
Abstract
The functional dichotomy of antibodies against interleukin-2 (IL-2) is thought to depend upon recognition of different cytokine epitopes. Beyond functional studies, the only molecular evidence obtained so far located the epitopes recognized by the immunoenhancing antibodies S4B6 and JES6–5H4 within the predicted interface of IL-2 with the α receptor subunit, explaining the preferential stimulation of effector cells displaying only β and γ receptor chains. A consistent functional map of the epitope bound by the immunoregulatory antibody JES6-1A12 has now been delineated by screening the interactions of phage-displayed antigen variants (with single and multiple mutations) and antigen mimotopes. The target determinant resides in a region between the predicted interfaces with α and β/γ receptor subunits, supporting the dual inhibitory role of the antibody on both interactions. Binding by JES6-1A12 would thus convert complexed IL-2 into a very weak agonist, reinforcing the advantage of T regulatory cells (displaying the high affinity αβγ heterotrimeric receptor) to capture the cytokine by competition and expand over effector cells, ultimately resulting in the observed strong tolerogenic effect of this antibody. Detailed knowledge of the epitopes recognized by anti-IL-2 antibodies with either immunoenhancing or immunoregulatory properties completes the molecular scenario underlying their use to boost or inhibit immune responses in multiple experimental systems. The expanded functional mapping platform now available could be exploited to study other interactions involving related molecular pairs with the final goal of optimizing cytokine and anti-cytokine therapies.
Introduction
The dual role of IL-2 as a master regulator of immune responses, promoting either tolerance or immunity depending on the context,Citation1,Citation2 has added complexity to the translation of the extensive basic knowledge about this cytokine into clinical benefit for patients.Citation3 The net effect of IL-2-based therapies is the result of the balance between its interactions with different cell populations within the immune system and the whole organism.Citation4
Since the description of a clear functional dichotomy of immune complexes formed between IL-2 and different monoclonal antibodies (mAbs),Citation5 the use of such complexes has expanded to multiple experimental settings for either enhancing the effector responsesCitation6-Citation9 or down modulating immunity.Citation10-Citation13 Besides the therapeutic potential of these approaches,Citation14 immune complexes highlight different features of IL-2 biology. Their divergent, but potent, activities have been interpreted as reflecting the ability of particular mAbs to mask specific IL-2 functions, allowing other roles to be fully evident. Selective functional blockade has been anticipated to depend upon recognition of diverse epitopes and subsequent inhibition of the interactions with the different subunits within the multi-chain IL-2 receptor (IL-2R).Citation5 Despite the wide use of IL-2-containing immune complexes and the biological and practical implications derived from such experience, the molecular bases of their actions remain largely unexplored.
Our recent epitope mapping studiesCitation15 provided for the first time molecular support to the prediction that immunostimulatory mAbs S4B6 and JES6-5H4 recognize antigenic determinants on mouse IL-2 overlapping with the interface with the α receptor subunit.Citation5 Because the identity of the epitope recognized by an antibody with opposite biological functions (the immunoregulatory JES6-1A12 mAb) was not known, delineation of a whole picture of the molecular phenomena underlying the divergent biological effects of IL-2-containing immune complexes has not been possible. The current work locates the JES6-1A12 epitope in the middle region between the interfaces with α and β/γ IL-2R subunits, supporting the dual inhibitory role of the antibody on both interactions. This model is consistent with the observed biological effects of the antibody.
Convergent results obtained through different experimental approaches (extensive single and multiple mutagenesis of phage-displayed mIL-2, epitope grafting on human IL-2, peptide selection from a random library, and direct binding assays) allowed a clear molecular definition of the JES6-1A12 functional epitope. The available epitope mapping platform was thus expanded and could be used to map multiple interactions within the complex network established by IL-2 and related cytokines, contributing to the understanding and optimization of cytokine and anti-cytokine therapies.
Results
Phage-displayed mouse IL-2 was shown to be biologically active
Phage-displayed mouse IL-2 (mIL-2) was the basic molecule used in our epitope mapping strategy, both for binding assays involving the wild-type protein and for the construction of new mutated variants. Although the antigenicity of mIL-2 in this format had been extensively shown,Citation15 an additional quality control criterion was introduced in the current work. Phage-displayed mIL-2 was shown to be biologically active, as proven by its ability to induce the proliferation of the IL-2-dependent CTLL-2 cell line, in contrast with phages displaying an unrelated single chain Fv antibody fragment (scFv), which were totally inert in the same experiment (). The c-myc tag fused to the C-terminal end of every displayed protein in our system provided a simple way to measure their relative levels by ELISA with the anti-tag 9E10 mAb, allowing use of equivalent amounts of each phage-displayed protein (measured in arbitrary units/mL) in the proliferation assay. Taking into account that between 1–10% of the filamentous phage particles display a single copy of the heterologous molecule in similar phagemid-based systems,Citation16 the concentration of phage-displayed mIL-2 inducing half-maximal cell proliferation could be roughly estimated to be between 0.6 and 6 pmol/L. Control in vitro refolded recombinant mIL-2 induced half-maximal proliferation at 20 pmol/L (). The above described assay was a very stringent test of the proper overall folding of the molecule and showed that its ability to bind the receptor in a natural context and to deliver cell signaling was preserved after periplasmic expression and attachment to phage particles. Although similar results had been obtained for human IL-2,Citation17,Citation18 this is the first time to our knowledge that phage-displayed mouse IL-2 is shown to be functional.
Figure 1. CTLL-2 cell proliferation induced by phage-displayed mouse IL-2. 104 cells/well were incubated with serial dilutions of purified phage preparations displaying either mIL-2 or an unrelated scFv antibody fragment (A) and soluble in vitro refolded recombinant mIL-2 (B) during 48h. Phages were diluted to reach equivalent levels of phage-displayed proteins (measured in units/mL in a previous ELISA using the anti-c-myc 9E10 mAb as coating antibody). Alamar blue dye was added to the cells and, after 12h incubation, the absorbances at 540 and 630 nm were measured. (C) In order to assess the effects of neutralizing anti-mIL-2 mAbs in this system, increasing concentrations of purified JES6–1A12, JES6–5H4 and S4B6 mAbs were added during the incubation of a sub-saturating concentration of phage-displayed mIL-2 with the cells. 9E10 was used as a control non-neutralizing mAb recognizing the phage-displayed mIL-2 through the c-myc epitope fused to its C-terminal end.
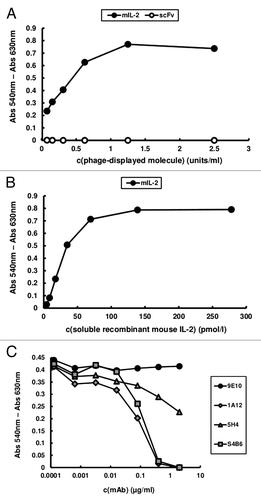
Inhibition of cell proliferation induced by phage-displayed mIL-2 by the three neutralizing anti-mIL-2 mAbs under study () served two purposes. Besides providing further confirmation of the specificity of the effects of the phage-displayed cytokine on CTLL-2 cells, the blocking activity of the antibodies could be directly compared on cells having the heterotrimeric IL-2R. Remarkably, the neutralizing ability of S4B6 and JES6-1A12 mAbs (immunostimulatory and immunoregulatory in vivo, respectively) was similar and higher than the one displayed by a second immunostimulatory mAb (JES6-5H4). Such behavior could depend upon their relative affinities because S4B6 and JES6-5H4 have been shown to have closely related fine specificities.Citation15 The absence of inhibitory effects of increasing concentrations of 9E10 mAb, which recognizes the phage-displayed IL-2 through the c-myc epitope fused to its C-terminal end, ruled out any non-specific interference related to the use of the bulky viral particles in neutralization experiments.
Competition assays defined two different antigenic regions on mIL-2 for immunoenhancing and immunoregulatory antibodies
Direct competition enzyme-linked immunosorbent assay (ELISA) experiments between coating mAbs and mAbs in solution for binding to phage-displayed mIL-2 resulted in a clear pattern of cross-competition between the two immunoenhancing antibodies S4B6 and JES6-5H4 (). This was consistent with their well-known shared biological effectsCitation5 and with previous molecular evidences of a large overlap between their target epitopes.Citation15 On the other hand, the immunoregulatory mAb JES6-1A12 was not able to inhibit mIL-2 capture by the two other antibodies and its binding was not inhibited by a large excess of any of them (), indicating that the two classes of antibodies recognize different antigenic regions on mIL-2 surface. Lack of competition between JES6-1A12 and S4B6 mAbs had been previously described,Citation5 based on their behavior in a sandwich ELISA. The specificity of competition effects in our display system was confirmed by the absence of any inhibitory effect of the 9E10 mAb.
Figure 2. Competition between mAbs for binding to mouse IL-2. Polyvinyl chloride microtiter plates were coated with anti-mIL-2 mAbs at 2.5 µg/mL. Diluted purified phages displaying mIL-2 (1011 viral particles/mL) were incubated on coated wells, after pre-incubating them in the presence or absence of competitor antibodies in solution at 100 µg/mL. 9E10 mAb was also used as a control non-competitor antibody recognizing the c-myc tag fused to IL-2 molecules in our display system. Bound phages were detected with an anti-M13 mAb conjugated to horseradish peroxidase. Arrows indicate cross-competition between the different antibodies. Cross-competition was defined as the decrease in capture by a given antibody (by more than 50%) after incubating phage-displayed mIL-2 with a second antibody in solution.
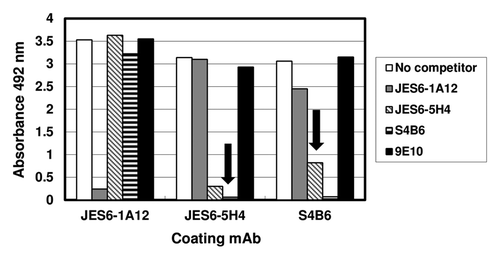
While competitor S4B6 and JES6-1A12 mAbs in solution fully abolished binding to mAbs-coated ELISA microplates, JES6-5H4 only produced partial inhibition even at the maximal tested concentration (100 µg/mL). This finding supported the idea of a relatively lower affinity of this antibody and was consistent with its lower neutralizing ability in cell proliferation assays. All the above described results pointed to shared biological effects of S4B6 and JES6-5H4 mAbsCitation5 being determined by similar recognition specificities regardless of divergent affinities.
Screening of partially humanized mIL-2 variants highlighted a region involved in recognition by the immunoregulatory antibody JES6-1A12
Despite the lack of cross-reactivity of JES6-1A12 with human IL-2, the individual replacement of every solvent-exposed amino acid by its human counterpart had previously failed to highlight any residue as contributor to epitope formation. This was in sharp contrast with the success to identify the epitopes recognized by other non-cross-reactive anti-IL-2 antibodies using this methodology.Citation15 The absence of cross-reactivity in the case of JES6-1A12 mAb could be the net result of several sequence differences between human and mouse IL-2, each one resulting in a minor effect difficult to underscore with single mutated variants. This idea led to the design of an alternative mapping strategy, also based on phage display and extensive mutagenesis of the whole antigen, but distinguished by the simultaneous introduction of multiple human residues in a selected region of the predicted mIL-2 structure. Eight discrete mIL-2 segments, covering all the differences between IL-2 from both species (), were thus separately humanized in order to determine their contributions to JES6-1A12 epitope formation (). In five of these segments, all the targeted residues were solvent-exposed (>20% relative solvent accessibility, RSA), being able to form clusters directly interacting with the antibody, and humanization implied replacement by their human counterparts. The remaining three segments also contained residues with non-exposed side chains that differ from human IL-2. The latter could affect the antigenicity by disturbing the relative orientation of the contiguous solvent-accessible amino acids (aa), or have a longer-range effect on epitopes that are not so close through changes in the core of the molecule. To distinguish between the two possibilities, these three segments were humanized to different degrees by constructing mini-libraries of variants where all the predicted solvent-exposed residues were replaced by their human counterparts, while some aa predicted to be non-exposed were substituted by an equimolar mixture of the original mIL-2 residue and its human equivalent ().
Figure 3. Sequence alignment of mouse and human IL-2. Eight discrete segments of mIL-2 covering all the differences between both molecules (selected for partial humanization) are underlined. The first two residues (Ala-Pro, shown in gray) are absent from the phage-displayed molecules. Residues Cys140 and Cys125 (unpaired Cys) in mouse and human IL-2 respectively were replaced by Ser (shaded in gray).

Table 1. Phage-displayed variants of mouse IL-2 containing humanized segments
ELISA screening of all the humanized phage-displayed variants revealed that the introduction of human residues in two contiguous segments of mIL-2, Q30-E43 and R46-P51, resulted in loss of recognition by JES6-1A12 mAb (). When the first segment was targeted, reactivity was totally abolished, while humanization of the second one led to a large reactivity decrease (by more than 90%). The effect was specific for this antibody and not due to the lack of protein display on viral particles or to any global folding defect caused by mutations, since the immunoreactivity with 9E10 mAb (recognizing the c-myc tag peptide fused to the carboxy-terminus of IL-2 molecules in our display system), and with S4B6 and JES6–5H4 anti-mIL-2 mAbs, was preserved in every case. There were no differences between the reactivity of the variants within the mini-library of partial/total humanization of the segment Q30-E43, indicating that at least some accessible residues within this segment (which were humanized in all the variants) contribute to epitope formation. The fortuitous appearance of a variant not recognized by JES6-1A12 mAb, in which only the segment comprised between E37 and E43 had been humanized (probably arising from a mutagenesis artifact), allowed narrowing the antigenic region recognized by the antibody.
Figure 4. Recognition of IL-2-derived mutated variants by monoclonal antibodies. The antigenicity of IL-2-derived variants with multiple mutations was tested by ELISA. Phages displaying the mutated molecules were rescued at a 96-well scale. Polyvinyl chloride microtiter plates were coated with anti-IL-2 mAbs and the control 9E10 mAb. Diluted phage-containing supernatants (1011 viral particles/mL) were incubated on coated wells. Bound phages were detected with an anti-M13 mAb conjugated to horseradish peroxidase. Phage-displayed wt mouse and human IL-2 were included as positive and negative controls respectively. The presence of the desired replacements, as well as the absence of undesired mutations along the rest of the gene, was confirmed by sequencing. (A) Discrete segments within the phagemid-inserted mIL-2 gene were humanized through Kunkel mutagenesis. Eight non-overlapping segments were modified, while an additional partially humanized variant (*) arose as a mutagenesis artifact. The names of the humanized variants (hum) indicate the limits of the humanized segment in each one. Arrows indicate the lack of specific reactivity of a particular mIL-2 variant with a given antibody. (B) The segment E37-P51 of mIL-2 was grafted on the hIL-2 scaffold to recapitulate the JES6–1A12 mAb epitope. Anti-hIL-2 mAbs (IL-2.1 and IL-2.2) were used to detect the human scaffold displayed on filamentous phages.
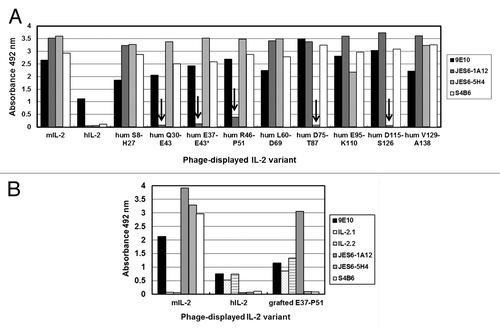
Lack of recognition of mIL-2 variants including humanized segments D75-T87 and D115-S126 by JES6-5H4 mAb () matched with the presence of individually critical residues contributing to the formation of its epitope in each of these segments,Citation15 and confirmed the accuracy of our mapping strategy based on molecules harboring multiple mutations. The fact that there was no overlap between the effects of humanizing mIL-2 regions on recognition by immunoenhancing and immunoregulatory mAbs was totally consistent with the lack of competition between them.
Unlike for JES6-5H4, none of the partially humanized variants lost reactivity toward the competitor S4B6 mAb. This was not surprising because three of the solvent-exposed residues contributing to the formation of the epitope recognized by S4B6 (Y59, E74, and E76) are conserved between human and mouse IL-2, while the two other critical aa, D75 and F122, were fully functionally replaced by the residues present at the equivalent positions of hIL-2 (Glu and Tyr).Citation15 The case of S4B6 illustrates the difficulties in associating the lack of cross-reactivity of some antibodies with differences in a single, or even in a few neighboring, critical aa. Species specificity in such cases could be related to global folding differences and to long-range cooperative effects of multiple sequence variations. This fact has practical implications: inter-species mutagenesis scanning is not always useful to locate a putative target region to start mapping. The functional map of S4B6 epitope was indeed revealed through the exploration of an antigenic region previously defined for another (competitor) mAb, JES6-5H4.Citation15
Successful recapitulation of JES6-1A12 mAb binding by grafting a mouse IL-2 segment on human IL-2 confirmed the relevance of the identified antigenic region
To further confirm the identity of the antigenic region identified as relevant for JES6-1A12 recognition of mIL-2, an additional gain-of-function experiment was performed. Grafting the previously identified mIL-2 segment (E37-P51) into the hIL-2 scaffold allowed the reconstruction of the epitope on the previously non-reactive human molecule (). The resulting chimeric protein was still non-reactive with the two other anti-mIL-2 mAbs and properly recognized by anti-hIL-2 mAbs (IL-2.1 and IL-2.2). All these control antibodies bind to regions in mouse/human IL-2 far from the segment involved in grafting,Citation15 and were useful to confirm the specificity of epitope recapitulation. On the other hand, individual replacement of each human residue within the selected segment by its mouse counterpart did not result in any detectable reactivity. The above described results highlighted the contribution of the mIL-2 E37-P51 segment as a whole to the formation of the epitope recognized by JES6-1A12 mAb.
Site-directed mutagenesis delineated a detailed picture of the epitope recognized by JES6-1A12 antibody
While molecules with multiple mutations were useful to define a broad antigenic region involved in recognition by JES6-1A12 mAb, the above described experiments did not shed light on the contribution of individual residues to the interaction. The observed reactivities were the net result of introducing several simultaneous changes. Screening of molecules with individual replacements within this antigenic region, not limited to positions that are different between mouse and human IL-2, would reveal details of the epitope. Since the effects of diverse conservative and non-conservative substitutions can be drastically different, depending on the precise nature of the interactions contributing to epitope formation, each position within the whole antigenic region (E37-P51) was comprehensively scanned by randomization (). Despite the potential emergence of 19 different replacements at each targeted position, the amount of characterized mutated variants was lower due to the overrepresentation of some substitutions within randomization products, the deleterious effects of other mutations on protein display (assessed with the anti-tag mAb), and the fortuitous appearance of additional undesired changes (mutations, deletions, insertions) in some of the variants. However, the number and diversity of characterized replacements allowed the definition of tolerance/sensitivity patterns to modifications at each position, overcoming the limitations of a conventional alanine scanning. The exploration was restricted to solvent-exposed residues (>20% RSA) presumed to establish direct interactions with the antibody. The antigenic region to be scanned was broadened to include the flanking solvent-exposed residues M33 and Q36. While the initial mapping step (humanization of continuous mIL-2 segments) dismissed the cooperative effects on binding of residues distant in the primary sequence, but close in the 3D structure, site-directed randomization in the search for individual contributions also included two residues located in the spatial neighborhood (<8Å) of the segments already shown to be relevant, although far from them in the primary sequence (K90 and Q93).
Table 2. Effects of individual mutations on recognition of mouse IL-2 by JES6–1A12 mAb
Antigenicity screening revealed two critical residues whose substitution by many other aa was enough to cause a drastic loss of recognition by JES6–1A12 mAb: Asn 44 and Arg 46 (). The acceptance of only Gln and Lys at positions 44 and 46, respectively, without decreasing antigenicity, underscored the critical side chain features directly involved in mIL-2 recognition by JES6-1A12 mAb. Both an amide moiety at position 44 and a positively charged group at position 46 are required for binding. An additional adjacent residue (Glu 43) also contributes to epitope formation, although to a lesser extent, since its replacement only caused a moderate antigenicity decrease (between 50 and 75% of the original reactivity was kept) in some cases. It is noteworthy that substituting this residue by the oppositely charged Arg (presumably creating a non-favorable electrostatic context at the interface) had an even greater effect on reactivity. The same happened with the replacements by Cys and Pro, which could cause the formation of non-natural disulfide bridges and the distortion of the backbone, respectively, resulting in additional changes in the vicinity of the epitope (). Position 45 in mIL-2 could be occupied by diverse aa with very different properties () without affecting antigenicity, despite being located between the two critical residues contributing to epitope formation. The orientation of Tyr 45, whose side chain points to an opposite direction compared with the ones of the flanking residues according to our structural model,Citation15 is consistent with the lack of a major individual contribution of this residue to the interaction. Tyr 45 side chain is indeed buried from the solvent, and was included in the screening only because of its close vicinity to the previously identified critical residues. Pro was the only aa able to disrupt binding when introduced at this position of mIL-2, presumably due to main chain structural constraints that disturbed the relative orientations of the two flanking critical residues (Asn 44 and Arg 46). All the above described mutations affected specifically the JES6-1A12 epitope because the variants displaying changes at these positions were well recognized by JES6-5H4 and S4B6 mAbs (>75% reactivity compared with phage-displayed wt mIL-2).
The fact that many residues within the explored antigenic region (either contained in the segment E37-P51 or spatially located in its close vicinity) had no detectable individual effects on reactivity (), did not rule out multiple minor contributions among them that influence antigenicity as a whole. Actually, such cooperative influence should be expected, given the drastic effect of introducing multiple replacements in the segments E37-E43 and R46-P51. These additional contributions would thus modulate the binding mediated by the individually critical residues Asn 44 (conserved between human and mouse IL-2) and Arg 46 (replaced by a Lys fully compatible with the interaction in the human molecule), resulting in the observed lack of cross-reactivity. The few examples of individual mutations at the surrounding positions causing a marked antigenicity decrease were limited to the introduction of Cys and Pro (), which could disturb the folding of the protein besides affecting the epitope itself. Some of them were indeed able to affect recognition by both JES6–1A12 and the non-competitor immunostimulatory mAbs, indicating a general effect on the antigen folding rather than specific epitope disruption.
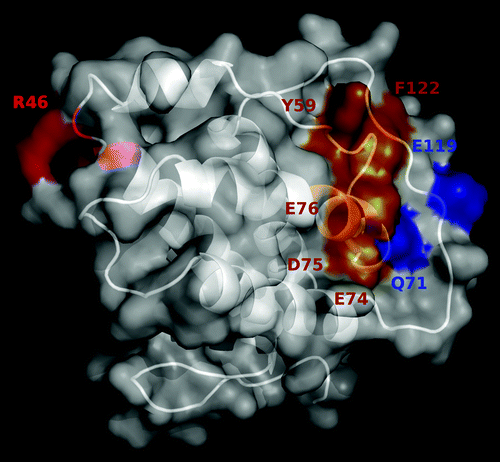
The location of the cluster of residues that contribute to the formation of the epitope recognized by JES6-1A12 mAb is shown in . They are far from the antigenic region comprising the overlapping epitopes previously defined for the immunoenhancing S4B6 and JES6-5H4 mAbs.Citation15 Recognition of two different antigenic regions by antibodies with divergent biological activities supported a role for fine specificity in the in vivo outcome of IL-2 binding by them, as it had been previously proposed.Citation5
Figure 5. Location of the antigenic regions recognized by immunoenhancing and immunoregulatory antibodies on mouse IL-2. The predicted 3D structure of mIL-2 is represented in white as a cartoon with semi-transparent surface. Residues belonging to functional epitopes are labeled. The functional epitope recognized by the immunoregulatory antibody JES6-1A12 is colored red. Residues contributing to the formation of the overlapping epitopes recognized by the immunoenhancing antibodies JES6-5H4 and S4B6 are highlighted in orange, while additional residues involved only in recognition by JES6-5H4 mAb are shown in blue. The figure was generated with Pymol.
Peptides selected from a random phage-displayed library showed a common motif resembling the functional epitope recognized by JES6-1A12 mAb
Additional information about the nature of the epitope recognized by JES6–1A12 mAb was obtained through selection of antigenic mimotopes from a phage-displayed random peptide library. A recurrent tripeptide motif (Q/N)X(R/K) was found in 44 of 47 unique peptides selected on JES6-1A12. This common motif was located in different positions along the 10-mer peptide sequences. The first motif position was predominantly occupied by Gln (34/44), while the ten remaining peptides contained Asn. The middle position (X) was quite variable, with Leu being the most frequent residue (found in 20 of the 44 (Q/N)X(R/K)-containing peptides), followed by Ile (5/44), Met (5/44), Val (3/44), Cys (2/44), and Ala, Phe, Trp, Tyr, Thr, Glu, Gln, Lys, or Arg (each one represented only once). Despite the preferential selection of hydrophobic residues at this position in the peptide context, the acceptance of aa with widely divergent chemical and physical properties indicated that the critical interactions underlying peptide selection by JES6-1A12 are restricted to Gln/Asn and Arg/Lys. The third position displayed almost exclusively Arg (43/44), although one peptide exhibited Lys instead.
Random peptide selection on antibodies, although routinely used for epitope mapping,Citation19 does not always result in the isolation of peptides that resemble the nominal antigen.Citation20,Citation21 Peptide mimotopes could establish totally new interactions with the selector paratope compared with the nominal antigen,Citation22 resulting in antigenic mimicry in the absence of structural or even functional similarities between their binding modes. In the case of anti-IL-2 antibodies, mixed results have been obtained. While the identity of one hIL-2 epitope predicted on the basis of peptide library screening was confirmed by the more reliable characterization of mutated antigen variants (IL-2.2 mAb), the set of peptides selected on another anti-hIL-2 mAb (IL-2.1) led to misidentification of its epitope.Citation15,Citation23 On the other hand, library screening with S4B6 resulted in a set of binding peptides without obvious similarities, neither between them, nor with the target antigen (unpublished results). In this complex scenario, the fact that the peptide motif selected on JES6–1A12 closely resembled the most critical antigen features contributing to epitope formation is particularly remarkable. The sequence (Q/N)X(R/K) was able to mimic the segment NYR in mIL-2 (positions 44–46). The coexistence of Asn/Gln and Arg/Lys in the first and third positions of the peptide motif was fully consistent with the acceptance of amide-containing and positively charged side chains at positions 44 and 46 of mIL-2, respectively (). The high variability at the middle motif position among peptides selected on JES6–1A12 mAb was in good agreement with the wide tolerance of replacements at mIL-2 position 45. Pro, which was the only aa able to disrupt binding when introduced in this location, was not found among the selected peptides.
The above described peptide selection experiments supported the direct involvement of Asn 44 and Arg 46 in binding of JES6-1A12 mAb to mIL-2. The coincidence of the critical motif identified through peptide mimicry with the one defined by site-directed mutagenesis on the native antigen confirmed the accuracy of our mapping studies. Despite the limitations of peptide library screening as an independent epitope mapping procedure, its combination with other experimental approaches can reveal molecular details of the interaction, as in the case of JES6-1A12 mAb.
Both immunoenhancing and immunoregulatory antibodies inhibit binding of mouse IL-2 to IL-2 receptor α chain
Beyond the mere identification of the epitopes recognized by anti-IL-2 mAbs, the most relevant output of mapping studies would be the description of the functional consequences of their binding for the complex interaction between IL-2 and its multi-chain receptor. The availability of biologically active soluble mouse IL-2R α chain allowed us to test the ability of neutralizing mAbs to block the interaction between IL-2 and this particular receptor chain. The fact that the immunoenhancing antibodies JES6-5H4 and S4B6 totally abolished IL-2/IL-2R α chain interaction () was fully consistent with their anticipated role as selective inhibitors of IL-2 binding to this chain constitutively present in T regulatory cells.Citation5 This blocking effect also matched with recent mapping studiesCitation15 that located the relevant residues contributing to the formation of their epitopes within the region of mouse IL-2 predicted to interact with the IL-2R α chain ().
Figure 6. Direct inhibition of the interaction between mouse IL-2 and IL-2 receptor α chain by anti-mIL-2 antibodies. Polyvinyl chloride microtiter plates were coated with soluble receptor α chain at 2.5 µg/mL. Diluted purified phages displaying mIL-2 (1011 viral particles/mL) were incubated on coated wells, after pre-incubating them in the presence or absence of competitor antibodies in solution at 1 µg/mL. 9E10 mAb was used as a control non-competitor antibody (recognizing the c-myc tag fused to IL-2 molecules in our display system). Bound phages were detected with an anti-M13 mAb conjugated to horseradish peroxidase. Arrows indicate inhibition of mIL-2 binding to the immobilized α chain.
Figure 7. Relative location of the epitopes recognized by anti-IL-2 antibodies and the predicted mouse IL-2 receptor interfaces. The surface of mouse IL-2 is shown in white and residues belonging to functional epitopes are labeled. The interfaces with the different receptor chains were predicted on the bases of the available knowledge about the structure of the complex between human IL-2 and its multi-chain receptor, and the structural alignment between human and mouse IL-2. Overlapping between the predicted α chain interface and the functional epitope(s) recognized by the immunostimulatory antibodies JES6-5H4/S4B6 is shown in panel (A). mIL-2 residues in contact with both these antibodies and the α chain are colored green. The rest of the α chain interface is represented in yellow, while additional residues contributing to JES6–5H4 epitope formation are shown in blue. Location of the JES6–1A12 functional epitope (adjacent to both α and β/γ chain interfaces) can be appreciated in panel (B). Residues in the β and γ chain interfaces are colored cyan and magenta respectively, and the epitope recognized by JES6-1A12 is represented in red. Panel (C) highlights with a dashed line the putative contact area of mIL-2 with JES6-1A12 mAb (surrounding the residues involved in functional epitope formation). Overlapping between this region (presumably buried in the immune complex with JES6–1A12) and both α and γ chain interfaces can be appreciated. The figures were generated with Pymol.
Remarkably, the immunoregulatory mAb JES6-1A12 was also a potent inhibitor of the interaction with the isolated α chain (). Although this finding is in contrast with the first speculations about selective blocking of interactions with β chain by immunoregulatory antibodies,Citation5 it is not difficult to understand from a molecular point of view because the cluster of residues shown to be relevant for mIL-2 recognition by JES6-1A2 in the current study lies in the region between the predicted interfaces with α and β/γ chains of the receptor (). The antibody could thus inhibit the interactions at both interfaces by virtue of steric hindrance. highlights the putative contact region between JES6-1A12 mAb and its target antigen. This area was proposed taking into account the location of mIL-2 residues involved in epitope formation (E43, N44, and R46) and the usual extension of the interfaces between antibodies and protein antigens, having between 600 and 900 Å2 contact areas.Citation24-Citation26 Arg 46 is adjacent to the α chain receptor interface residues, particularly to Leu 48. Glu 43 is very close to the gamma chain interface (mainly to Gln 36, Val 130, and Arg 134). Residues from both α and γ chain receptor interfaces of mIL-2 are thus included in the putative contact area that should be buried in the complex with JES6–1A12, providing a structural rationale for the observed dual inhibition. Our results did not support the simplest view of different classes of antibodies blocking binding to either α or β/γ chains alone. However, they provided the first clues to molecular dissection of biological activity of different antibodies on IL-2/IL-2R interaction, and strongly supported the idea of a link between anti-IL-2 antibodies fine specificity and functional effects.Citation5
Discussion
The differential ability of anti-IL-2 mAbs in immune complexes to either boost or inhibit immune responses has been anticipated to depend upon selective blockade of IL-2 binding to the receptor α chain (constitutively present in T regulatory cells) or to β/γ receptor subunits.Citation5 The latter subunits form the intermediate affinity receptor on effector CD8+ T cells and NK cells, and are also components of the high affinity heterotrimeric receptor on T regulatory cells (together with the α chain). Speculations about such preferential blocking effects have been raised based on functional studiesCitation5 and are supported by the recent mapping of the epitopes recognized by the immunoenhancing antibodies JES6-5H4 and S4B6.Citation15 Residues contributing to the formation of their epitopes are located on a surface patch of mIL-2 that is predicted to contact directly the receptor α chain, being far from the interfaces with β and γ chains. The prediction was based on the current knowledge about the structure of the human IL-2/IL-2R complexCitation27,Citation28 and the structural alignment between IL-2 from both species. Binding by this kind of antibody would thus impair the ability of IL-2 to stimulate preferentially T regulatory cells (due to the presence of the high affinity αβγ heterotrimeric receptor on them), while leaving unaffected its potential to induce the expansion of CD8+ T cells and NK cells through the heterodimeric βγ receptor. This interpretation of mapping results is fully consistent with the finding that the activity of immunoenhancing IL-2/anti-IL-2 immune complexes in vivo does not require their dissociation to render free IL-2 and is even reinforced by a large excess of antibody strongly displacing the equilibrium to the bound form.Citation29 An extreme example of the bioactivity of the bound form is the recent report of a chimeric protein including IL-2 and the S4B6 mAb light chain in a single polypeptide able to bind the S4B6 heavy chain.Citation30 Such chimera stabilizes the immune complex between IL-2 and the antibody, because the physical linkage promotes reassociation. A related, and even more drastic, approach is the use of an IL-2-derived mutein in which the interface with the chain was severely disrupted by mutagenesis, resulting in permanent abrogation of this particular interaction. The mutein is strongly immunostimulatory and evokes potent antitumor responses.Citation31 In total, the evidence indicates that masking a particular IL-2 antigenic region by immunoenhancing antibodies (the α chain interface) would result in functional masking of a crucial property of IL-2: its ability to promote tolerance,Citation1 allowing its other facet (enhancement of immune responses) to be fully evident. In such a way, the balance between regulatory and effector cells would be altered, resulting in exacerbated effector responses, as has been described for the immunostimulatory antibody S4B6 in the context of anti-viral vaccination,Citation7 and anti-tumor responses.Citation6,Citation8,Citation9
On the other hand, the simultaneous blockade of the interactions with α and β/γ chains by JES6-1A12 mAb provides a rationale for its strong immunoregulatory activity observed in diverse scenarios like autoimmune diseases therapy and transplantation,Citation10,Citation11,Citation13 and tolerance induction against therapeutic proteins.Citation12 Such dual inhibition of receptor binding would result in complexed IL-2 becoming a very weak agonist with negligible activity on effector cells characterized by the presence of the intermediate affinity receptor, together with selective delivery to T regulatory cells that would still be able to compete with the antibody (due to the extremely high affinity of the heterotrimeric receptor) and capture IL-2 from immune complexes. This model explains the weak, but significant, in vitro stimulation of cells expressing the high affinity αβγ receptor, as well as the total abrogation of the response of CD25- memory phenotype T cells (displaying the dimeric βγ receptor) in the presence of immune complexes formed by JES6-1A12 mAb and IL-2.Citation5 Despite the observed neutralizing effects of JES6-1A12, preferential stimulation of T regulatory cells over effector populations would even be reinforced in vivo. The proposed model explaining the functional dichotomy of anti-mIL-2 antibodies based on our mapping results is closely related, although not identical, to the original one that was anticipated solely from functional studies.Citation5
Remarkably, the current results are compatible with theoretical predictions arising from in silico simulations of therapeutic intervention with different classes of anti-IL-2 antibodies. According to mathematical modeling results, either mAbs inhibiting IL-2 binding to the receptor β chain or mAbs blocking the interactions with both α and β/γ subunits would be useful to treat autoimmunity.Citation32 On the other hand, our results argue against an alternative interpretation of the functional dichotomy of anti-IL-2 antibodies based on the idea that immunoregulatory antibodies like JES6–1A12 could leave exposed the regions of IL-2 that engage IL-2R α, β, and γ chains, augmenting the natural tolerance-promoting potential of IL-2 through a substantial increase of its half-life in immune complexes, without significantly inhibiting any interaction with the receptor.Citation33 This possibility was not supported by direct competition experiments with the α chain in our experimental system, and is also in contradiction with the total in vitro blocking activity of JES6-1A12 mAb on CD25- memory phenotype CD8+ T cells.Citation5
The current work differs from previous mapping studies in several issues. First, mutagenesis experiments were complemented by direct inhibition assays of IL-2 binding to the IL-2R α chain. The latter confirmed the proposed blocking activity of immunoenhancing antibodies and revealed the unexpected ability of the JES6-1A12 immunoregulatory mAb to block this particular interaction as well. Even though similar direct binding inhibition assays focused on the interaction between IL-2 and the isolated β and γ IL-2R subunits would challenge the JES6-1A12 dual inhibition idea in a conclusive way, the repeated failure to detect both interactions in several experimental formats, presumably due to their lower affinities (mainly of gamma chain binding)Citation34 precluded the generation of this kind of data in our hands. However, the direct inhibitory effect on α chain binding, together with the well-known neutralizing activity of the antibody on cells having only the β/γ IL-2R subunits,Citation5 gives experimental support to the dual inhibition notion.
A second difference from previous mapping studies is that the mIL-2 region previously thought to be important for S4B6 binding (Q26-Y45) was now shown to contribute to the non-competing antibody JES6-1A12 epitope formation instead. The explanation for this apparent contradiction should reside in the fact that this initial studyCitation35 was performed with deletion variants, and removal of large segments within the molecule could result in long-range effects on antigenicity. Our approach, based on replacing individual residues or discrete sets of aa taking into account the available structural information, rendered consistent results that locate the S4B6 epitope on a totally different region, which is in agreement not only with the lack of competition between both antibodies, but also with their in vivo functions. Immunoregulatory and immunoenhancing activities of JES6-1A12 and S4B6 antibodies are better explained by the current results locating their epitopes in a region close to the interfaces with α and β/γ subunits, and in the middle of the contact region with the α chain, respectively, as it was described above.
The mapping strategy exploited in the current study is based on the previously reported methodology used to map other anti-IL-2 antibodies.Citation15 The choice of the phage-displayed whole antigen as the basic molecule for mutagenesis studies instead of antigen fragments was due to the versatility of this approach, which could be useful to explore both linear and conformational epitopes through the introduction of any array of mutations located in different regions of the target. The original procedure was modified to circumvent the failure to identify individual critical residues for JES6-1A12 recognition. The first step of the new protocol was to introduce multiple simultaneous mutations in defined regions of the mIL-2 molecule in order to find a broad antigenic region, which was subsequently narrowed by randomizing individual positions. Both strategies are complementary and allow delineating a detailed picture of the target epitope(s). The current study thus expands the usefulness of the previously described phage display-based platform to map the epitopes on IL-2 and related molecules.Citation15 The choice of one or another protocol depends upon the characteristics of each antigen/antibody pair because some interactions would be easier to map starting with the identification of single critical residues, while other cases would require simultaneous disruption of multiple contacts to locate an antigenic region to be deeply explored.
Taken as a whole, our mapping platform should allow determination of the epitopes recognized by other anti-IL-2 antibodies, including those against human IL-2, and establishment of a global correlation between fine specificity and biological effects. Such information would result in the rational selection of novel antibodies targeting pre-defined Il-2 regions to increase their therapeutic efficacy. Detailed knowledge of their interactions should shed new light on our understanding of cytokine and anti-cytokine therapies.
Materials and Methods
Phage display of IL-2
Mouse and human IL-2 were displayed on filamentous phages as previously described.Citation15 Briefly, mIL-2/hIL-2 genes were cloned into the phagemid vector pCSM (fused to the M13 gene 3), and phage particles displaying the corresponding proteins were rescued with M13KO7 helper phage on a 50 mL scale and purified.Citation36
Biological activity of phage-displayed mouse IL-2 on CTLL-2 cells
Purified phages displaying either mIL-2 or an unrelated scFv, similarly fused to M13 Protein III, were filter-sterilized. The amounts of the displayed proteins were estimated by ELISA on polyvinyl chloride microtiter plates coated with the anti-tag 9E10 mAb. An anti-M13 mAb conjugated to horseradish peroxidase (HRP) was used to detect bound phages. An available standard phage preparation was assumed to have 100 units/mL of the displayed protein and taken as the reference to calculate the relative display levels of the other molecules. Both phage preparations were appropriately diluted to use the same amount of displayed proteins (measured in units/mL) in the cell proliferation assay. CTLL-2 cells were grown at 37° C under a humidified 5% CO2 atmosphere in RPMI 1640 (Gibco) supplemented with 10% heat-inactivated fetal bovine serum, 50 IU/mL of human recombinant IL-2 (CIGB), 2 mmol/L L-glutamine, 50 u/mL penicillin and 50 µg/mL streptomycin. Before the proliferation assay, cells were harvested by centrifugation, washed twice with RPMI without IL-2, and incubated in this medium during 5h. After that, 104 cells/well were incubated with serial dilutions of the filtered purified phage preparations diluted in the same medium during 48h. Cells were separately incubated with diluted control recombinant mouse IL-2 (specific activity 3 × 106 IU/mg). 20 µL of Alamar blue dye (Invitrogen) were then added to each well and the plates were incubated during 12h. The absorbances (Abs) at 540 nm and 630 nm were measured.
To assess the effects of neutralizing anti-mIL-2 mAbs in this system, increasing concentrations of purified JES6-1A12, JES6-5H4, and S4B6 mAbs were added during the incubation of a sub-saturating concentration of phage-displayed mIL-2 (1 unit/mL) with the cells. 9E10 was used as a control non-neutralizing mAb recognizing the phage-displayed mIL-2 through the c-myc epitope fused to its C-terminal end.
Phage ELISA screening of IL-2 interactions
Polyvinyl chloride microtiter plates were coated with anti-mIL-2 mAbs (JES6-1A12, JES6-5H4 or S4B6), the anti-tag 9E10 mAb, or soluble IL-2 receptor mouse α chain at 2.5 µg/mL in phosphate-buffered saline (PBS). Plates were blocked for 1h at room temperature (RT) with PBS containing 4% skim powder milk (M-PBS). Purified phages displaying mIL-2 or phage-containing supernatants from deep-well plates (see below), were diluted in M-PBS and incubated on coated/blocked plates during 2h at RT. For competition experiments only, purified mIL-2-displaying phages were diluted in M-PBS with or without the competitor mAbs (at concentrations between 1 µg/mL and 100 µg/mL) and pre-incubated 2h at RT before being added to the plates. After phage incubation on the plates, they were washed with PBS containing 0.1% Tween 20 (PBS-T) and an anti-M13 mAb conjugated to HRP (GE Healthcare), appropriately diluted in M-PBS, was added. Plates were incubated for 1h at RT and washed. Substrate solution (500 µg/mL ortho-phenylenediamine and 0.015% hydrogen peroxide in 0.1 mol/L citrate-phosphate buffer, pH 5.0) was added. The reaction was stopped after 15 min with 2.5 mol/L sulfuric acid. Absorbances were measured at 492 nm.
Site-directed mutagenesis and randomization
Partially humanized mIL-2 variants () were constructed by site-directed mutagenesisCitation37 using described procedures.Citation38 Briefly, single strand DNA (from mIL-2 gene-containing pCSM phagemid) obtained from phages produced by the CJ236 E. coli strain (dut-ung-thi-1 relA1 spoT1 mcrA/pCJ105 (F’ camr)) was used as the template. Antisense mutagenic oligonucleotides included 15 nucleotides complementary to the template at each end, flanking the region to be modified. Such target region contained one or more triplets (adjacent or not) to be replaced to obtain the desired mutations. Degenerate mutagenic oligonucleotides were used to introduce a mixture of two amino acids at some positions ().
Randomization of selected positions resulted in a panel of additional mIL-2 variants with single mutations. This was accomplished through the introduction by site-directed mutagenesis of the degenerate triplet NNK at a given position of the mIL-2 gene.
The antigenic region E37-P51 (from mIL-2) was grafted by similar site-directed mutagenesis procedures onto the hL-2 scaffold. The mutagenesis reaction was performed using single strand DNA from the hIL-2 gene-containing pCSM phagemid as the template.
Screening and molecular characterization of new IL-2 variants
TG1 E. coli cells (K12_(lac-pro), supE, thi, hsdD5/F’ traD36, proA+B+, lacIq, lacZ_M15) were transformed with mutagenesis products. Phages displaying the mutated IL-2 variants were rescued with M13KO7 helper phage at a 96-well scale.Citation36 Phage-containing supernatants were used for binding screening by ELISA as described above. Only those IL-2 variants that were successfully displayed (as assessed with the anti-c-myc tag 9E10 mAb) were included in any reactivity analysis. XL1-Blue E. coli cells (recA1 endA1 gyrA96 thi-1 hsdR17 supE44 relA1 lac F´ proAB lacIqZ_M15 Tn10 Tetr) were infected with the corresponding phage-containing supernatants and used to purify plasmid DNA with the QIAprep Spin Miniprep kit (Qiagen). Phagemid inserts were sequenced by Macrogen. Protein sequences of phage-displayed IL-2 variants were deduced from DNA sequences.
Phage-displayed peptide selection on JES6-1A12 mAb
A 10-mer random phage-displayed peptide library (composed by 109 molecules fused to M13 PVIII) was used. Immunotubes were coated overnight at 4° C with JES6-1A12 mAb at 10 µg/mL in PBS. Purified phages (5 × 1012 viral particles) rescued from the library with M13KO7 helper phage as describedCitation36 and coated immunotubes were blocked with M-PBS during 1h at RT. Blocked phages were incubated on blocked immunotubes 1h at RT. Tubes were washed 20 times with PBS-T and twice with PBS. Bound phages were eluted with 100 mmol/L triethylamine during 10 min at RT, and neutralized with 1 mol/L Tris, pH 7.5. Selected phages were used to infect exponentially growing TG1 cells, rescued and purified. The resulting phage preparations were used as starting material for a new selection round in the same conditions.
After two-three selection rounds, phages were individually rescued in 96-deep-well plates from phagemid–transformed TG1 colonies as described.Citation36 Phage-containing supernatants (diluted 1/5 in M-PBS) were tested on JES6–1A12-coated ELISA microplates as described above. Clones producing Abs values above 2-fold those obtained on plates coated with a non-related control mAb were considered to be positive. XL1-Blue E. coli cells were infected with the corresponding phage-containing supernatants and used to purify plasmid DNA to sequence the phagemid inserts. Peptide sequences were deduced from DNA sequences.
| Abbreviations: | ||
| aa | = | amino acid |
| ELISA | = | enzyme-linked immunosorbent assay |
| hIL-2 | = | human IL-2 |
| IL-2 | = | Interleukin-2 |
| IL-2R | = | Interleukin-2 receptor |
| IU | = | international units |
| mAbs | = | monoclonal antibodies |
| mIL-2 | = | mouse IL-2 |
| PBS | = | phosphate-buffered saline |
| RSA | = | relative solvent accessibility |
| RT | = | room temperature |
| scFv | = | single chain Fv |
| wt | = | wild-type |
Acknowledgments
We thank Kalet Leon for helpful discussions and Yaquelin Marichal for her excellent technical assistance.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Related Research Data
References
- Malek TR, Bayer AL. Tolerance, not immunity, crucially depends on IL-2. Nat Rev Immunol 2004; 4:665 - 74; http://dx.doi.org/10.1038/nri1435; PMID: 15343366
- Hoyer KK, Dooms H, Barron L, Abbas AK. Interleukin-2 in the development and control of inflammatory disease. Immunol Rev 2008; 226:19 - 28; http://dx.doi.org/10.1111/j.1600-065X.2008.00697.x; PMID: 19161413
- Liao W, Lin J-X, Leonard WJ. Interleukin-2 at the crossroads of effector responses, tolerance, and immunotherapy. Immunity 2013; 38:13 - 25; http://dx.doi.org/10.1016/j.immuni.2013.01.004; PMID: 23352221
- Cesana GC, DeRaffele G, Cohen S, Moroziewicz D, Mitcham J, Stoutenburg J, Cheung K, Hesdorffer C, Kim-Schulze S, Kaufman HL. Characterization of CD4+CD25+ regulatory T cells in patients treated with high-dose interleukin-2 for metastatic melanoma or renal cell carcinoma. J Clin Oncol 2006; 24:1169 - 77; http://dx.doi.org/10.1200/JCO.2005.03.6830; PMID: 16505437
- Boyman O, Kovar M, Rubinstein MP, Surh CD, Sprent J. Selective stimulation of T cell subsets with antibody-cytokine immune complexes. Science 2006; 311:1924 - 7; http://dx.doi.org/10.1126/science.1122927; PMID: 16484453
- Kamimura D, Sawa Y, Sato M, Agung E, Hirano T, Murakami M. IL-2 in vivo activities and antitumor efficacy enhanced by an anti-IL-2 mAb. J Immunol 2006; 177:306 - 14; PMID: 16785526
- Mostböck S, Lutsiak ME, Milenic DE, Baidoo K, Schlom J, Sabzevari H. IL-2/anti-IL-2 antibody complex enhances vaccine-mediated antigen-specific CD8+ T cell responses and increases the ratio of effector/memory CD8+ T cells to regulatory T cells. J Immunol 2008; 180:5118 - 29; PMID: 18354238
- Tomala J, Chmelova H, Mrkvan T, Rihova B, Kovar M. In vivo expansion of activated naive CD8+ T cells and NK cells driven by complexes of IL-2 and anti-IL-2 monoclonal antibody as novel approach of cancer immunotherapy. J Immunol 2009; 183:4904 - 12; http://dx.doi.org/10.4049/jimmunol.0900284; PMID: 19801515
- Kohyama K, Sugiura H, Kozawa E, Wasa J, Yamada K, Nishioka A, Kamei Y, Taguchi O. Antitumor activity of an interleukin-2 monoclonal antibody in a murine osteosarcoma transplantation model. Anticancer Res 2012; 32:779 - 82; PMID: 22399592
- Webster KE, Walters S, Kohler RE, Mrkvan T, Boyman O, Surh CD, Grey ST, Sprent J. In vivo expansion of T reg cells with IL-2-mAb complexes: induction of resistance to EAE and long-term acceptance of islet allografts without immunosuppression. J Exp Med 2009; 206:751 - 60; http://dx.doi.org/10.1084/jem.20082824; PMID: 19332874
- Liu R, Zhou Q, La Cava A, Campagnolo DI, Van Kaer L, Shi F-D. Expansion of regulatory T cells via IL-2/anti-IL-2 mAb complexes suppresses experimental myasthenia. Eur J Immunol 2010; 40:1577 - 89; http://dx.doi.org/10.1002/eji.200939792; PMID: 20352624
- Liu C-L, Ye P, Yen BC, Miao CH. In vivo expansion of regulatory T cells with IL-2/IL-2 mAb complexes prevents anti-factor VIII immune responses in hemophilia A mice treated with factor VIII plasmid-mediated gene therapy. Mol Ther 2011; 19:1511 - 20; http://dx.doi.org/10.1038/mt.2011.61; PMID: 21468007
- Lee S-Y, Cho M-L, Oh H-J, Ryu J-G, Park M-J, Jhun J-Y, Park M-K, Stone JC, Ju J-H, Hwang S-Y, et al. Interleukin-2/anti-interleukin-2 monoclonal antibody immune complex suppresses collagen-induced arthritis in mice by fortifying interleukin-2/STAT5 signalling pathways. Immunology 2012; 137:305 - 16; http://dx.doi.org/10.1111/imm.12008; PMID: 23167249
- Boyman O, Surh CD, Sprent J. Potential use of IL-2/anti-IL-2 antibody immune complexes for the treatment of cancer and autoimmune disease. Expert Opin Biol Ther 2006; 6:1323 - 31; http://dx.doi.org/10.1517/14712598.6.12.1323; PMID: 17223740
- Rojas G, Pupo A, Leon K, Avellanet J, Carmenate T, Sidhu S. Deciphering the molecular bases of the biological effects of antibodies against Interleukin-2: a versatile platform for fine epitope mapping. Immunobiology 2013; 218:105 - 13; http://dx.doi.org/10.1016/j.imbio.2012.02.009; PMID: 22459271
- Chasteen L, Ayriss J, Pavlik P, Bradbury AR. Eliminating helper phage from phage display. Nucleic Acids Res 2006; 34:e145; http://dx.doi.org/10.1093/nar/gkl772; PMID: 17088290
- Buchli PJ, Wu Z, Ciardelli TL. The functional display of interleukin-2 on filamentous phage. Arch Biochem Biophys 1997; 339:79 - 84; http://dx.doi.org/10.1006/abbi.1996.9853; PMID: 9056236
- Vispo NS, Callejo M, Ojalvo AG, Santos A, Chinea G, Gavilondo JV, Araña MJ. Displaying human interleukin-2 on the surface of bacteriophage. Immunotechnology 1997; 3:185 - 93; http://dx.doi.org/10.1016/S1380-2933(97)00012-2; PMID: 9358271
- Felici F, Castagnoli L, Musacchio A, Jappelli R, Cesareni G. Selection of antibody ligands from a large library of oligopeptides expressed on a multivalent exposition vector. J Mol Biol 1991; 222:301 - 10; http://dx.doi.org/10.1016/0022-2836(91)90213-P; PMID: 1720463
- Deroo S, Muller CP. Antigenic and immunogenic phage displayed mimotopes as substitute antigens: applications and limitations. Comb Chem High Throughput Screen 2001; 4:75 - 110; http://dx.doi.org/10.2174/1386207013331309; PMID: 11281827
- Menendez A, Scott JK. The nature of target-unrelated peptides recovered in the screening of phage-displayed random peptide libraries with antibodies. Anal Biochem 2005; 336:145 - 57; http://dx.doi.org/10.1016/j.ab.2004.09.048; PMID: 15620878
- Saphire EO, Montero M, Menendez A, van Houten NE, Irving MB, Pantophlet R, Zwick MB, Parren PWHI, Burton DR, Scott JK, et al. Structure of a high-affinity “mimotope” peptide bound to HIV-1-neutralizing antibody b12 explains its inability to elicit gp120 cross-reactive antibodies. J Mol Biol 2007; 369:696 - 709; http://dx.doi.org/10.1016/j.jmb.2007.01.060; PMID: 17445828
- Vispo NS, Araña MJ, Chinea G, Ojalvo AG, Cesareni G. Characterization of epitopes on human interleukin-2 using phage displayed-peptide libraries: insights into antibody-peptide interactions. Hybridoma 1999; 18:251 - 5; http://dx.doi.org/10.1089/027245799315907; PMID: 10475239
- Davies DR, Padlan EA, Sheriff S. Antibody-antigen complexes. Annu Rev Biochem 1990; 59:439 - 73; http://dx.doi.org/10.1146/annurev.bi.59.070190.002255; PMID: 2197980
- Braden BC, Poljak RJ. Structural features of the reactions between antibodies and protein antigens. FASEB J 1995; 9:9 - 16; PMID: 7821765
- Davies DR, Cohen GH. Interactions of protein antigens with antibodies. Proc Natl Acad Sci U S A 1996; 93:7 - 12; http://dx.doi.org/10.1073/pnas.93.1.7; PMID: 8552677
- Wang X, Rickert M, Garcia KC. Structure of the quaternary complex of interleukin-2 with its α, β, and gammac receptors. Science 2005; 310:1159 - 63; http://dx.doi.org/10.1126/science.1117893; PMID: 16293754
- Stauber DJ, Debler EW, Horton PA, Smith KA, Wilson IA. Crystal structure of the IL-2 signaling complex: paradigm for a heterotrimeric cytokine receptor. Proc Natl Acad Sci U S A 2006; 103:2788 - 93; http://dx.doi.org/10.1073/pnas.0511161103; PMID: 16477002
- Phelan JD, Orekov T, Finkelman FD. Cutting edge: mechanism of enhancement of in vivo cytokine effects by anti-cytokine monoclonal antibodies. J Immunol 2008; 180:44 - 8; PMID: 18097002
- Tomala J, Kovarova J, Kabesova M, Votavova P, Chmelova H, Dvorakova B, Rihova B, Kovar M. Chimera of IL-2 linked to light chain of anti-IL-2 mAb mimics IL-2/anti-IL-2 mAb complexes both structurally and functionally. ACS Chem Biol 2013; 8:871 - 6; http://dx.doi.org/10.1021/cb3007242; PMID: 23419043
- Carmenate T, Pacios A, Enamorado M, Moreno E, Garcia-Martínez K, Fuente D, León K. Human IL-2 mutein with higher antitumor efficacy than wild type IL-2. J Immunol 2013; 190:6230 - 8; http://dx.doi.org/10.4049/jimmunol.1201895; PMID: 23677467
- García-Martínez K, León K. Modeling the role of IL2 in the interplay between CD4+ helper and regulatory T cells: studying the impact of IL2 modulation therapies. Int Immunol 2012; 24:427 - 46; http://dx.doi.org/10.1093/intimm/dxr120; PMID: 22371423
- Malek TR, Castro I. Interleukin-2 receptor signaling: at the interface between tolerance and immunity. Immunity 2010; 33:153 - 65; http://dx.doi.org/10.1016/j.immuni.2010.08.004; PMID: 20732639
- Nakamura M, Asao H, Takeshita T, Sugamura K. Interleukin-2 receptor heterotrimer complex and intracellular signaling. Semin Immunol 1993; 5:309 - 17; http://dx.doi.org/10.1006/smim.1993.1037; PMID: 8260647
- Zurawski SM, Mosmann TR, Benedik M, Zurawski G. Alterations in the amino-terminal third of mouse interleukin 2: effects on biological activity and immunoreactivity. J Immunol 1986; 137:3354 - 60; PMID: 2430019
- Marks JD, Hoogenboom HR, Bonnert TP, McCafferty J, Griffiths AD, Winter G. By-passing immunization. Human antibodies from V-gene libraries displayed on phage. J Mol Biol 1991; 222:581 - 97; http://dx.doi.org/10.1016/0022-2836(91)90498-U; PMID: 1748994
- Kunkel TA. Rapid and efficient site-specific mutagenesis without phenotypic selection. Proc Natl Acad Sci U S A 1985; 82:488 - 92; http://dx.doi.org/10.1073/pnas.82.2.488; PMID: 3881765
- Fellouse FA, Sidhu SS. Making antibodies in bacteria. In Making and using antibodies. A practical handbook. G.C. Howard, and M.R.Kaser, CRC Press, Boca Raton, Florida, 2007, pp 157-177.