensemble averages), oligosaccharide moieties exert their influence indirectly through modification of the Fc conformation. As previously stated, glycan truncation can reduce FcR (I‒III) binding by decreasing the distance between CH2 domain, and thereby causing the FcR binding site to close. Therefore, contacts between oligosaccharides from glycans attached to different heavy chains (inter-glycan contacts) are crucial to maintain the Fc in an active state. Fc fragment crystal structures with intact glycans, however, show very few inter-glycan contacts with MAN8 mediating most of the inter-glycan contacts.Citation34,Citation46,Citation47 Moreover, Fc crystal structures obtained under different crystallization conditions have different inter-glycan interactions.Citation35 Thus, there is a need to investigate inter-glycan interactions and their dynamics to understand their effect on Fc conformation.
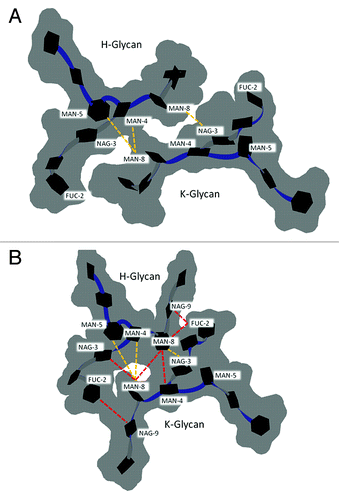
Here, the inter-glycan contacts from the crystal structure (1HZH) are compared with the preferred glycan-glycan conformation found during G2F simulations at 300 K (). The preferred glycan (chain H) – glycan (chain K) conformation, labeled 8240, was identified by clustering all G2F trajectory samples from both 100 ns simulations (see Methods). In total, 85% of all structures submitted for clustering had the preferred glycan-glycan conformation. The crystal structure (1HZH) with modeled GAL10(K) and FUC(H) showed only three inter-glycan contacts MAN4(H)-MAN8(K), MAN5(H)-MAN8(K), MAN8(H)-NAG3(K), (; ). The glycan-glycan conformation has both α1–3 branches facing down and away from the FcR binding site, and both α1–6 branch termini have GAL7 bound to the protein surface.Citation48 In the preferred glycan-glycan conformation from simulations, both α1–3 branches were extended and faced outwards (). The outward position of the α1–3 branch may allow sialylation. Compared with the crystal structure, several new inter-glycan contacts were observed in the preferred glycan-glycan structure. These are FUC2(H)-NAG9(K), NAG3(H)-MAN8(K), MAN8(H)-MAN8(K), MAN8(H)-MAN4(K), MAN8(H)-FUC2(K), NAG9(H)-FUC2(K), (). Three of these new contacts were near the glycan branching point. The new FUC2 contacts in the preferred structure, FUC2(H)-NAG9(K), MAN8(H)-FUC2(K), NAG9(H)-FUC2(K), result from α1–3 branches (MAN8, NAG9, GAL10) facing in an outward direction. Overall, the inter-glycan contacts in the preferred conformation were shifted compared with those in the crystal (), and crystal contacts MAN4(H)-MAN8(K) and MAN5(H)-MAN8(K) were not well maintained (). The critical stabilizing CH2 domain contacts NAG6-F243, and MAN4-F241 were ~100% retained for both glycans. Comparatively, fucose-C’E loop contacts (FUC2-Y296 [H], FUC2-Q295 [K]) were partially lost during simulations, indicating much weaker interactions. Taken together, the preferred oligosaccharide conformation had significantly more inter-glycan contacts than observed in crystal structure (1HZH) and inter-glycan contacts were crucial to maintain Fc quaternary structure and/or activity. These observations may have important implications not only for basic understanding of Fc structure-function relationships, but also toward rational design of glycoengineered antibodies.
Figure 6. Glycan-glycan conformation for G2F in the (A) crystal structure of PDB entry 1HZH with GAL10 of chain K and FUC2 of chain H modeled in using MOE. (B) The preferred glycan-glycan conformation in 300 K simulations (see Materials and Methods). Carbohydrates (black hexagons) are represented using Twister and Paper Chain representations in VMD.Citation57,Citation58 Gray outlines show the surface of carbohydrates. Yellow dotted lines indicate inter-glycan contacts (identified at ≤ 4.5 Å for non-hydrogen atoms) between chains H and K in the crystal structure. Red dotted lines indicate inter-glycan contacts in the preferred glycan-glycan conformation that were formed during simulations at 300 K and are not formed in the crystal structure for 1HZH.
Discussion
In addition to simulations performed at 300 K, this study reports findings for molecular dynamics performed at an elevated temperature (375 K) used to enhance conformational sampling and perturb glycoform variants during simulations. In these elevated temperature simulations, the goal was not to unfold the IgG Fc molecular system. Protein folding/unfolding transitions occur over the millisecond to second timescales. One hundred nanoseconds of all-atom explicit solvent simulation time cannot achieve this for a molecular system as large as the Fc fragment, even when such studies are performed at an elevated temperature. Instead, the elevated temperature simulations in this study were performed to perturb the Fc molecular system as a means to mimic the thermal stress experienced by biopharmaceuticals as a result of occasional temperature incursions that may happen during manufacture, shipping and storage. These simulations are analogous to accelerated stability studies, which are routinely performed in pharmaceutical development. Importantly, the temperature values in molecular dynamics simulations should be carefully interpreted. In simulations, temperature is simply a measure of the amount of kinetic energy in the system. As such, elevating the temperature is a commonly used strategy to allow molecular systems to escape from deep energy minima.Citation49 However, elevating the simulation temperature is not a reliable strategy to reproduce experimental temperature-dependent phenomena such as changes in protein-protein binding affinity. On the other hand, the molecular origins that underpin protein conformational changes upon thermal stress can be reliably accelerated by arbitrarily increasing the temperature.Citation50-Citation52 The molecular origins of these changes remain similar when simulations are performed at different elevated temperatures, but the time interval necessary to observe these conformational changes is different due to varying the kinetic energy.
In the presented simulation analyses, glycoform variants were evaluated for differences in domain ensemble averages, FcR (I‒III) binding site closure, local structure perturbations, and sugar dynamics/conformational preferences. Upon glycan truncation/removal, we find that simulations can recapitulate the thermodynamic trends for CH2 and CH3 domains. The closure of the FcR binding site was dependent on the degree of glycan truncation. During simulations at both 300 K and 375 K, most local structure deformation differences among glycoform variants were confined to the C'E loop, which contains the oligosaccharide attachment point, Asn297. In simulations at elevated temperature, Helix-1 and Helix-2 were the most perturbed local structures and were more perturbed for the deglycosylated glycoform variant than for the other variants. Within the glycan, the oligosaccharides of the α1–3 branch were more mobile than those of the α1–6 at room temperature (300K); however, at an elevated temperature (375K), the α1–6 branch was more mobile than the α1–3 branch. Furthermore, the preferred glycan-glycan conformation, identified by clustering glycan-glycan conformations in simulation trajectories, had significantly more inter-glycan contacts than those found in the crystal structure.
Observations of structural differences among glycoform variants suggest that glycan truncation and removal can cause quaternary structural deformation of the Fc region by CH2 domain closure which results from the loss (DEGLYCO) or disruption (M3N2) of a significant number of inter-glycan contacts revealed by the preferred glycan-glycan conformation observed in simulations. These inter-glycan contacts are not formed in the crystal structure, PDB entry 1HZH. Glycan truncation also destabilized CH2 domains by deforming the C'E loop at 300 K, suggesting that the absence of glycans can increase protein entropy, leading to domain destabilization. Glycan truncation also appears to affect structure deformation in locations (Helix-1 and Helix-2) that are far from the oligosaccharide attachment point in simulations at elevated temperature. Deformation of these local structures could affect FcRn binding if these regions are unable to refold after temperature normalization. Observations from these glycan truncation simulations have improved our understanding of how glycan composition can affect mAb stability and demonstrated the usefulness of molecular dynamics simulations in biopharmaceutical development studies.
Materials and Methods
Fc coordinates were taken from Protein Data Bank (PDB) entry 1HZH (heavy chains labeled H and K) that contains atomic coordinates for the full length human IgG1 b12 antibody.Citation48 The N and C termini were acetylated and amidated, respectively, to neutralize their charges. All inter-chain and intra-domain cysteine (Cys) pairs were disulfide bonded. Coordinates for the hinge (225-TCPPCPAPELLGG-237, Kabat numbering schemeCitation4) were included in all Fc glycoform variant simulations. All residues noted here are numbered according to the Kabat scheme. The inter-heavy chain disulfide bond formed by Cys229 residues, which is not formed in the 1HZH crystal structure, was modeled in using MOE.Citation53 Glycans for each glycoform variant were identical (M3N2 and G2F). GAL10 of chain K and FUC2 of chain H (), which are absent in the 1HZH crystal structure, were modeled for the G2F variant using MOE, with identical glycosidic dihedral angles to their corresponding carbohydrate, which is present in the opposite glycan. Histidines were doubly protonated, simulating a pH near or below 6, which is commonly used in formulation development. Each Fc system was solvated in a TIP3P water box with an initial box size of ~786 348 Å3 for all glycoform variants. Chloride ions were added to neutralize all systems, resulting in average Cl- ion concentration of ~16 mM per bath of water molecules.
Energy minimizations and molecular dynamics simulations were performed using the NAMD packageCitation54 with the AMBER99 all-atom forcefieldCitation55 and the GLYCAM06 forcefield for carbohydrates.Citation56 Molecular systems were first subjected to 20 000 energy minimization steps with protein heavy atoms fixed, followed by 5 000 steps of energy minimization without any restraints. Production runs were performed at 300 K and 375 K and each system was simulated for 100 ns. Trajectory samples were taken every 10 ps. For all glycoform variants, DEGLYCO, M3N2, G2F, two production simulations were performed at each temperature (). Overall, 12 production runs were performed during this study for a total simulation time of 1.2 μs.
Ensemble averages were calculated using the last 90 ns of each production run. The first 10 ns were used as an equilibration phase. System equilibration was assessed by monitoring energy, temperature, and pressure profiles. Domain ensemble averages were computed without the hinge or linker between CH2-CH3 domains. Reported ensemble averages are values averaged over simulation time (trajectory samples taken 10 ps apart) and between the two production runs at each temperature. RMSD and Rg values were calculated using all heavy atoms. HpASA was calculated using all carbon atoms and carbon bonded hydrogens in the absence of the glycan. The CH2-CH2 distance was computed between Gly237 (β-strand A) pairs, which is near the FcR (I‒III) binding site ().
For all trajectory samples, CH2 structural domains were superimposed separately onto their corresponding crystal structure coordinates (chain H onto H, chain K onto K) and residue-wise RMSD values were computed using all atoms. Superimposing individual structural domains limits residue-wise RMSD values to the tertiary structure changes and excludes the effect of quaternary structural changes. Residue-wise RMSDs were then averaged across all trajectory structure samples (10 ps apart). Residue-wise RMSDs were also averaged between CH2 domains (H and K) and across the two production runs at the same temperature (300 K and 375 K). RMSF values for glycans were computed by first superimposing glycans individually (chain H onto H, chain K onto K) onto the glycan coordinates found in the crystal structure (1HZH). RMSF values for glycan carbohydrates were also averaged between chains and production runs.
Glycan clustering for G2F was done in VMDCitation57 using all trajectory structure samples (10 ps apart) in both production runs at 300 K. Coordinates for both oligosaccharides were used in the superposition for clustering. Clusters were defined using an arbitrarily chosen cut-off value of 2.0 Å. The largest cluster had 85% of all trajectory samples at 300 K. Glycan coordinates found in the largest cluster center were used for contact analysis. In all contact analyses, including inter-glycan and glycan-protein contacts, a cut-off value of 4.5 Å for the distance between heavy atoms was used to define contacts.
| Abbreviations: | ||
| mAb | = | monoclonal antibody |
| RMSD | = | root mean square deviation |
| RMSF | = | root mean square fluctuation |
| Rg | = | radius of gyration |
| HpASA | = | hydrophobic accessible surface area |
| GAL | = | galactose |
| FUC | = | fucose, MAN, mannose |
| NAG | = | N-acetyl glucosamine |
Acknowledgments
The authors thank Pfizer Business Technology for computing resources. This work was supported by a postdoctoral fellowship awarded to PMB by Pfizer Inc.
Disclosure of Potential Conflicts of Interest
No potential conflict of interest was disclosed.
References
- Aggarwal S. Targeted cancer therapies. Nat Rev Drug Discov 2010; 9:427 - 8; http://dx.doi.org/10.1038/nrd3186; PMID: 20514063
- Reichert JM. Trends in the development and approval of monoclonal antibodies for viral infections. BioDrugs 2007; 21:1 - 7; http://dx.doi.org/10.2165/00063030-200721010-00001; PMID: 17263584
- Jefferis R. Isotype and glycoform selection for antibody therapeutics. Arch Biochem Biophys 2012; 526:159 - 66; http://dx.doi.org/10.1016/j.abb.2012.03.021; PMID: 22465822
- Kabat EA, Te Wu T, Gottesman KS, Foeller C. Sequences of proteins of immunological interest. Darby (PA); Diane Publishing; 1992.
- Raju S. Glycosylation variations with expression systems. Westborough (MA); BioProcess International; 2003:44-53.
- Raju TS, Briggs JB, Borge SM, Jones AJ. Species-specific variation in glycosylation of IgG: evidence for the species-specific sialylation and branch-specific galactosylation and importance for engineering recombinant glycoprotein therapeutics. Glycobiology 2000; 10:477 - 86; http://dx.doi.org/10.1093/glycob/10.5.477; PMID: 10764836
- Jefferis R. Glycosylation as a strategy to improve antibody-based therapeutics. Nat Rev Drug Discov 2009; 8:226 - 34; http://dx.doi.org/10.1038/nrd2804; PMID: 19247305
- Raju TS. Terminal sugars of Fc glycans influence antibody effector functions of IgGs. Curr Opin Immunol 2008; 20:471 - 8; http://dx.doi.org/10.1016/j.coi.2008.06.007; PMID: 18606225
- Boyd PN, Lines AC, Patel AK. The effect of the removal of sialic acid, galactose and total carbohydrate on the functional activity of Campath-1H. Mol Immunol 1995; 32:1311 - 8; http://dx.doi.org/10.1016/0161-5890(95)00118-2; PMID: 8643100
- Hodoniczky J, Zheng YZ, James DC. Control of recombinant monoclonal antibody effector functions by Fc N-glycan remodeling in vitro. Biotechnol Prog 2005; 21:1644 - 52; http://dx.doi.org/10.1021/bp050228w; PMID: 16321047
- Matsumiya S, Yamaguchi Y, Saito J, Nagano M, Sasakawa H, Otaki S, Satoh M, Shitara K, Kato K. Structural comparison of fucosylated and nonfucosylated Fc fragments of human immunoglobulin G1. J Mol Biol 2007; 368:767 - 79; http://dx.doi.org/10.1016/j.jmb.2007.02.034; PMID: 17368483
- Kellner C, Derer S, Valerius T, Peipp M. Boosting ADCC and CDC activity by Fc engineering and evaluation of antibody effector functions. Methods 2013; http://dx.doi.org/10.1016/j.ymeth.2013.06.036; PMID: 23851282
- Rose RJ, van Berkel PH, van den Bremer ET, Labrijn AF, Vink T, Schuurman J, Heck AJ, Parren PW. Mutation of Y407 in the CH3 domain dramatically alters glycosylation and structure of human IgG. MAbs 2013; 5:219 - 28; http://dx.doi.org/10.4161/mabs.23532; PMID: 23406897
- Beck A, Reichert JM. Marketing approval of mogamulizumab: a triumph for glyco-engineering. MAbs 2012; 4:419 - 25; http://dx.doi.org/10.4161/mabs.20996; PMID: 22699226
- Beck A, Reichert JM. Therapeutic Fc-fusion proteins and peptides as successful alternatives to antibodies. MAbs 2011; 3:415 - 6; http://dx.doi.org/10.4161/mabs.3.5.17334; PMID: 21785279
- Ayoub D, Jabs W, Resemann A, Evers W, Evans C, Main L, Baessmann C, Wagner E, Suckau D, Beck A. Correct primary structure assessment and extensive glyco-profiling of cetuximab by a combination of intact, middle-up, middle-down and bottom-up ESI and MALDI mass spectrometry techniques. MAbs 2013; 5; http://dx.doi.org/10.4161/mabs.25423; PMID: 23924801
- Burnina I, Hoyt E, Lynaugh H, Li H, Gong B. A cost-effective plate-based sample preparation for antibody N-glycan analysis. J Chromatogr A 2013; 1307:201 - 6; http://dx.doi.org/10.1016/j.chroma.2013.07.104; PMID: 23932029
- Stöckmann H, Adamczyk B, Hayes J, Rudd PM. Automated, high-throughput IgG-antibody glycoprofiling platform. Anal Chem 2013; http://dx.doi.org/10.1021/ac402068r; PMID: 23919734
- Mimura Y, Church S, Ghirlando R, Ashton PR, Dong S, Goodall M, Lund J, Jefferis R. The influence of glycosylation on the thermal stability and effector function expression of human IgG1-Fc: properties of a series of truncated glycoforms. Mol Immunol 2000; 37:697 - 706; http://dx.doi.org/10.1016/S0161-5890(00)00105-X; PMID: 11275255
- Zheng K, Bantog C, Bayer R. The impact of glycosylation on monoclonal antibody conformation and stability. MAbs 2011; 3:568 - 76; http://dx.doi.org/10.4161/mabs.3.6.17922; PMID: 22123061
- Latypov RF, Hogan S, Lau H, Gadgil H, Liu D. Elucidation of acid-induced unfolding and aggregation of human immunoglobulin IgG1 and IgG2 Fc. J Biol Chem 2012; 287:1381 - 96; http://dx.doi.org/10.1074/jbc.M111.297697; PMID: 22084250
- Solá RJ, Griebenow K. Effects of glycosylation on the stability of protein pharmaceuticals. J Pharm Sci 2009; 98:1223 - 45; http://dx.doi.org/10.1002/jps.21504; PMID: 18661536
- Rhodes G. Crystallography made crystal clear: a guide for users of macromolecular models. Philadelphia (PA); Elsevier Academic Press; 2010.
- Houde D, Arndt J, Domeier W, Berkowitz S, Engen JR. Characterization of IgG1 Conformation and Conformational Dynamics by Hydrogen/Deuterium Exchange Mass Spectrometry. Anal Chem 2009; 81:5966; http://dx.doi.org/10.1021/ac9009287; PMID: 19606834
- Caflisch A. Computational models for the prediction of polypeptide aggregation propensity. Curr Opin Chem Biol 2006; 10:437 - 44; http://dx.doi.org/10.1016/j.cbpa.2006.07.009; PMID: 16880001
- Chaudhri A, Zarraga IE, Yadav S, Patapoff TW, Shire SJ, Voth GA. The role of amino acid sequence in the self-association of therapeutic monoclonal antibodies: insights from coarse-grained modeling. J Phys Chem B 2013; 117:1269 - 79; http://dx.doi.org/10.1021/jp3108396; PMID: 23316912
- Chennamsetty N, Voynov V, Kayser V, Helk B, Trout BL. Prediction of aggregation prone regions of therapeutic proteins. J Phys Chem B 2010; 114:6614 - 24; http://dx.doi.org/10.1021/jp911706q; PMID: 20411962
- Voynov V, Chennamsetty N, Kayser V, Helk B, Forrer K, Zhang H, Fritsch C, Heine H, Trout BL. Dynamic fluctuations of protein-carbohydrate interactions promote protein aggregation. PLoS One 2009; 4:e8425; http://dx.doi.org/10.1371/journal.pone.0008425; PMID: 20037630
- Wu C, Shea JE. Coarse-grained models for protein aggregation. Curr Opin Struct Biol 2011; 21:209 - 20; http://dx.doi.org/10.1016/j.sbi.2011.02.002; PMID: 21371882
- Wang X, Kumar S, Buck PM, Singh SK. Impact of deglycosylation and thermal stress on conformational stability of a full length murine IgG2a monoclonal antibody: observations from molecular dynamics simulations. Proteins 2013; 81:443 - 60; http://dx.doi.org/10.1002/prot.24202; PMID: 23065923
- Buck PM, Kumar S, Singh SK. Insights into the potential aggregation liabilities of the b12 Fab fragment via elevated temperature molecular dynamics. Protein Eng Des Sel 2013; 26:195 - 205; http://dx.doi.org/10.1093/protein/gzs099; PMID: 23188804
- Wang X, Kumar S, Singh SK. Disulfide scrambling in IgG2 monoclonal antibodies: insights from molecular dynamics simulations. Pharm Res 2011; 28:3128 - 44; http://dx.doi.org/10.1007/s11095-011-0503-9; PMID: 21671135
- Yamaguchi Y, Nishimura M, Nagano M, Yagi H, Sasakawa H, Uchida K, Shitara K, Kato K. Glycoform-dependent conformational alteration of the Fc region of human immunoglobulin G1 as revealed by NMR spectroscopy. Biochim Biophys Acta 2006; 1760:693 - 700; http://dx.doi.org/10.1016/j.bbagen.2005.10.002; PMID: 16343775
- Deisenhofer J. Crystallographic refinement and atomic models of a human Fc fragment and its complex with fragment B of protein A from Staphylococcus aureus at 2.9- and 2.8-A resolution. Biochemistry 1981; 20:2361 - 70; http://dx.doi.org/10.1021/bi00512a001; PMID: 7236608
- Krapp S, Mimura Y, Jefferis R, Huber R, Sondermann P. Structural analysis of human IgG-Fc glycoforms reveals a correlation between glycosylation and structural integrity. J Mol Biol 2003; 325:979 - 89; http://dx.doi.org/10.1016/S0022-2836(02)01250-0; PMID: 12527303
- Roopenian DC, Akilesh S. FcRn: the neonatal Fc receptor comes of age. Nat Rev Immunol 2007; 7:715 - 25; http://dx.doi.org/10.1038/nri2155; PMID: 17703228
- Anthony RM, Ravetch JV. A novel role for the IgG Fc glycan: the anti-inflammatory activity of sialylated IgG Fcs. J Clin Immunol 2010; 30:Suppl 1 S9 - 14; http://dx.doi.org/10.1007/s10875-010-9405-6; PMID: 20480216
- Anthony RM, Nimmerjahn F, Ashline DJ, Reinhold VN, Paulson JC, Ravetch JV. Recapitulation of IVIG anti-inflammatory activity with a recombinant IgG Fc. Science 2008; 320:373 - 6; http://dx.doi.org/10.1126/science.1154315; PMID: 18420934
- Kaneko Y, Nimmerjahn F, Madaio MP, Ravetch JV. Pathology and protection in nephrotoxic nephritis is determined by selective engagement of specific Fc receptors. J Exp Med 2006; 203:789 - 97; http://dx.doi.org/10.1084/jem.20051900; PMID: 16520389
- Sondermann P, Pincetic A, Maamary J, Lammens K, Ravetch JV. General mechanism for modulating immunoglobulin effector function. Proc Natl Acad Sci U S A 2013; 110:9868 - 72; http://dx.doi.org/10.1073/pnas.1307864110; PMID: 23697368
- Wormald MR, Rudd PM, Harvey DJ, Chang SC, Scragg IG, Dwek RA. Variations in oligosaccharide-protein interactions in immunoglobulin G determine the site-specific glycosylation profiles and modulate the dynamic motion of the Fc oligosaccharides. Biochemistry 1997; 36:1370 - 80; http://dx.doi.org/10.1021/bi9621472; PMID: 9063885
- Yamaguchi Y, Kato K, Shindo M, Aoki S, Furusho K, Koga K, Takahashi N, Arata Y, Shimada I. Dynamics of the carbohydrate chains attached to the Fc portion of immunoglobulin G as studied by NMR spectroscopy assisted by selective 13C labeling of the glycans. J Biomol NMR 1998; 12:385 - 94; http://dx.doi.org/10.1023/A:1008392229694; PMID: 9835046
- Barb AW, Meng L, Gao Z, Johnson RW, Moremen KW, Prestegard JH. NMR characterization of immunoglobulin G Fc glycan motion on enzymatic sialylation. Biochemistry 2012; 51:4618 - 26; http://dx.doi.org/10.1021/bi300319q; PMID: 22574931
- Barb AW, Prestegard JH. NMR analysis demonstrates immunoglobulin G N-glycans are accessible and dynamic. Nat Chem Biol 2011; 7:147 - 53; http://dx.doi.org/10.1038/nchembio.511; PMID: 21258329
- Sondermann P, Huber R, Oosthuizen V, Jacob U. The 3.2-A crystal structure of the human IgG1 Fc fragment-Fc gammaRIII complex. Nature 2000; 406:267 - 73; http://dx.doi.org/10.1038/35018508; PMID: 10917521
- Deisenhofer J, Colman PM, Huber R, Haupt H, Schwick G. Crystallographic structural studies of a human Fc-fragment. I. An electron-density map at 4 A resolution and a partial model. Hoppe Seylers Z Physiol Chem 1976; 357:435 - 45; http://dx.doi.org/10.1515/bchm2.1976.357.1.435; PMID: 955567
- Huber R, Deisenhofer J, Colman PM, Matsushima M, Palm W. Crystallographic structure studies of an IgG molecule and an Fc fragment. Nature 1976; 264:415 - 20; http://dx.doi.org/10.1038/264415a0; PMID: 1004567
- Saphire EO, Parren PW, Pantophlet R, Zwick MB, Morris GM, Rudd PM, Dwek RA, Stanfield RL, Burton DR, Wilson IA. Crystal structure of a neutralizing human IGG against HIV-1: a template for vaccine design. Science 2001; 293:1155 - 9; http://dx.doi.org/10.1126/science.1061692; PMID: 11498595
- Perez D, Uberuaga BP, Voter AF. Accelerated Molecular Dynamics Methods. Hierarchical Methods for Dynamics in Complex Molecular Systems 2012; 10:329
- Caflisch A, Karplus M. Molecular dynamics simulation of protein denaturation: solvation of the hydrophobic cores and secondary structure of barnase. Proc Natl Acad Sci U S A 1994; 91:1746 - 50; http://dx.doi.org/10.1073/pnas.91.5.1746; PMID: 8127876
- Caflisch A, Karplus M. Acid and thermal denaturation of barnase investigated by molecular dynamics simulations. J Mol Biol 1995; 252:672 - 708; http://dx.doi.org/10.1006/jmbi.1995.0528; PMID: 7563082
- Li A, Daggett V. Identification and characterization of the unfolding transition state of chymotrypsin inhibitor 2 by molecular dynamics simulations. J Mol Biol 1996; 257:412 - 29; http://dx.doi.org/10.1006/jmbi.1996.0172; PMID: 8609633
- Molecular Operating Environment (MOE). Chemical Computing Group Inc., 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7, 2011.
- Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kalé L, Schulten K. Scalable molecular dynamics with NAMD. J Comput Chem 2005; 26:1781 - 802; http://dx.doi.org/10.1002/jcc.20289; PMID: 16222654
- Wang J, Cieplak P, Kollman PA. How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules?. J Comput Chem 2000; 21:1049 - 74; http://dx.doi.org/10.1002/1096-987X(200009)21:12<1049::AID-JCC3>3.0.CO;2-F
- Kirschner KN, Yongye AB, Tschampel SM, González-Outeiriño J, Daniels CR, Foley BL, Woods RJ. GLYCAM06: a generalizable biomolecular force field. Carbohydrates. J Comput Chem 2008; 29:622 - 55; http://dx.doi.org/10.1002/jcc.20820; PMID: 17849372
- Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph 1996; 14:33 - 8, 27-8; http://dx.doi.org/10.1016/0263-7855(96)00018-5; PMID: 8744570
- Cross S, Kuttel MM, Stone JE, Gain JE. Visualisation of cyclic and multi-branched molecules with VMD. J Mol Graph Model 2009; 28:131 - 9; http://dx.doi.org/10.1016/j.jmgm.2009.04.010; PMID: 19473861