
Abstract
Antibody drug conjugates (ADCs) are an emerging class of targeted therapeutics with the potential to improve therapeutic index over traditional chemotherapy. Drugs and linkers have been the current focus of ADC development, in addition to antibody and target selection. Recently, however, the importance of conjugate homogeneity has been realized. The current methods for drug attachment lead to a heterogeneous mixture, and some populations of that mixture have poor in vivo performance. New methods for site-specific drug attachment lead to more homogeneous conjugates and allow control of the site of drug attachment. These subtle improvements can have profound effects on in vivo efficacy and therapeutic index. This review examines current methods for site-specific drug conjugation to antibodies, and compares in vivo results with their non-specifically conjugated counterparts. The apparent improvement in pharmacokinetics and the reduced off target toxicity warrant further development of this site-specific modification approach for future ADC development.
Introduction
Cancer has long been treated with a variety of cytotoxic drugs in an attempt to destroy the malignant cells without causing significant harm to the host cells.Citation1 Most existing chemotherapy drugs enter cells non-specifically through lipophilic interaction with the cell membrane.Citation2 Many of these drugs preferentially kill cell types with higher proliferation or metabolic rates, a class that includes most tumor cells but also many healthy cells such as those in the digestive system epithelium. In an attempt to increase the therapeutic index of these cytotoxic drugs, the drugs were attached to targeting groups that preferentially delivered the payload to tumor tissue. Monoclonal antibodies against cell surface antigens have been among the most popular targeting moieties. The attached antibody can significantly decrease non-specific uptake of the drug and increase specific uptake of the conjugate by tumor cells.Citation3 Assuming the antigen shows a significantly elevated expression on the target cells vs. healthy cells, the therapeutic index of the drug should increase. Early attempts to construct antibody drug conjugates (ADCs) utilized doxorubicin as the cytotoxic drug, but those conjugates showed insufficient potency (IC50 = 100 nM) and were ineffective anti-cancer agents.Citation4 In response to these results, new conjugates with more potent drugs such as calicheamicinCitation5 or maytansineCitation6 proved to have significantly higher potencies (IC50 = 0.01–0.04 nM, for maytansine conjugate), yet were still well-tolerated in mouse models. These conjugates showed promising results when used to treat mouse xenograft tumors, as complete regressions of the tumors were observed at non-toxic doses. This success prompted the development of a variety of drugs from the maytansine and dolastatin familiesCitation7,Citation8 along with both cleavable and non-cleavable linkers.Citation9 The method for chemical attachment of drug to antibody, however, remained relatively unchanged. More recently, substantial efforts have gone toward investigation of new techniques that result in a more homogeneous mixture with greater control over the site of drug attachment, which could potentially result in an improved therapeutic index of the conjugates. This review will focus on these new techniques for site-specific drug attachment, as there are numerous other reviews that have already explained the important factors for selection of antibody, drug, and linker.Citation10-Citation19
Current ADC Conjugation Methods
There are many techniques for chemical modification of proteins,Citation20-Citation22 but two in particular are notable because they were used to construct the two ADCs with current FDA marketing approvals. Brentuximab vedotin, developed by Seattle Genetics, Inc., consists of an anti-CD30 monoclonal antibody conjugated to the highly cytotoxic drug monomethylauristatin E (MMAE) via modification of native cysteine side chain thiols.Citation23,Citation24 This method involves reduction of the solvent-exposed disulfides with dithiothreitol (DTT) or tris(2-carboxyethyl)phosphine (TCEP) followed by modification of the resulting thiols with maleimide-containing drugs (). For brentuximab vedotin, the thiols were modified with MC-VC-PAB-MMAE, which incorporates a cathepsin B protease cleavage siteCitation25 (VC: valine, citrulline) and a self-immolative linker (PAB: para-aminobenzyloxycarbonyl) between the maleimide group (MC: maleimidocaproyl) and the cytotoxic drug (MMAE).Citation26 The cysteine attachment strategy resulted in two drugs per reduced disulfide; most human IgG molecules have four solvent-exposed disulfide bonds, and so a range of zero to eight drugs per antibody was possible. The exact number of drugs per antibody was determined by the extent of disulfide reduction. Full reduction of all four disulfide bonds gave a homogeneous construct with eight drugs per antibody while a partial reduction resulted in a heterogeneous mixture with zero, two, four, six, or eight drugs per antibody. Though the mixture is heterogeneous, there are still only eight potential sites for conjugation, which gives some measure of site control compared with the lysine modification strategy described below. Although these disulfide bonds aid in the stability of the antibody, there have been many examples of antibodies that bind their antigen with similar affinity after reduction.Citation26,Citation27 Overall, this method has proven to be reliable and robust for many different antibody substrates. In addition to brentuximab vedotin, several other conjugates in clinical trials have also utilized this method.Citation28-Citation31
Figure 1. Schemes for non-specific (A and B) and site-specific (C, D, and E) drug conjugation to an antibody molecule. (A) After reduction of the interchain disulfide bonds with TCEP or DTT, MC-VC-PAB-MMAE (maleimidocaproyl-valinecitrulline-p-amino-benzyloxycarbonyl-monomethylauristatin E) was attached to the resulting cysteine side chain thiols using the maleimide functional group. The resulting conjugate was a heterogeneous mixture of antibodies with different numbers of drugs attached to the eight cysteine residues involved in interchain disulfide bonds. (B) DM1 (mertansine) was connected to the antibody with a maleimidocyclohexanecarboxylate bifunctional linker (SMCC) through the antibody lysine side chain amines. The resulting conjugate was a heterogeneous mixture with zero to eight drugs per antibody attached to as many as 40 different lysine residues.Citation34 (C) Site directed mutagenesis was utilized to install one additional cysteine residue in each heavy chain. Upon antibody expression this cysteine was found in a disulfide bond with glutathione. Reduction of all solvent exposed disulfide bonds followed by re-oxidation of the native disulfide bonds with CuSO4 resulted in a single thiol on each heavy chain which could be modified with a maleimide-containing drug such as MC-VC-PAB-MMAE. This gave a homogeneous product with precisely two drugs attached at the sites of the mutant cysteines. (D) AcLys-VC-PAB-MMAD (acetyllysine-valinecitrulline-p-aminobenzyloxycarbonyl-monomethyldolastatin 10) was attached using the enzyme microbial transglutaminase to catalyze the site-specific reaction between the drug lysine and engineered glutamine side chains in the antibody. This method produced a homogeneous conjugate with two drugs per antibody, attached to the engineered glutamines. (E) The unnatural amino acid p-acetylphenylalanine was genetically incorporated into an antibody using an amber stop codon suppressor tRNA/aaRS pair. The resulting antibody contained one p-acetylphenylalanine in each heavy chain at the location of the genetically encoded amber stop codon. Site-specific modification of p-acetylphenylalanine with AF-oxyamine (auristatin F-oxyamine) resulted in a homogeneous conjugate with precisely two drugs per antibody. (F) Chemical structures and abbreviations of drugs drawn with relevant antibody amino acid side chain attached.
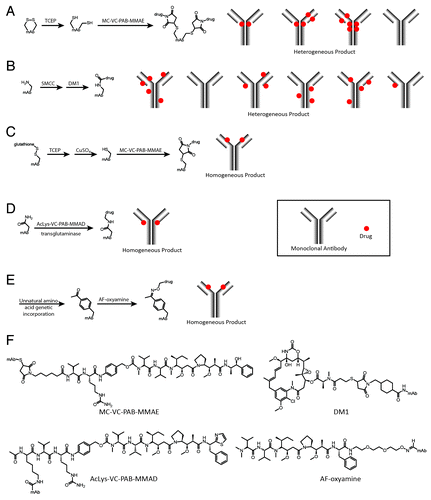
The other ADC with current FDA approval is ado-trastuzumab emtansine (Genentech, Inc.), which was constructed by coupling an anti-HER2 monoclonal antibody to the cytotoxic drug maytansine through modification of lysine side chain amines.Citation32 This version of maytansine (DM1) was modified to include a thiol that could be attached to a maleimide linker. A bifunctional linker (SMCC) with a maleimide at one end and an N-hydroxysuccinimidyl ester (NHS ester) at the other was reacted with lysine primary amine side chains to form a stable amide bond. The modified maytansine (DM1) was then attached to the antibody through conjugation to the maleimide end of the bifunctional linker (). In contrast to the linker utilized in brentuximab vedotin, this linker had no protease cleavage site and thus required lysosomal degradation of the antibody to liberate the active DM1-linker-lysine metabolite. The attachment method resulted in a heterogeneous mixture of conjugates with an average of 3.0–3.6 drugs per antibody, but a range of zero to approximately six.Citation33 Compared with the cysteine method described above, this strategy gave a more heterogeneous mixture because 20Citation33 to 40Citation34 different lysine residues were found to be modified, while only 8 different cysteine residues are modified using the native cysteine modification method. Nonetheless, this method has proven effective as evidenced by the success of ado-trastuzumab emtansine and the numerous other conjugates in advanced trials that use this technology.Citation35-Citation37
Limitations of Current Conjugation Methods
Though the previously described methods have led to FDA-approved ADCs, and they are being used for most of the conjugates in clinical trials, there is still substantial room for improvement in the areas of therapeutic index, toxicity, and pharmacokinetics. While the exact mechanisms of ADC clearance and toxicity are still not fully understood, it has become clear from empirical evidence that the number of drugs per antibody can have a large effect on the important in vivo parameters of the conjugate. Hamblett et al. constructed ADCs using the cysteine modification strategy, and then used non-denaturing hydrophobic interaction chromatography to isolate conjugates with exactly four drugs per antibody from the heterogeneous mixture. While this method allowed comparison of different drug loading levels, it was not scalable. When equal concentrations of antibody were tested in cell toxicity assays, ADCs with eight drugs per antibody showed a lower IC50 than those with four drugs per antibody.Citation38 This trend did not translate to in vivo mouse xenograft experiments, however, as, at equivalent antibody doses, the conjugate with four drugs per antibody was equally potent as the conjugate with eight drugs per antibody. Further, on a per drug basis, the antibody with four drugs was twice as potent as the eight drugs per antibody conjugate. The differences observed between in vitro and in vivo potency was due to an increased rate of clearance for the more heavily modified conjugates (). These experiments led to the conclusion that the optimal loading was two to four drugs per antibody to maximize potency while avoiding rapid clearance from circulation. While the cysteine and lysine attachment methods can be adjusted to give an average drug loading of two to four per antibody, the resulting mixture will still be heterogeneous and contain species with both less and more drug loading than desired. It is sub-optimal to have a non-potent portion of antibody (no drug loading) and a portion that has the potential to be rapidly cleared and could contribute to toxicity (high loading).Citation34,Citation38 Therefore, a conjugation strategy that results in a homogeneous mixture with two or four drugs per antibody would be ideal for maximizing the therapeutic index.
Table 1. Relevant pharmacokinetic comparisons between antibody drug conjugates with different drug loading or site of drug attachment
In addition to the issues with heterogeneous mixtures, there have been reports that the maleimide-thiol bond may have stability issues in circulation. Alley et al. were the first to show that drug transfer from antibody to cysteine-34 of albumin occurred slowly in serum.Citation39 They postulated that the maleimide was released via retro-Michael reaction and the high concentration of thiol-containing albumin scavenged the free maleimide drug. Transfer to the albumin was blocked by either adding an excess of thiol to the serum or alkylating albumin cysteine-34. The conjugate was stable in buffered aqueous solution as long as there were no thiols present to scavenge the maleimide drug before it could re-attach to the antibody. The rate of this reaction in serum is slow, with a similar half-life to that of the IgG in circulation, and resulted in an approximately 25% drop in the exposure of drug to the tumor according to area under curve calculations. These experiments were performed using ADCs with the drug attached to the interchain disulfide cysteines, and do not necessarily represent the rate of maleimide exchange for thiols incorporated at different positions. Alley et al. showed that replacing the maleimidocaproyl group with bromoacetylcaproyl decreased the rate of drug release, but there is always a need for new conjugation chemistry that results in high stability linkages.
Site Specific Methods for Drug Attachment
To solve some of the issues described above, several research groups developed methods to site-specifically attach drugs at precise locations on the antibody. This strategy imparts advantages such as well-defined, homogeneous conjugates and, in some cases, new chemistry for attachment. With a tunable, homogeneous system, the pharmacokinetics of the conjugate can be optimized by changing the site of attachment of the drug, the linker chemistry, and the overall number of drugs. These factors are difficult to change with the standard attachment strategies and the resulting heterogeneous mixtures would require significant separation efforts to analyze each component.
THIOMAB drug conjugates
The first site-specific approach was developed by Junutula et al. at Genentech by introducing extra cysteine residues using site-directed mutagenesis.Citation34 This common practice in protein modificationCitation40 was more complicated in an antibody because of the numerous native disulfide bonds already present. Introducing the extra cysteine residue in an unsuitable position could result in improper formation of the native disulfide bonds and therefore improper folding of the antibody. A further complication arose when the cysteine mutants were expressed, as the mutant residues were found as disulfides with cellular thiols such as glutathione or cysteine. In addition, a small portion of the antibodies were found to have three light chains due to a disulfide bond between engineered cysteine residues in the light chain, but this issue was resolved through purification and adjustment of cell culture methods.Citation41 Drug attachment to the mutant residues required reduction first, but there was no way to selectively reduce only the mutant cysteine disulfides without also reducing the native disulfide bonds. This problem was solved by reducing both the interchain and mutant disulfides, purifying the antibody away from the small molecule thiols, then reoxidizing the native cysteines using a gentle oxidant such as CuSO4 or dehydro-ascorbic acid. The resulting antibody had all of its native disulfide bonds intact, and a single reduced thiol in each heavy or light chain depending on the position of the mutation ().
In addition to the mixed disulfide issue, some of the mutants were rendered unreactive by formation of a disulfide between the two mutant thiol side chains (light chain V110C). Other mutants (heavy chain A114C) showed excellent reactivity toward maleimide reagents, thus giving a mostly homogeneous conjugate with additions only at the mutated residues. MC-VC-PAB-MMAE was then attached to an anti-MUC16 antibody (heavy chain A114C – now referred to as a THIOMAB) to form a THIOMAB drug conjugate (TDC). This particular conjugate was studied with 1.6 drugs per antibody but later preparations achieved approximately 2.0 drugs per antibody. In comparison to the corresponding ADC constructed through the standard cysteine reduction strategy, the TDC showed slightly decreased efficacy in vitro and this difference was attributed to the 2-fold lower drug loading (1.6 for TDC, 3.1 for ADC). However, similar to the four vs. eight drugs per antibody case described by Hamblett et al.,Citation38 the TDC was equally or more potent than the ADC in vivo when equivalent antibody doses were injected. Additionally, the TDC showed much lower toxicity at equivalent doses compared with the ADC. In rats, the ADC at 16.6 mg/kg (1,500 µg/m2 MMAE) showed depletion of circulating neutrophils five days after injection with a compensatory rebound by day 12, while the TDC at 36.4 mg/kg (1,500 µg/m2 MMAE) showed no difference from the vehicle injection. Similar results were also observed in cynomolgus monkeys. In summary, the TDC was equally potent to the ADC in vivo when the injections were equivalent by antibody concentration, and the TDC showed fewer adverse effects when the injections were equivalent by total drug concentration. The serum clearance rate of the TDC was also slower than the ADC (), and a higher percentage of the TDC retained at least one drug. That the TDC both stayed in circulation longer and had fewer adverse effects strongly suggested that the source of much of the toxicity comes from the portion of drug conjugate that is degraded or removed from circulation. In addition to the MUC16 TDC, a TDC version of ado-trastuzumab emtansine showed similar efficacy to the corresponding ADC, but with fewer adverse effects, and thus it has a 2-fold higher therapeutic index.Citation42 The promising results of these studies demonstrate the importance of developing new methods to precisely control the drug loading of ADCs.
After showing the benefits of the controlled drug loading, the researchers examined how changing the site of drug attachment could alter the properties of the conjugates. To further investigate the role of the site of attachment on the properties of a TDC, Shen et al. made several TDC preparations with the mutant cysteine in different locations.Citation43 The three constructs compared were light chain V205C, heavy chain A114C, and Fc-S396C. Drug attachment to each resulted in conjugates with two drugs per antibody. All showed similar in vitro potency, but dramatic differences when tested in vivo. The light chain mutant showed slightly better anti-tumor efficacy than the heavy chain mutant, while the Fc mutant was significantly less potent than the other two.
In experiments meant to explain these results, the three TDC samples were incubated in serum at 37°C and the ratio of two-drug TDC to total antibody was determined by liquid chromatography-mass spectrometry (LC-MS). An increased rate of drug loss from the antibody correlated well with diminished efficacy of the conjugates. The less potent Fc mutant had little remaining two-drug TDC after only 24 h, while the other conjugates showed a much more gradual loss of drug. Other mutations in the Fc region did not have the same potency-reducing effect, so the effect was site-specific and not antibody region-specific. After cleavage from the antibody, the drug was found covalently attached to albumin, in agreement with observations by Alley et al.Citation39 Upon further examination of the LC-MS data, it was observed that the light chain V205C mutant showed mass shifts of +17 Da and +33 Da upon incubation in serum. These mass shifts corresponded to hydrolysis of one or both maleimide rings, which prevented the retro-Michael reaction that separates the drug from the antibody. It was then postulated that the high concentration of positive charges near the mutant cysteine catalyzed this hydrolysis, and this trend was observed on several different TDC mutants. It should be noted that these difficult LC-MS experiments were made significantly easier by the homogeneity of the sample, and might not be possible when analyzing a heterogeneous conjugate. Clearly the site of attachment plays a role in the pharmacokinetics of these conjugates, although these experiments do not allow determination of whether the solvent accessibility or the maleimide hydrolysis plays the larger role.
The engineered cysteine method proved rigorous enough to be used for the site-specific ADC SGN-CD33A from Seattle Genetics, Inc., which recently entered a Phase 1 study as a treatment of acute myeloid leukemia (AML). It was constructed using a proprietary engineered cysteine method to attach a dimer of the novel DNA crosslinking drug pyrrolobenzodiazepine.Citation44 The resulting conjugate mixture consisted of approximately 95% antibody with two linked drugs and an average loading of 1.9 drugs per antibody. No pharmacokinetic data has been released to date, but the conjugate showed full xenograft tumor regressions with single doses as low as 0.1 mg/kg.
Antibody drug conjugates via transglutaminase
In addition to the THIOMAB conjugation strategy, other methods for site-specific attachment of drugs have been developed. Strop et al. from Rinat-Pfizer demonstrated a new technique for conjugation using microbial transglutaminase to couple an amine-containing drug to an engineered glutamine on the antibody.Citation45 Transglutaminase is an enzyme that catalyzes bond formation between the acyl group of a glutamine side chain and the primary amine of a lysine side chain.Citation46 Previous experiments by other groups suggested that this method would result in site-specific conjugates. Jeger et al. showed that the enzyme does not modify any of the native glutamine residues in the antibody,Citation47 thus allowing selective modification of glutamine residues incorporated via site-directed mutagenesis. In addition, Ohtsuka et al. demonstrated that microbial transglutaminase could catalyze bond formation using a wide range of amino substrates, provided a four carbon linker separated the amine group from the rest of the molecule.Citation48 The group at Rinat-Pfizer utilized these observations to develop a strategy that efficiently coupled cytotoxic drugs functionalized with a lysine or amino-PEG spacer to glutamine side chain residues (). They incorporated the short sequence LLQG into 90 locations throughout the antibody, and 12 sites showed high levels of conjugation when exposed to microbial transglutaminase and a suitable amine substrate. Two conjugates were carried forward for analysis in animal models, both utilizing an anti-M1S1 antibody (C16). One contained the extra glutamine sequence at the C-terminus of the light chain (C16-LC) while the other incorporated the glutamine at the C-terminus of the heavy chain (C16-HC). Upon conjugation to AcLys-VC-MMAD (lysine with N-terminal amine acetylated, valine-citrulline protease cleavage site, and monomethyl dolastatin 10) the mutants had a loading of 1.9 (C16-HC) and 1.8 (C16-LC) drugs per antibody out of a maximum of 2.0, and peptide mapping demonstrated that the drugs were attached to the mutant glutamine residues. The resulting peptide bond between the glutamine side chain and lysine of the drug moiety did not have the stability issue found with the cysteine-maleimide conjugates described in the THIOMAB section above. This allowed the effect of the conjugation site on pharmacokinetics to be probed independent of chemical stability.
C16-LC and C16-HC were first compared with a traditional C16 ADC (3.6 drugs per antibody) in vitro. Both conjugates showed IC50 values in the 0.050–0.075 nM range for M1S1 overexpressing cell lines BxPC3 and A431, while the traditional C16 ADC showed values in the 0.030–0.040 nM range for the same cell lines. The difference in potency roughly corresponded to the ~2-fold difference in drug loading. Further, the site-specific conjugates showed similar in vivo efficacy to the traditional ADC in a mouse model with BxPC3 xenograft. Treatment with each of the three agents at a single dose of 3 mg/kg conjugate caused significant tumor regression that was stable over the 120 d of the study. Similar to the findings of Junutula et al.,Citation34 these site-specific conjugates showed similar efficacy at equivalent antibody doses, but double the potency when the drug loading was taken into consideration. While similar in mice, C16-LC and C16-HC showed significant pharmacokinetic differences in rats (). The C16-HC conjugate showed a much accelerated rate of clearance in rats as 83% of the antibody was lost from serum 24 h post-injection. In rats, the clearance of the C16-HC conjugate was similar to the anti-Muc1 ADC described by Junutula et al., whereas the C16-LC conjugate showed similar clearance to the anti-Muc1 site-specific THIOMAB drug conjugate. Because the C16-LC and C16-HC conjugates do not utilize maleimide chemistry and therefore do not suffer from the maleimide transfer issues, the marked differences between the two conjugates are likely due to the position of the attached drug. To verify that the C-terminal modification of the heavy chain did not interfere with FcRn interactions, the binding of both conjugates was tested and showed no difference from the respective naked antibodies. However, the C-terminal heavy chain mutation may affect other cellular interactions that impact conjugate clearance. Taken together, these experiments suggest that the placement of the drug on the antibody can have a significant effect on the pharmacokinetics, though the mechanism of this effect is unknown. Further experiments with the drug attached in a wider variety of positions could help elucidate some of these unknowns.
Unnatural amino acids in antibody drug conjugates
In addition to enzymatic conjugation, orthogonal chemistry conjugation has also been used to site-specifically modify a wide variety of proteins using unnatural amino acids.Citation49-Citation51 One method for inserting unnatural amino acids into proteins uses an evolved tRNA/tRNA synthetase pair that specifically recognizes the unnatural amino acid and incorporates it as the 21st amino acid in place of the amber stop codon.Citation52,Citation53 Recent experiments by Axup et al. demonstrated that this method was viable for producing IgG with unnatural amino acids.Citation54 They chose to introduce p-acetylphenylalanine as the unnatural amino acid because it contains a ketone functional group that is not found in any of the 20 natural amino acid side chains. This allows for specific modification of the ketone groups without interference from other amino acids, hence the term orthogonal conjugation. As a proof of concept, p-acetylphenylalanine was incorporated into an anti-Her2 IgG1. This was accomplished by first stably integrating the evolved tRNA and corresponding p-acetylphenylalanine-tRNA synthetase genes into Chinese hamster ovary cells. The light and heavy chain genes were then incorporated, with the heavy chain bearing a mutation that changed alanine-121 to the amber stop codon recognized by the evolved tRNA. The resulting cells produced over 300 mg/L of the mutant antibody from stable clones, which showed that the yield did not suffer from this technique. The mutant antibody showed similar affinity for its ligand as trastuzumab (Herceptin®), and electrospray ionization mass spectrometry (ESI-MS) of the deglycosylated heavy chain showed the expected mass given the mutation of one alanine to p-acetylphenylalanine.
With mutant antibody in hand, a drug containing the appropriate chemical moiety was required to make the desired ADC. Ketone groups can be modified with an alkoxyamine to form an oxime bond that is stable under physiological conditions ().Citation55,Citation56 Therefore, an auristatin F derivative attached to an oxyamine group using a short polyethylene glycol linker (AF-oxyamine) was synthesized. Similar to the DM1 molecule from ado-trastuzumab emtansine, this drug contained a non-cleavable linker so release of the drug was dependent on lysosomal degradation of the adjoining antibody.Citation32,Citation57 After reaction of AF-oxyamine with the mutant antibody, the resulting conjugate (anti-Her2-IgG-nAF) contained two drugs per antibody, with less than 5% unreacted antibody as detected by ESI-MS. When tested in vitro the conjugate showed excellent potency against various Her2+ cell lines with EC50 values in the 0.11–0.35 nM range, while showing significantly less potency (> 40 nM) against MCF-7 cells (Her2-). Similarly impressive results were observed in vivo as the conjugate showed nearly full regressions of MDA-MB-435 Her2+ xenograft tumors with a single injection at a dose of 5 mg/kg. To evaluate the pharmacokinetics, anti-Her2-IgG-nAF was injected into rats and the serum concentration of antibody was observed over time. The conjugate was indistinguishable from the naked mutant antibody (), thus suggesting that this conjugation method and placement of drug were effective. In addition, the conjugate was incubated in mouse serum at 37°C for 3 d before use in cell toxicity assays and showed nearly identical in vitro potency and selectivity as non-incubated conjugate. No toxicity was observed in Her2- cells for the incubated conjugate, which suggested there was no cleavage of drug from the antibody during incubation. This method provided an additional route for constructing ADCs with precisely two drugs per antibody. In addition, the oxime chemistry resulted in drug-antibody linkages with excellent biological stability.
This work only included pharmacokinetic data for one mutant, so it was not possible to draw any conclusions about the site of drug attachment. However, the unnatural amino acid method is particularly well suited for scanning potential positions of attachment on the antibody. Simple site-directed mutagenesis to incorporate an amber stop codon is all that is necessary to survey new mutants. The importance and potential of this method is illustrated by the number of companies pursuing various technologies for unnatural amino acid incorporation. Ambrx, Inc. (associated with Axup et al.), Allozyne, Inc., and Sutro Biopharma, Inc. are all developing ADC platforms using unnatural amino acids. Questions remain, however, about the potential immunogenicity of unnatural amino acids and their bio-orthogonal linkages. Though no evidence of immunogenicity was reported by Axup et al., further investigation is required to ensure the safety of these compounds.
Conclusion
The methods described in this review have provided three new routes to homogeneous ADCs with two drugs per antibody attached at precise locations. Compared with traditional methods, the site-specific methods showed an improvement in therapeutic index. Since ADC toxicity correlates roughly with the total amount of drug, the antibodies with high drug loading contribute more to toxicity, but are also more rapidly cleared from circulation than the low drug loading antibodies, so they not only contribute more to toxicity but also less to tumor killing efficacy. These site-specific methods eliminate the high drug loading antibodies while also limiting the amount of unconjugated antibody present. In addition, these site-specific methods will allow for further optimization of pharmacokinetic properties based on the site of attachment of the drug. Finally, these methods have proven to be relatively scalable, which makes them viable for clinical exploration.
Although the site-specific ADCs appear to improve upon the non-specific ADCs currently used in the clinic, these conclusions are based on fairly limited preclinical data and require more rigorous testing in clinical trials. Additionally, site-specific ADCs are still in the nascent stage of development. Further improvements are still possible, such as homogeneous conjugates with greater than two drugs per antibody, which could be more potent. While higher drug loading has been correlated with increased rate of clearance and toxicity, those conclusions were drawn from ADCs constructed using non-specific lysine or cysteine modification methods and may not be valid for site-specific ADCs. As the mechanisms for toxicity and ADC clearance are better understood, these site-specific methods can be engineered to produce ADCs with the desired properties. The area of ADCs is rapidly expanding and the contributions of site-specific chemistry will help fuel that expansion.
| Abbreviations: | ||
| ADC | = | antibody drug conjugate |
| VC | = | valine citrulline |
| TDC | = | THIOMAB drug conjugate |
| MMAE | = | monomethylauristatin E |
| MMAD | = | monomethyl dolastatin 10 |
| LC-MS | = | liquid chromatography-mass spectrometry |
| ESI-MS | = | electrospray ionization mass spectrometry |
| AUC | = | area under the curve |
Disclosure of Potential Conflicts of Interest
No potential conflict of interest was disclosed.
Acknowledgements
We thank the National Institutes of Health/National Cancer Institute for financial support (R01 CA118919, R01 CA129491, and R01 CA171315).
Related Research Data
References
- Chabner BA, Roberts TG Jr.. Timeline: Chemotherapy and the war on cancer. Nat Rev Cancer 2005; 5:65 - 72; http://dx.doi.org/10.1038/nrc1529; PMID: 15630416
- Minchinton AI, Tannock IF. Drug penetration in solid tumours. Nat Rev Cancer 2006; 6:583 - 92; http://dx.doi.org/10.1038/nrc1893; PMID: 16862189
- Wu AM, Senter PD. Arming antibodies: prospects and challenges for immunoconjugates. Nat Biotechnol 2005; 23:1137 - 46; http://dx.doi.org/10.1038/nbt1141; PMID: 16151407
- Yang HM, Reisfeld RA. Doxorubicin conjugated with a monoclonal antibody directed to a human melanoma-associated proteoglycan suppresses the growth of established tumor xenografts in nude mice. Proc Natl Acad Sci USA 1988; 85:1189 - 93; http://dx.doi.org/10.1073/pnas.85.4.1189; PMID: 3422487
- Hinman LM, Hamann PR, Wallace R, Menendez AT, Durr FE, Upeslacis J. Preparation and characterization of monoclonal antibody conjugates of the calicheamicins: a novel and potent family of antitumor antibiotics. Cancer Res 1993; 53:3336 - 42; PMID: 8324745
- Chari RV, Martell BA, Gross JL, Cook SB, Shah SA, Blättler WA, McKenzie SJ, Goldmacher VS. Immunoconjugates containing novel maytansinoids: promising anticancer drugs. Cancer Res 1992; 52:127 - 31; PMID: 1727373
- Koehn FE. Natural Product Cytotoxins as Payloads for Antibody Drug Conjugates. Natural Products and Cancer Drug Discovery. 2013. 97–119.
- Anderl J, Faulstich H, Hechler T, Kulke M. Antibody-drug conjugate payloads. Methods Mol Biol 2013; 1045:51 - 70; http://dx.doi.org/10.1007/978-1-62703-541-5_4; PMID: 23913141
- Nolting B. Linker technologies for antibody-drug conjugates. Methods Mol Biol 2013; 1045:71 - 100; http://dx.doi.org/10.1007/978-1-62703-541-5_5; PMID: 23913142
- Thudium K, Bilic S, Leipold D, Mallet W, Kaur S, Meibohm B, Erickson H, Tibbitts J, Zhao H, Gupta M. American Association of Pharmaceutical Scientists National Biotechnology Conference Short Course: Translational Challenges in Developing Antibody-Drug Conjugates: May 24, 2012, San Diego, CA. MAbs 2013; 5:5 - 12; http://dx.doi.org/10.4161/mabs.22909; PMID: 23255090
- Sievers EL, Senter PD. Antibody-drug conjugates in cancer therapy. Annu Rev Med 2013; 64:15 - 29; http://dx.doi.org/10.1146/annurev-med-050311-201823; PMID: 23043493
- Teicher BA, Chari RV. Antibody conjugate therapeutics: challenges and potential. Clin Cancer Res 2011; 17:6389 - 97; http://dx.doi.org/10.1158/1078-0432.CCR-11-1417; PMID: 22003066
- Chari RV. Targeted cancer therapy: conferring specificity to cytotoxic drugs. Acc Chem Res 2008; 41:98 - 107; http://dx.doi.org/10.1021/ar700108g; PMID: 17705444
- Harper J, Mao S, Strout P, Kamal A. Selecting an optimal antibody for antibody-drug conjugate therapy: internalization and intracellular localization. Methods Mol Biol 2013; 1045:41 - 9; http://dx.doi.org/10.1007/978-1-62703-541-5_3; PMID: 23913140
- Bander NH. Antibody-drug conjugate target selection: critical factors. Methods Mol Biol 2013; 1045:29 - 40; http://dx.doi.org/10.1007/978-1-62703-541-5_2; PMID: 23913139
- Gerber H-P, Koehn FE, Abraham RT. The antibody-drug conjugate: an enabling modality for natural product-based cancer therapeutics. Nat Prod Rep 2013; 30:625 - 39; http://dx.doi.org/10.1039/c3np20113a; PMID: 23525375
- Carter PJ, Senter PD. Antibody-drug conjugates for cancer therapy. Cancer J 2008; 14:154 - 69; http://dx.doi.org/10.1097/PPO.0b013e318172d704; PMID: 18536555
- Ducry L, Stump B. Antibody-drug conjugates: linking cytotoxic payloads to monoclonal antibodies. Bioconjug Chem 2010; 21:5 - 13; http://dx.doi.org/10.1021/bc9002019; PMID: 19769391
- Schrama D, Reisfeld RA, Becker JC. Antibody targeted drugs as cancer therapeutics. Nat Rev Drug Discov 2006; 5:147 - 59; http://dx.doi.org/10.1038/nrd1957; PMID: 16424916
- Stephanopoulos N, Francis MB. Choosing an effective protein bioconjugation strategy. Nat Chem Biol 2011; 7:876 - 84; http://dx.doi.org/10.1038/nchembio.720; PMID: 22086289
- Hermanson GT. Bioconjugate Techniques. Elsevier; 2008.
- Baslé E, Joubert N, Pucheault M. Protein chemical modification on endogenous amino acids. Chem Biol 2010; 17:213 - 27; http://dx.doi.org/10.1016/j.chembiol.2010.02.008; PMID: 20338513
- Senter PD, Sievers EL. The discovery and development of brentuximab vedotin for use in relapsed Hodgkin lymphoma and systemic anaplastic large cell lymphoma. Nat Biotechnol 2012; 30:631 - 7; http://dx.doi.org/10.1038/nbt.2289; PMID: 22781692
- van de Donk NW, Dhimolea E. Brentuximab vedotin. MAbs 2012; 4:458 - 65; http://dx.doi.org/10.4161/mabs.20230; PMID: 22684302
- Dubowchik GM, Firestone RA, Padilla L, Willner D, Hofstead SJ, Mosure K, Knipe JO, Lasch SJ, Trail PA. Cathepsin B-labile dipeptide linkers for lysosomal release of doxorubicin from internalizing immunoconjugates: model studies of enzymatic drug release and antigen-specific in vitro anticancer activity. Bioconjug Chem 2002; 13:855 - 69; http://dx.doi.org/10.1021/bc025536j; PMID: 12121142
- Doronina SO, Toki BE, Torgov MY, Mendelsohn BA, Cerveny CG, Chace DF, DeBlanc RL, Gearing RP, Bovee TD, Siegall CB, et al. Development of potent monoclonal antibody auristatin conjugates for cancer therapy. Nat Biotechnol 2003; 21:778 - 84; http://dx.doi.org/10.1038/nbt832; PMID: 12778055
- Sun MM, Beam KS, Cerveny CG, Hamblett KJ, Blackmore RS, Torgov MY, Handley FG, Ihle NC, Senter PD, Alley SC. Reduction-alkylation strategies for the modification of specific monoclonal antibody disulfides. Bioconjug Chem 2005; 16:1282 - 90; http://dx.doi.org/10.1021/bc050201y; PMID: 16173809
- Pollack VA, Alvarez E, Tse KF, Torgov MY, Xie S, Shenoy SG, MacDougall JR, Arrol S, Zhong H, Gerwien RW, et al. Treatment parameters modulating regression of human melanoma xenografts by an antibody-drug conjugate (CR011-vcMMAE) targeting GPNMB. Cancer Chemother Pharmacol 2007; 60:423 - 35; http://dx.doi.org/10.1007/s00280-007-0490-z; PMID: 17541593
- Ma D, Hopf CE, Malewicz AD, Donovan GP, Senter PD, Goeckeler WF, Maddon PJ, Olson WC. Potent antitumor activity of an auristatin-conjugated, fully human monoclonal antibody to prostate-specific membrane antigen. Clin Cancer Res 2006; 12:2591 - 6; http://dx.doi.org/10.1158/1078-0432.CCR-05-2107; PMID: 16638870
- Li D, Poon KA, Yu S-F, Dere R, Go M, Lau J, Zheng B, Elkins K, Danilenko D, Kozak KR, et al. DCDT2980S, an anti-CD22-monomethyl auristatin E antibody-drug conjugate, is a potential treatment for non-Hodgkin lymphoma. Mol Cancer Ther 2013; 12:1255 - 65; http://dx.doi.org/10.1158/1535-7163.MCT-12-1173; PMID: 23598530
- Govindan SV, Cardillo TM, Sharkey RM, Tat F, Gold DV, Goldenberg DM. Milatuzumab-SN-38 conjugates for the treatment of CD74+ cancers. Mol Cancer Ther 2013; 12:968 - 78; http://dx.doi.org/10.1158/1535-7163.MCT-12-1170; PMID: 23427296
- Lewis Phillips GD, Li G, Dugger DL, Crocker LM, Parsons KL, Mai E, Blättler WA, Lambert JM, Chari RV, Lutz RJ, et al. Targeting HER2-positive breast cancer with trastuzumab-DM1, an antibody-cytotoxic drug conjugate. Cancer Res 2008; 68:9280 - 90; http://dx.doi.org/10.1158/0008-5472.CAN-08-1776; PMID: 19010901
- Wang L, Amphlett G, Blättler WA, Lambert JM, Zhang W. Structural characterization of the maytansinoid-monoclonal antibody immunoconjugate, huN901-DM1, by mass spectrometry. Protein Sci 2005; 14:2436 - 46; http://dx.doi.org/10.1110/ps.051478705; PMID: 16081651
- Junutula JR, Raab H, Clark S, Bhakta S, Leipold DD, Weir S, Chen Y, Simpson M, Tsai SP, Dennis MS, et al. Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat Biotechnol 2008; 26:925 - 32; http://dx.doi.org/10.1038/nbt.1480; PMID: 18641636
- Tassone P, Gozzini A, Goldmacher V, Shammas MA, Whiteman KR, Carrasco DR, Li C, Allam CK, Venuta S, Anderson KC, et al. In vitro and in vivo activity of the maytansinoid immunoconjugate huN901-N2′-deacetyl-N2′-(3-mercapto-1-oxopropyl)-maytansine against CD56+ multiple myeloma cells. Cancer Res 2004; 64:4629 - 36; http://dx.doi.org/10.1158/0008-5472.CAN-04-0142; PMID: 15231675
- Ikeda H, Hideshima T, Fulciniti M, Lutz RJ, Yasui H, Okawa Y, Kiziltepe T, Vallet S, Pozzi S, Santo L, et al. The monoclonal antibody nBT062 conjugated to cytotoxic Maytansinoids has selective cytotoxicity against CD138-positive multiple myeloma cells in vitro and in vivo. Clin Cancer Res 2009; 15:4028 - 37; http://dx.doi.org/10.1158/1078-0432.CCR-08-2867; PMID: 19509164
- Blanc V, Bousseau A, Caron A, Carrez C, Lutz RJ, Lambert JM. SAR3419: an anti-CD19-Maytansinoid Immunoconjugate for the treatment of B-cell malignancies. Clin Cancer Res 2011; 17:6448 - 58; http://dx.doi.org/10.1158/1078-0432.CCR-11-0485; PMID: 22003072
- Hamblett KJ, Senter PD, Chace DF, Sun MMC, Lenox J, Cerveny CG, Kissler KM, Bernhardt SX, Kopcha AK, Zabinski RF, et al. Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate. Clin Cancer Res 2004; 10:7063 - 70; http://dx.doi.org/10.1158/1078-0432.CCR-04-0789; PMID: 15501986
- Alley SC, Benjamin DR, Jeffrey SC, Okeley NM, Meyer DL, Sanderson RJ, Senter PD. Contribution of linker stability to the activities of anticancer immunoconjugates. Bioconjug Chem 2008; 19:759 - 65; http://dx.doi.org/10.1021/bc7004329; PMID: 18314937
- Chalker JM, Bernardes GJL, Lin YA, Davis BG. Chemical modification of proteins at cysteine: opportunities in chemistry and biology. Chem Asian J 2009; 4:630 - 40; http://dx.doi.org/10.1002/asia.200800427; PMID: 19235822
- Gomez N, Vinson AR, Ouyang J, Nguyen MD, Chen X-N, Sharma VK, Yuk IH. Triple light chain antibodies: factors that influence its formation in cell culture. Biotechnol Bioeng 2010; 105:748 - 60; PMID: 19845001
- Junutula JR, Flagella KM, Graham RA, Parsons KL, Ha E, Raab H, Bhakta S, Nguyen T, Dugger DL, Li G, et al. Engineered thio-trastuzumab-DM1 conjugate with an improved therapeutic index to target human epidermal growth factor receptor 2-positive breast cancer. Clin Cancer Res 2010; 16:4769 - 78; http://dx.doi.org/10.1158/1078-0432.CCR-10-0987; PMID: 20805300
- Shen B-Q, Xu K, Liu L, Raab H, Bhakta S, Kenrick M, Parsons-Reponte KL, Tien J, Yu S-F, Mai E, et al. Conjugation site modulates the in vivo stability and therapeutic activity of antibody-drug conjugates. Nat Biotechnol 2012; 30:184 - 9; http://dx.doi.org/10.1038/nbt.2108; PMID: 22267010
- Kung Sutherland MS, Walter RB, Jeffrey SC, Burke PJ, Yu C, Kostner H, Stone I, Ryan MC, Sussman D, Lyon RP, et al. SGN-CD33A: a novel CD33-targeting antibody-drug conjugate using a pyrrolobenzodiazepine dimer is active in models of drug-resistant AML. Blood 2013; 122:1455 - 63; http://dx.doi.org/10.1182/blood-2013-03-491506; PMID: 23770776
- Strop P, Liu S-H, Dorywalska M, Delaria K, Dushin RG, Tran T-T, Ho W-H, Farias S, Casas MG, Abdiche Y, et al. Location matters: site of conjugation modulates stability and pharmacokinetics of antibody drug conjugates. Chem Biol 2013; 20:161 - 7; http://dx.doi.org/10.1016/j.chembiol.2013.01.010; PMID: 23438745
- Lorand L, Graham RM. Transglutaminases: crosslinking enzymes with pleiotropic functions. Nat Rev Mol Cell Biol 2003; 4:140 - 56; http://dx.doi.org/10.1038/nrm1014; PMID: 12563291
- Jeger S, Zimmermann K, Blanc A, Grünberg J, Honer M, Hunziker P, Struthers H, Schibli R. Site-specific and stoichiometric modification of antibodies by bacterial transglutaminase. Angew Chem Int Ed Engl 2010; 49:9995 - 7; http://dx.doi.org/10.1002/anie.201004243; PMID: 21110357
- Ohtsuka T, Sawa A, Kawabata R, Nio N, Motoki M. Substrate specificities of microbial transglutaminase for primary amines. J Agric Food Chem 2000; 48:6230 - 3; http://dx.doi.org/10.1021/jf000302k; PMID: 11141280
- Kim CH, Axup JY, Schultz PG. Protein conjugation with genetically encoded unnatural amino acids. Curr Opin Chem Biol 2013; 17:412 - 9; http://dx.doi.org/10.1016/j.cbpa.2013.04.017; PMID: 23664497
- Sletten EM, Bertozzi CR. Bioorthogonal chemistry: fishing for selectivity in a sea of functionality. Angew Chem Int Ed Engl 2009; 48:6974 - 98; http://dx.doi.org/10.1002/anie.200900942; PMID: 19714693
- Best MD. Click chemistry and bioorthogonal reactions: unprecedented selectivity in the labeling of biological molecules. Biochemistry 2009; 48:6571 - 84; http://dx.doi.org/10.1021/bi9007726; PMID: 19485420
- Wang L, Brock A, Herberich B, Schultz PG. Expanding the genetic code of Escherichia coli. Science 2001; 292:498 - 500; http://dx.doi.org/10.1126/science.1060077; PMID: 11313494
- Wang L, Schultz PG. Expanding the genetic code. Angew Chem Int Ed Engl 2004; 44:34 - 66; http://dx.doi.org/10.1002/anie.200460627; PMID: 15599909
- Axup JY, Bajjuri KM, Ritland M, Hutchins BM, Kim CH, Kazane SA, Halder R, Forsyth JS, Santidrian AF, Stafin K, et al. Synthesis of site-specific antibody-drug conjugates using unnatural amino acids. Proc Natl Acad Sci USA 2012; 109:16101 - 6; http://dx.doi.org/10.1073/pnas.1211023109; PMID: 22988081
- Jencks WP. Mechanism and Catalysis of Simple Carbonyl Group Reactions. Prog Phys Org Chem 1964; 2:63 - 128; http://dx.doi.org/10.1002/9780470171813.ch2
- Kalia J, Raines RT. Hydrolytic stability of hydrazones; oximes. Angew Chem Int Ed Engl 2008; 47:7523 - 6; http://dx.doi.org/10.1002/anie.200802651; PMID: 18712739
- Doronina SO, Mendelsohn BA, Bovee TD, Cerveny CG, Alley SC, Meyer DL, Oflazoglu E, Toki BE, Sanderson RJ, Zabinski RF, et al. Enhanced activity of monomethylauristatin F through monoclonal antibody delivery: effects of linker technology on efficacy and toxicity. Bioconjug Chem 2006; 17:114 - 24; http://dx.doi.org/10.1021/bc0502917; PMID: 16417259