
Abstract
Various constructs of bispecific antibodies (bsAbs) to redirect effector T cells for the targeted killing of tumor cells have shown considerable promise in both preclinical and clinical studies. The single-chain variable fragment (scFv)-based formats, including bispecific T-cell engager (BiTE) and dual-affinity re-targeting (DART), which provide monovalent binding to both CD3 on T cells and to the target antigen on tumor cells, can exhibit rapid blood clearance and neurological toxicity due to their small size (~55 kDa). Herein, we describe the generation, by the modular DOCK-AND-LOCKTM (DNLTM) method, of novel T-cell redirecting bispecific antibodies, each comprising a monovalent anti-CD3 scFv covalently conjugated to a stabilized dimer of different anti-tumor Fabs. The potential advantages of this design include bivalent binding to tumor cells, a larger size (~130 kDa) to preclude renal clearance and penetration of the blood-brain barrier, and potent T-cell mediated cytotoxicity. These prototypes were purified to near homogeneity, and representative constructs were shown to provoke the formation of immunological synapses between T cells and their target tumor cells in vitro, resulting in T-cell activation and proliferation, as well as potent T-cell mediated anti-tumor activity. In addition, in vivo studies in NOD/SCID mice bearing Raji Burkitt lymphoma or Capan-1 pancreatic carcinoma indicated statistically significant inhibition of tumor growth compared with untreated controls.
Introduction
Cancer therapy with T cells has been an active area of research for more than two decades. One major approach, referred to as adoptive T-cell therapy, is being tested clinically in various strategies,Citation1 including therapy with tumor-specific cytotoxic T cells,Citation2 adoptive transfer therapy with tumor-infiltrating lymphocytes,Citation3 and therapy with T cells engineered to express chimeric receptors that target cell-surface molecules on cancer cells.Citation4,Citation5 Another method, which may be more convenient for clinical translation and more amenable to commercialization, is the exploration of bispecific antibodies (bsAbs) for redirecting a patient?s T cells to destroy the target cancer. Since the proof-of-concept studies in the mid-1980s using bsAbs produced by chemical crosslinkingCitation6,Citation7 or hybrid hybridomas,Citation8 the continued pursuit of bsAbs that are capable of triggering T-cell mediated lysis of target cancer cells has resulted in a plethora of recombinant constructs with differences in structure, valency, constituents, size, and tumor specificity.Citation9-Citation12 These are exemplified by bispecific T-cell engagers (BiTE)Citation13-Citation15 comprising two monovalent single-chain variable fragments (scFvs) linked in tandem; dual-affinity re-targeting (DART) moleculesCitation16 composed of a diabody format stabilized by a C-terminal disulfide bridge, as well as other variations.Citation17-Citation21 With the approval in 2009 of catumaxomab (anti-EpCAM x anti-CD3), a quadroma-produced, mouse-rat monoclonal antibody, in the European Union for the intraperitoneal (i.p.) treatment of malignant ascites in patients with EpCAM-positive carcinomas,Citation22 and the acquisition of Micromet for its BiTE technology by Amgen in 2012,Citation23 efforts to develop optimal bsAbs for cancer therapy by redirecting T cells have intensified. One notable goal is to prolong the serum half-life of BiTE antibodies without compromising their potency.Citation24
The elevation of proinflammatory cytokines in the sera of patients treated with antibodies that target immune cells is an adverse event known as cytokine release syndrome, and has been observed during infusion of, for example, rituximab (anti-CD20),Citation25 alemtuzumab (anti-CD52),Citation26 and OKT3 (anti-CD3).Citation27 Although clinically manageable, such target-mediated infusion reactions could be life-threatening, as revealed in the Phase 1 trial of TGN1412 (anti-CD28), during which all 6 healthy volunteers suffered a ?cytokine storm? from massive release of multiple cytokines and required weeks of hospitalization to recover.Citation28 It should be noted that the release of proinflammatory cytokines dominated by interleukin (IL-10), IL-6 and interferon (IFN)-? has been reported in about half of the 20 patients treated with continuous infusion of blinatomomab (anti-CD19 x anti-CD3 BiTE) at the start of the first, but not subsequent, treatment cycles.Citation29 Likewise, elevated levels of IL-6, IFN-?, and tumor necrosis factor-? (TNF-?) were observed in more than half of the 17 patients receiving three weekly doses of ertumaxomab (anti-HER2 x anti-CD3) intravenously (i.v.) upon the second and third infusions.Citation30 Systemic elevation of IL-6 and TNF-? in serum was also associated with the i.p. treatment of catumaxomab.Citation31 Thus, the development of agents or clinical regimens (for example, a stepwise dosing schemeCitation29) to minimize the adverse events, particularly cytokine release syndrome, is desirable for increasing the safety of such bsAbs.
The DOCK-AND-LOCKTM (DNLTM) method is a modular approach that yields multivalent, multifunctional complexes of antibodies with defined structures and retained activities.Citation32,Citation33 One example is the construction of a trivalent bsAb by linking a stabilized dimer of Fab specific for one antigen to a monomeric Fab with specificity for a different antigen.Citation34 Another example is the generation of a hexavalent bsAb by derivatizing a divalent IgG, at the carboxyl termini of either the heavy chain or the light chain, so that it contains two stabilized dimers of Fab with a different specificity from the IgG.Citation35 We have now applied the DNL method to generate a panel of novel T-cell redirecting trivalent bsAbs, each of which comprises an anti-CD3 scFv covalently conjugated to a stabilized dimer of a distinct antitumor Fab. Collectively designated as (X)-3s, where X and 3s denote the specific antitumor antibody and the anti-CD3 scFv, respectively, these constructs enable bivalent binding to tumor cells, while their larger size (~130 kDa) should preclude renal clearance, extend the circulating half-life, and allow less frequent dosing. The present study shows that (X)-3s mediate the formation of immunological synapses between T cells and cognate target cells, induce T-cell activation and proliferation in the presence of target cells, redirect potent T-cell mediated killing of target cells in vitro, and inhibit growth of human tumor xenografts in vivo. Studies showing improved pharmacokinetics are in progress.
Results
Biochemical analysis of (X)-3s
We applied the DNL method to generate a panel of (X)-3s () by combining Okt3-scFv-AD2 with a CH1-DDD2-Fab-X module derived from a humanized antibody specific for CD19, CD20, CD22, HLA-DR, Trop-2, CEACAM5, CEACAM6, IGF-1R, or MUC5AC. The cDNA sequence encoding the Okt3-scFv-AD2 polypeptide is provided in Figure S1. Separate production cell lines were developed in SpESFX-10 mouse myeloma cells for each of the DNL modules.Citation36 Following purification, the Okt3-scFv-AD2 module was combined with a CH1-DDD2-Fab-X module of interest to obtain (X)-3s, as illustrated in , and described in Supplementary Methods. Representative data showing the purity and composition of these bsAbs were provided in Figure S2 for SDS-PAGE of (19)-3s, Okt3-scFv-AD2, and CH1-DDD2-Fab-hA19, and in Figure S3 for the corresponding SE-HPLC. Similar SE-HPLC results are shown in Figure S4 for (22)-3s, (20)-3s, and (C2)-3s. A further validation of the molecular entity of (X)-3s is supported by the LC-MS analysis of (19)-3s (Fig. S5), which identified a single RP-HPLC peak having a deconvoluted mass of 137433.93 Da within 11 ppm of the calculated mass of 137432.37 Da from its deduced amino acid sequence, including the predicted N-terminal pyroglutamates on the Okt3scFv-AD2 and each of the two CH1-DDD2-hA19 Fd chains.
Table 1. A list of (X)-3s generated by the modular DNLTM method
Figure 1. Schematic showing the DNL formation of (X)-3s between a dimer of anti-X Fab and a monomer of anti-CD3 scFv via the site-specific conjugation of fused DDD2 and AD2 peptides.
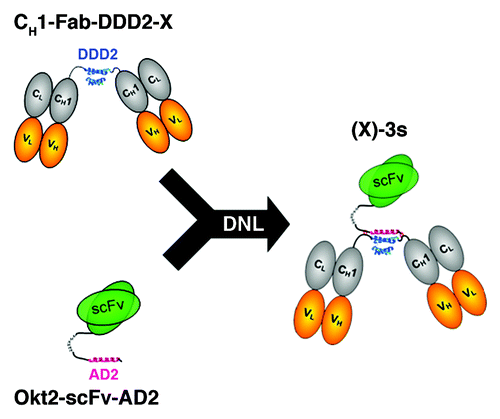
Association of T cells with target cells
To evaluate the ability of (X)-3s to facilitate the association of T cells to its target tumor cells, Daudi cells were incubated with a 10-fold excess of purified T cells (96.7%; Fig. S6A) for 30 min at room temperature (RT) in the presence and absence of (19)-3s, and, following staining with anti-CD3-FITC and anti-CD22-APC to identify T cells and Daudi cells, respectively, the cell mixtures were evaluated by flow cytometry. Similar results were obtained using anti-CD20-PE staining, but anti-CD22-APC was used preferably because of the presence of CD20+ T cells,Citation37 which complicate the interpretation of the results. As shown in , the number of CD3/CD22 double-positive events increased to 10.7% in the sample containing 1 µg/mL of (19)-3s (right panel), compare with 0.6% in the control sample in which (19)-3s was omitted (left panel). Gating of the CD3+ events show that 11.9% of the T cells were conjugated with Daudi cells with the addition of (19)-3s, compared with 0.6% in the control mixture (). Gating of CD22+ (or CD20+) events indicates that most (87.2%) of the Daudi cells were associated with T cells in the presence of (19)-3s, compared with 4.9% in the absence of (19)-3s (). Further studies using Jurkat cells and Daudi cells with sequentially increasing concentrations of (19)-3s from 0 to 0.333 µg/mL revealed that cell-cell association was already apparent with 0.0005 µg/mL of (19)-3s (), which progressed to formation of large cell aggregates at higher concentrations of (19)-3s (Fig. S7). The flow cytometric data were corroborated by the results of fluorescent microscopy, showing the evident formation of immunological synapses following treatment with 0.1 ?g/mL of (19)-3s for 30 min at RT (). No synapse formation was seen in the absence of (19)-3s ().
Figure 2. Association of target and effector cells mediated by (X)-3s. (A?C) Purified T cells (96.7%) were mixed with Daudi cells at a 10:1 ratio in the absence (left panels) or presence (right panels) of 1 ?g/mL (19)-3s. After 30 min at RT, the cell mixture was stained for 15 min on ice with anti-CD22-APC and anti-CD3-FITC to identify Daudi and T cells, respectively. (A) Anti-CD3 and anti-CD22 staining of the cell mixture. (B) T cells (CD3+) were gated and analyzed by anti-CD22 and forward scattering. (C) Daudi cells (CD22+) were gated and analyzed by anti-CD3 and forward scattering. For each dot plot, the indicated events in the upper right quadrant represent Daudi-T cell conjugates. (D) Daudi cells and Jurkat cells were mixed with various concentrations of (19)-3s and analyzed by flow cytometry following addition of anti-CD20-FITC and anti-CD3-PE. The percentage of CD3+/CD20+ double positive events was plotted vs. (19)-3s concentration.
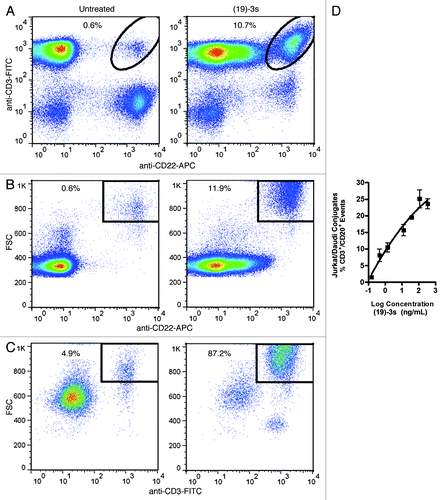
Figure 3. Images of Daudi cells (labeled with anti-CD20-FITC) and Jurkat cells (labeled with anti-CD3-PE) taken with fluorescent microscopy in three different fields, showing the formation of immunological synapse in the presence (A), but not in the absence (B), of (19)-3s. The prominent clustering of CFSE-labeled Capan-1 (green) cells with PKH26-labeled Jurkat cells (red) was observed in (C) with either (E1)-3s or (M1)-3s, but not (19)-3s, as Capan-1 cells express the antigens targeted by (E1)-3s (Trop-2) and (M1)-3s (MUC5AC), but not (19)-3s (CD19).
Because Capan-1 cells express both Trop-2, the target of (E1)-3s, and MUC5AC, the target of (M1)-3s, but not CD19, the target of (19)-3s, immunological synapses were observed upon adding either (E1)-3s or (M1)-3s, but not (19)-3s, to a mixture of CFSE-labeled Capan-1 cells and PKH26-labeled Jurkat cells ().
T-cell activation and proliferation induced by (19)-3s
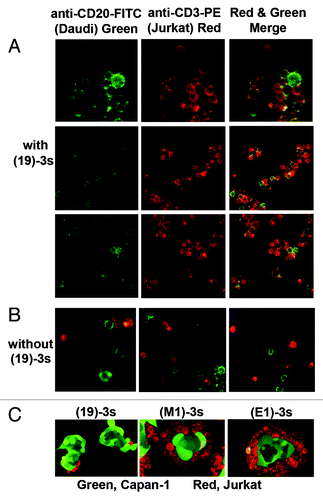
The ability of (19)-3s to activate T cells was evaluated either in peripheral blood mononuclear cells (PBMCs), or in purified T cells mixed with Daudi cells, by measuring the expression levels of CD69, an early marker of T-cell activation. An overnight treatment with 3 ng/mL of (19)-3s, but not (19)-DDD2 or (M1)-3s, induced a >50-fold increase in CD69 expression in T cells mixed with Daudi cells (). The requirement for the presence of Daudi cells to activate T cells by (19)-3s is shown in . Similar results () were observed in PBMCs incubated with (19)-3s, but not with (19)-DDD2 or (M1)-3s.
Figure 4. T-cell activation and proliferation as assessed by the neo-expression of CD69 (A?D) and the increase in CD7+ cells (E, F), respectively. (A, B) Daudi and purified T cells were treated with (19)-3s, 19-DDD2, (M1)-3s, or none; (C) purified T cells were treated with (19)-3s or none; (D) PBMCs were treated with (19)-3s, 19-DDD2, (M1)-3s, or none; (E) PBMCs were treated with (19)-3s, (14)-3s, IL-2/PHA, or none; (F) PBMCs or PBMCs depleted in B cells were treated with (19)-3s or IL-2/PHA and the number of CD7+ viable cells in each sample was determined after 1, 4 and 7 d. The untreated PBMCs served as the control.
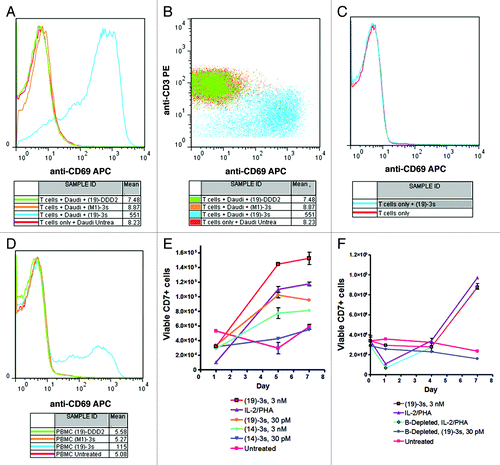
T-cell proliferation, which may follow T-cell activation, was evaluated in PBMCs after treatment with (19)-3s or (14)-3s for 5 and 7 d. shows that (19)-3s at 3 nM or 30 pM stimulated T-cell proliferation to a similar extent as that of IL-2/PHA, when measured on both days. In comparison, (14)-3s, which targets CEACMA5 not expressed on PBMCs, could induce some T-cell proliferation at 3 nM, but not at 30 pM. Importantly, whereas T-cell proliferation stimulated by IL-2/PHA was independent of B cells, it was aborted when PBMCs depleted with B cells were treated with (19)-3s (), suggesting that the linking of target cells to effector cells by (X)-3s is essential for the induction of T cells to proliferate.
Specific killing of tumor cells by a targeting (X)-3s via T-cell engagement
The cytotoxicity of (X)-3s was evaluated by its ability to engage T cells to lyse specific tumor cells to which it targets, with the observed potency expressed as IC50 (the concentration to achieve 50% specific lysis) and Lysismax (the maximal amount of specific lysis achieved at the highest test concentration). We used unstimulated T cells as effector cells for the hematologic tumor cell lines and established the optimal conditions of the assay to comprise a 10:1 ratio of effector-to-target cells (E/T) following incubation for 18 to 24 h. As shown in , (19)-3s potently induced the specific killing of four CD19-expressing cell lines, Ramos (IC50 = 0.17 pM, Lysismax = 79%), Nalm-6 (IC50 = 6 pM, Lysismax = 93%), Namalwa (IC50 = 63 pM, Lysismax = 60%), and Raji (IC50 = 3 nM, Lysismax = 41%). The sigmoidal dose-response curves (variable slope) were compared for statistical significance (P < 0.05) by F-test using Prism software. For each cell line, both the IC50 and Lysismax were significantly (P < 0.0001) different from the control treatments with (14)-3s. Results from additional studies () also demonstrated potent and specific T cell-mediated lysis by (22)-3s in Daudi (IC50 = 5 pM, Lysismax = 60%) and Namalwa cells (IC50 > 3 nM; Lysismax = 42%); by (C2)-3s in Jeko-1 (IC50 = 20 pM, Lysismax = 88%) and Ramos (IC50 = 2.3 pM, Lysismax = 79%); and by (20)-3s in Daudi (IC50 = < 0.3 pM, Lysismax = 90%), Jeko-1 (IC50 = 1 pM, Lysismax = 90%), Ramos (IC50 = 0.4 pM, Lysismax = 88%), and Namalwa (IC50 = 30 pM, Lysismax = 53%) cells. With Ramos, Jeko-1 and Daudi, (20)-3s was significantly (P < 0.0001 for EC50) more potent than all other treatments.
Figure 5. In vitro cytotoxicity of (X)-3s as determined from the dose-response curves: (A) comparison of (19)-3s and (14)-3s in Ramos, Nalm-6, Namalwa, and Raji cells; (B) comparison of (19)-3s, (20)-3s, and (22)-3s in Namalwa and Daudi cells, and (19)-3s, (20)-3s and (C2)-3s in Jeko-1 cells; (C) comparison of (14)-3s and (19)-3s in LS 174T cells, (E1)-3s and (19)-3s in Capan-1 cells, and (E1)-3s, (15)-3s and (19)-3s in NCI-N87 cells. For the hematologic tumor cell lines (Ramos, Nalm-6, Namalwa, Raji, Daudi, and Jeko-1), the indicated target cells (5 × 106) were labeled with PKH67, washed, combined with unstimulated, isolated T cells (5 × 107) as effector cells, and dispensed into 48-well plates containing serial dilutions of (19)-3s or (14)-3s such that each well contained 5 × 105 effector cells and 5 × 104 target cells at an E/T ratio of 10 to 1. Plates were incubated for 18?24 h in a 37 °C incubator containing 5% CO2. Following incubation, cells were processed and analyzed as described in the Materials and Methods. For the solid tumor cell lines (LS 174T, Capan-1, and NCI-N87), effector cells (as specified in the Materials and Methods) and PKH67-labeled target cells were combined at an E/T ratio of 3 to 1 (1.5 × 105 effector cells and 5 × 104 target cells) and dispensed onto 48-well plates containing serial dilutions of (E1)-3s, (14)-3s, or (19)-3s. Plates were incubated for 42?48 h in a 37 °C incubator containing 5% CO2. Following incubation, cells were processed and analyzed as described in the Materials and Methods.
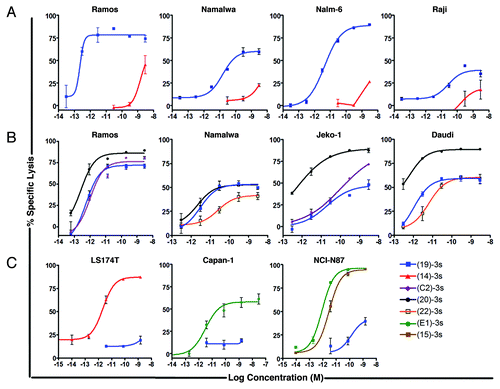
For the solid tumor cell lines, optimal assay conditions were determined to be at an E/T ratio of 3 to 1 using stimulated T cells as effector cells, following an incubation for 42 to 48 h. shows potent and specific T-cell mediated lysis by (14)-3s in the CEACAM5-expressing LS 174T colonic cancer cells (IC50 = 2 pM, Lysismax = 90%) and by (E1)-3s in Trop-2-expressing Capan-1 pancreatic cancer cells (IC50 = 29 pM, Lysismax = 60%), and by both (E1)-3s (IC50 = 0.85 pM, Lysismax > 90%) and (15)-3s (IC50 = 3 pM, Lysismax > 90%) in NCI-N87 human gastric cancer cells, which express high levels of both Trop-2 and CEACAM6. In these experiments, (19)-3s, the non-targeting control induced < 20% of lysis in Capan-1 and LS 174T, and ~40% of lysis in NCI-N87 cells, at 1 nM. For each solid tumor cell line, both the IC50 and Lysismax achieved with (E1)-3s or (14)-3s were significantly (P < 0.0001) different from the control treatment (19)-3s.
Anti-tumor activity of (19)-3s and (E1)-3s demonstrated in vivo
To investigate the in vivo activity of (19)-3s, we used a Burkitt lymphoma model in which the outgrowth of tumors was monitored in NOD/SCD mice injected subcutaneously (s.c.) with a mixture of human PBMCs (5 × 106) and Raji cells (1 × 106) at an E/T ratio of 5 to 1. A total of 260 ?g of (19)-3s was administered to each treated animal over two weeks in one of three different schedules: 130 ?g weekly, 65 ?g twice weekly, and 43 ?g three times per week. Animals that received only PBMCs and Raji cells served as the untreated controls. Measurable tumors became evident in most of the mice by day 14 (). Tumors progressed in all the untreated mice with no long-term survivors and a median survival time (MST) of 50.5 d (). In contrast, more than half of the mice in each of the three treatment groups were alive and tumor-free when the experiment ended on day 77, resulting in a significant survival benefit when compared with the untreated control mice (P < 0.0014). This study also indicated that weekly dosing of (19)-3s proved to be as beneficial as twice- or thrice-weekly dosing, since there was no significant difference between the three treatment groups.
Figure 6. In vivo activity of (19)-3s and (E1)-3s in female NOD/SCID mice bearing Raji and Capan-1 xenografts, respectively. Female NOD/SCID mice were injected s.c. with a mixture of Raji cells (1 × 106) and human PBMCs (5 × 106) combined with an equal volume of matrigel, or injected s.c. with a mixture of Capan-1 cells (5 × 106) and T cells (2.5 × 106) combined with an equal volume of matrigel. Therapy began 1 h later with i.v. injections of the indicated (X)-3s at the dosage and schedule as described in the Results. (A) Growth of Raji xenografts in individual mice of each cohort; (B) survival curves obtained for (19)-3s in the Raji model; (C) survival curves obtained for (E1)-3s in the Capan-1 model.
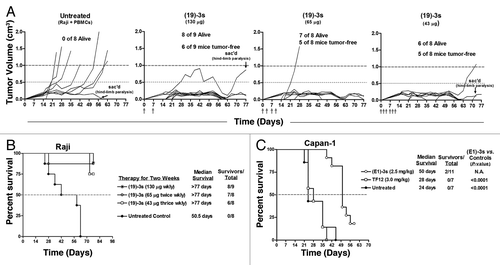
The potential of (E1)-3s to redirect T cells for solid cancer therapy was tested in three groups of NOD/SCID mice, with each mouse receiving a mixture of human T cells (2.5 × 106) and Capan-1 cells (5 × 106) at an E/T ratio of 1 to 2. One group of animals was treated daily with a 50 µg dose of (E1)-3s for 5 d. Another cohort was given 5 daily doses of an equimolar amount (60 µg) of TF12 (33), which is a trivalent, anti-Trop-2/anti-HSG (histamine-succinyl-glycine) bsAb of 160 kDa generated by linking CH1-DDD2-Fab-hRS7 to CH1-Fab-AD2 h679 (instead of Okt3-scFv-AD2), thus capable of targeting Trop-2+ cells but incapable of redirecting CD3+ T cells. The third group was left untreated. Measurable tumors were visible in the untreated group by day 7. A 2-fold increase in the MST was observed in mice treated with (E1)-3s compared with the untreated mice (50 d vs. Twenty-four days; P < 0.0001) and the group treated with the tri-Fab bsAb, TF12 (MST = 28 d; P < 0.0001). This experiment demonstrates not only the in vivo activity of (E1)-3s, but also the need to bind both the target tumor and the T cells to achieve effective therapy.
Discussion
Since its inception in 2005, the DNL method, by combining DDD2- and AD2 modules derived from assorted classes of molecules that encompass IgG,Citation38-Citation41 Fab,Citation34,Citation42,Citation43 cytokines,Citation44-Citation46 polyethylene glycols,Citation47 and enfuvirtide,Citation48 has been used to produce more than 100 different complexes with potential applications for targeted therapies of malignant, autoimmune, and infectious diseases. The generation of a functional Okt3-scFv-AD2 and the demonstration of the in vitro and in vivo activities of various (X)-3s to kill target tumor cells via T cells, as described in this study, expands the applications of the DNL repertoire to now include scFv as a viable building block for future exploration.
Whereas the impetus for continuing the surge of T-cell mediated, bsAb-based drug development can be attributed to the success of blinatumomab in treating patients with relapsed non-Hodgkin lymphoma and relapsed/refractory B-cell acute lymphoblastic leukemia,Citation49,Citation50 issues with blinatumomab, such as the need to extend its short half-life, which requires continuous i.v. infusion using a portable minipump, and to reduce the incidence of leukemic patients experiencing central nervous system (CNS) events, are well-recognized. Thus, although the concept of redirecting T cells for selective lysis of tumor cells is not new, and has been substantiated by existing anti-tumor/anti-CD3 bsAbs of various formats, the construction of (X)-3s to comprise a dimer of anti-tumor Fab site-specifically linked to a monomeric anti-CD3 scFv represents our first endeavor to address the two major problems associated with blinatumomab; namely, the requirement for continuous infusion and the notable adverse effect of CNS toxicity, which may occur with other products of similar size and valency, such as DARTs. We believe the larger size of (X)-3s (130 kDa), with its inherent features of a bsAb that efficiently mediates target-specific lysis of tumor cells by redirecting T cells, may hold promise for allowing bolus injections and prevention of blood-brain-barrier penetration, which now need to be confirmed experimentally.
A more thorough characterization of each (X)-3s generated to date, including their pharmacokinetic properties, is now being conducted. We contend, however, that the preclinical data presented here for (X)-3s, which include all the essential aspects of BiTE, DART or other related constructs, such as the induction of T-cell activation and proliferation only in the presence of target cells, the absence of immunological synapse formed between T cells and target cells without the addition of a cognate (X)-3s, the potent in vitro cytotoxicity in the picomolar ranges exhibited by (X)-3s against targeted hematological or epithelial cancer cell lines using unstimulated T cells, and the statistically significant inhibition of human tumor growth in NOD/SCID mice bearing Raji Burkitt lymphoma or Capan-1 pancreatic carcinoma compared with untreated controls, firmly establish (X)-3s among the most potent bsAbs for cancer therapy via redirecting, activation and proliferation of T cells. The current results also indicate that (20)-3s displays a consistently higher cytotoxicity than (19)-3s, (22)-3s or (C2)-3s when evaluated in the same tumor cells, which is in agreement with the notion that a T-cell-redirecting bsAb targeting an antigenic epitope in closer proximity to the cell surface would be more potent than those that bind to a distant epitope.Citation51 Therefore, an anti-CD20 bsAb construct for retargeting T cells for immunotherapy of CD20+ hematological tumors should be of interest to evaluate clinically.
Materials and Methods
Cell lines and reagents
Human cell lines of Burkitt lymphoma (Raji, Ramos, Daudi, Namalwa), acute T cell leukemia (Jurkat), mantle cell lymphoma (Jeko-1), colonic cancer (LS 174T), pancreatic cancer (Capan-1), and gastric cancer (NCI-N87) were purchased from the American Type Cell Culture Collection (ATCC). The B cell precursor leukemia cell line, Nalm-6, was purchased from Deutsche Sammlung von Mikroorganismen und Zellinien (DSMZ). All cell lines, except Capan-1, were maintained in RPMI-1640 containing 10% FBS, 1% L-glutamine, 1% penicillin-streptomycin, and 1% MEM non-essential amino acids. Capan-1 cells were maintained in 20% FBS. Each cell line was cultured according to the method specified by its supplier, and upon receipt, 5 to 10 vials containing cells from an early passage (typically the fifth passage) were prepared and kept as a frozen stock, from which only cells with fewer than 50 passages (less than 6 mo) were used for the experiments. Cells were tested for the presence of mycoplasma before being frozen. All cell culture media and supplements were purchased from Life Technologies. Recombinant human IL-2 and phytohemagglutinin (PHA) were acquired from Gibco and Calbiochem, respectively.
Cell analysis by flow cytometry
All flow cytometric studies were performed on a FACSCalibur flow cytometer (BD Bioscience) with the data analyzed by FlowJo (Tree Star). Fluorochrome-conjugated antibodies used for flow cytometry included anti-CD20-FITC (fluorescein isothiocyanate), anti-CD3-PE (phycoerythrin), anti-CD3-FITC, anti-CD7-APC (allophycocyanine), anti-CD22-APC, anti-CD69-APC, and anti-CD19-Alexa Fluor 488, all from Biolegend; and anti-CD14-PE from Miltenyi Biotec. Other cell-staining agents purchased were PKH26 Red Fluorescent Cell Linker Kit and PKH67 Green Fluorescent Cell Linker Kit from Sigma, and 7-AAD (amino-actinomycin D) and CFSE (carboxyfluorescein succinimidyl ester) from Life Technologies.
Isolation of PBMCs and T cells
Human PBMCs were prepared from buffy coats (Blood Center of New Jersey) using UNI-SEPMAXI tubes (Novamed) under a protocol approved by the New England Institutional Review Board. CD3-positive T cells were isolated from PBMCs by negative selection using the Pan T Cell Isolation Kit (Miltenyi Biotec) according to the manufacturer?s protocol. The efficiency of T cell isolation was assessed by flow cytometry after staining the enriched T cells with anti-CD3-PE or anti-CD3-FITC. In some cases, further staining with anti-CD19-Alexa Fluor 488 and anti-CD14-PE was performed to identify contaminating cells.
T-cell activation
Isolated T cells were plated in 6-well tissue culture plates at a final density of 2.25 × 106 cells/well. Daudi cells were added to designated wells at a final density of 1.5 × 106 cells/well; other wells were left to contain only T cells. Alternatively, PBMCs were added to 6-well tissue culture plates at a final cell density of 6 × 106 cells/well. The volume of each well was brought up to 3 mL. To the appropriate wells, (19)-3s, (M1)-3s, or CH1-DDD2-Fab-hA19 (abbreviated as (19)-DDD2 hereafter), each at 3 ng/mL, was added (designations explained in ). After incubation overnight at 37 °C, 1 mL of each sample was removed; the cells were pelleted and labeled on ice with anti-CD69-APC and anti-CD3-PE for 20 min. Cells were washed twice with 1% BSA in PBS and analyzed by flow cytometry.
T-cell proliferation
PBMCs were seeded in T25 flasks at a concentration of 1 × 106 cells/mL and the specified reagents were added. For B-cell-depleted flasks, B cells were removed by negative selection using a B-cell isolation kit (Miltenyi Biotec) according to manufacturer?s protocol. On selected days, duplicate samples of 100 ?L were removed from each flask, labeled with anti-CD7-APC for 20 min on ice, washed once, and resuspended in 300 ?L of 1% BSA/PBS containing 7-AAD. The entire volume of each sample was then evaluated by flow cytometry with the data analyzed by FlowJo for live CD7+ cells after excluding dead (7-AAD+) cells and debris (based on forward vs. side scatter).
Cell-cell association by flow cytometry
Daudi cells were mixed with 10 times the number of purified T cells or Jurkat cells, in the presence or absence of (19)-3s. Following a 30-min incubation at RT, the cell mixture was washed once and stained with a cocktail containing anti-CD22-APC and anti-CD3-FITC, or anti-CD20-FITC and anti-CD3-PE. Binding of (19)-3s does not interfere with detection by anti-CD3 (Fig. S6B).
Formation of immunological synapses
Jurkat and Capan-1 cells were stained with PKH26 Red Fluorescent Cell Linker Kit and 5 ?M CFSE, respectively. Labeled Capan-1 cells were added to 8-well chamber slides (ThermoWaltham) and allowed to attach overnight. The following day, media was removed, and PKH26-labeled Jurkat cells were added in media containing 0.1 ?g/mL of (E1)-3s, (M1)-3s or (19)-3s. Following 1 h incubation at 37 °C, slides were washed with PBS to remove any unbound cells and examined by fluorescence microscopy.
In a separate experiment, Jurkat and Daudi cells were labeled with anti-CD3-PE and anti-CD20-FITC, respectively. The labeled cells were mixed at a 2.5 to 1 ratio of Jurkat to Daudi, incubated with (19)-3s at 0.1 ?g/mL for 30 min at RT, and examined by fluorescence microscopy.
In vitro cytotoxicity of hematologic tumor cell lines
Target cells were labeled with PKH67 Green Fluorescent Cell Linker Kit according to Manufacturer?s protocol. Briefly, 5 × 106 target cells were resuspended in 250 ?L of diluent C. In a second tube, 1 ?L of PKH67 dye was added to 250 ?L of diluent C. The cell suspension was then added to the dye solution, mixed thoroughly and incubated at RT for 2 min. The reaction was quenched by adding an equal volume of FBS. The labeled cells were then washed 3 times with complete RPMI tissue culture medium. Unstimulated, isolated T cells were used as effector cells. Effector cells and PKH67-labeled target cells were combined at a 10:1 ratio and dispensed into 48-well plates containing serial dilutions of (X)-3s. Each well contained 5 × 104 target cells and 5 × 105 effector cells. Plates were incubated for 18?24 h in a 37 °C incubator containing 5% CO2. Following incubation, all cells were transferred from 48-well plates into flow cytometer tubes and resuspended in 1% BSA/PBS containing 30,000 CountBright Absolute Counting Beads (Life Technologies) and 1 ?g/mL of 7-AAD. Cells were evaluated by flow cytometry and the data analyzed by FlowJo for total live target cells. For each sample, 8,000 CountBright beads were counted as a normalized reference. The specific lysis (%) was calculated for each sample using the formula: [1-(A1/A2)] × 100, where A1 and A2 represent the number of viable target cells in the test sample and the number of viable target cells in the untreated sample, respectively.
In vitro cytotoxicity of solid tumor cell lines
Target cells were labeled with PKH67 Green Fluorescent Cell Linker Kit. For Capan-1 assays, CD8+ enriched T cells were used, following purification from a CD8+ enrichment column (R&D Systems). For LS 174T cells (human colonic cancer), stimulated T cells were used after incubation of PBMC for 5 d in media containing IL-2 (25 U/mL) and Okt3 (50 ng/mL), followed by 2-d incubation in media containing IL-2 (25 U/mL). Effector cells and PKH67-labeled target cells were combined at a 3:1 ratio (5 × 104 target cells and 1.5 × 105 effector cells/well) and dispensed onto 48-well plates containing serial dilutions of (E1)-3s, (14)-3s, or (19)-3s. Plates were incubated for 42?48 h in a 37 °C incubator containing 5% CO2. Following incubation, suspension cells were combined with trypsinized cells from all wells and transferred into flow cytometer tubes. Cells were washed one time, resuspended in 1% BSA/PBS containing 30,000 CountBright Absolute Counting Beads and 1 ?g/mL of 7-AAD.ells, and analyzed by flow cytometry, as describe above for hematologic tumor cell lines.
In vivo studies
Female NOD/SCID mice, 4 to 8 wk of age, were purchased from Charles River (Wilmington, MA). All animal studies were approved by the University of Medicine and Dentistry of New Jersey Institutional Animal Care and Use Committee. Mice were injected s.c. with a mixture of Raji cells (1 × 106) and human PBMCs (5 × 106) combined with an equal volume of matrigel. Other studies were evaluated in animals injected s.c. with a mixture of Capan-1 cells (5 × 106) and T cells (2.5 × 106) combined with an equal volume of matrigel. Therapy began 1 h later with i.v. injections given daily for 5 d. Treatment regimens, dosages, and number of animals in each experiment, as adapted from the protocols of others,Citation52,Citation53 are described in the Results section. Animals were monitored daily for signs of tumor outgrowth. Once tumors appeared, they were measured twice weekly. Tumor volume (TV) was determined by measurements in two dimensions using calipers, with volumes defined as: L × w2/2, where L is the longest dimension of the tumor and w the shortest.
Efficacy was determined by a log-rank test with Prism GraphPad software (v5) on Kaplan-Meier curves using survival surrogate endpoints as time for tumor progression (TTP) to 1.0 cm3. Significance was considered at P < 0.05.
| Abbreviations: | ||
| bsAb | = | bispecific antibody |
| Fab | = | antigen-binding fragment |
| scFv | = | single-chain variable domain fragment |
| BiTE | = | bispecific T-cell engager |
| DART | = | Dual-Affinity Re-Targeting |
| DNL | = | DOCK-AND-LOCK |
| i.p. | = | intraperitoneal |
| i.v. | = | intravenous |
| (X)-3s | = | bispecific trivalent antibody comprising a stabilized (Fab)2 of X and an Okt3-derived scFv |
| Lysismax | = | maximal cell killing |
| IC50 | = | concentration resulting in 50% cell killing |
Additional material
Download Zip (632.5 KB)Disclosure of Potential Conflicts of Interest
All authors are employees of Immunomedics, Inc, or IBC Pharmaceuticals, Inc, or both, and have stocks or stock options of Immunomedics, Inc. IBC Pharmaceuticals, Inc is a fully own subsidiary of Immunomedics, Inc.
Acknowledgments
The authors thank John Kopinski, Maria Zalath, and Gabi Terracina for excellent technical assistance.
References
- June CH. Adoptive T cell therapy for cancer in the clinic. J Clin Invest 2007; 117:1466 - 76; http://dx.doi.org/10.1172/JCI32446; PMID: 17549249
- Yee C, Thompson JA, Byrd D, Riddell SR, Roche P, Celis E, Greenberg PD. Adoptive T cell therapy using antigen-specific CD8+ T cell clones for the treatment of patients with metastatic melanoma: in vivo persistence, migration, and antitumor effect of transferred T cells. Proc Natl Acad Sci U S A 2002; 99:16168 - 73; http://dx.doi.org/10.1073/pnas.242600099; PMID: 12427970
- Rosenberg SA. Cell transfer immunotherapy for metastatic solid cancer--what clinicians need to know. Nat Rev Clin Oncol 2011; 8:577 - 85; http://dx.doi.org/10.1038/nrclinonc.2011.116; PMID: 21808266
- Gilham DE, Debets R, Pule M, Hawkins RE, Abken H. CAR-T cells and solid tumors: tuning T cells to challenge an inveterate foe. Trends Mol Med 2012; 18:377 - 84; http://dx.doi.org/10.1016/j.molmed.2012.04.009; PMID: 22613370
- Liu L, Sun M, Wang Z. Adoptive T-cell therapy of B-cell malignancies: conventional and physiological chimeric antigen receptors. Cancer Lett 2012; 316:1 - 5; http://dx.doi.org/10.1016/j.canlet.2011.10.027; PMID: 22099879
- Perez P, Hoffman RW, Shaw S, Bluestone JA, Segal DM. Specific targeting of cytotoxic T cells by anti-T3 linked to anti-target cell antibody. Nature 1985; 316:354 - 6; http://dx.doi.org/10.1038/316354a0; PMID: 3160953
- Staerz UD, Kanagawa O, Bevan MJ. Hybrid antibodies can target sites for attack by T cells. Nature 1985; 314:628 - 31; http://dx.doi.org/10.1038/314628a0; PMID: 2859527
- Staerz UD, Bevan MJ. Hybrid hybridoma producing a bispecific monoclonal antibody that can focus effector T-cell activity. Proc Natl Acad Sci U S A 1986; 83:1453 - 7; http://dx.doi.org/10.1073/pnas.83.5.1453; PMID: 2869486
- Choi BD, Cai M, Bigner DD, Mehta AI, Kuan CT, Sampson JH. Bispecific antibodies engage T cells for antitumor immunotherapy. Expert Opin Biol Ther 2011; 11:843 - 53; http://dx.doi.org/10.1517/14712598.2011.572874; PMID: 21449821
- Koristka S, Cartellieri M, Theil A, Feldmann A, Arndt C, Stamova S, Michalk I, Töpfer K, Temme A, Kretschmer K, et al. Retargeting of human regulatory T cells by single-chain bispecific antibodies. J Immunol 2012; 188:1551 - 8; http://dx.doi.org/10.4049/jimmunol.1101760; PMID: 22184723
- Lum LG, Thakur A. Targeting T cells with bispecific antibodies for cancer therapy. BioDrugs 2011; 25:365 - 79; http://dx.doi.org/10.2165/11595950-000000000-00000; PMID: 22050339
- Segal DM, Weiner GJ, Weiner LM. Bispecific antibodies in cancer therapy. Curr Opin Immunol 1999; 11:558 - 62; http://dx.doi.org/10.1016/S0952-7915(99)00015-1; PMID: 10508714
- Baeuerle PA, Reinhardt C. Bispecific T-cell engaging antibodies for cancer therapy. Cancer Res 2009; 69:4941 - 4; http://dx.doi.org/10.1158/0008-5472.CAN-09-0547; PMID: 19509221
- Nagorsen D, Baeuerle PA. Immunomodulatory therapy of cancer with T cell-engaging BiTE antibody blinatumomab. Exp Cell Res 2011; 317:1255 - 60; http://dx.doi.org/10.1016/j.yexcr.2011.03.010; PMID: 21419116
- Wolf E, Hofmeister R, Kufer P, Schlereth B, Baeuerle PA. BiTEs: bispecific antibody constructs with unique anti-tumor activity. Drug Discov Today 2005; 10:1237 - 44; http://dx.doi.org/10.1016/S1359-6446(05)03554-3; PMID: 16213416
- Moore PA, Zhang W, Rainey GJ, Burke S, Li H, Huang L, Gorlatov S, Veri MC, Aggarwal S, Yang Y, et al. Application of dual affinity retargeting molecules to achieve optimal redirected T-cell killing of B-cell lymphoma. Blood 2011; 117:4542 - 51; http://dx.doi.org/10.1182/blood-2010-09-306449; PMID: 21300981
- Asano R, Ikoma K, Sone Y, Kawaguchi H, Taki S, Hayashi H, Nakanishi T, Umetsu M, Katayose Y, Unno M, et al. Highly enhanced cytotoxicity of a dimeric bispecific diabody, the hEx3 tetrabody. J Biol Chem 2010; 285:20844 - 9; http://dx.doi.org/10.1074/jbc.M110.120444; PMID: 20444691
- Bühler P, Wolf P, Gierschner D, Schaber I, Katzenwadel A, Schultze-Seemann W, Wetterauer U, Tacke M, Swamy M, Schamel WW, et al. A bispecific diabody directed against prostate-specific membrane antigen and CD3 induces T-cell mediated lysis of prostate cancer cells. Cancer Immunol Immunother 2008; 57:43 - 52; http://dx.doi.org/10.1007/s00262-007-0348-6; PMID: 17579857
- Feldmann A, Stamova S, Bippes CC, Bartsch H, Wehner R, Schmitz M, Temme A, Cartellieri M, Bachmann M. Retargeting of T cells to prostate stem cell antigen expressing tumor cells: comparison of different antibody formats. Prostate 2011; 71:998 - 1011; http://dx.doi.org/10.1002/pros.21315; PMID: 21541976
- Kipriyanov SM, Moldenhauer G, Strauss G, Little M. Bispecific CD3 x CD19 diabody for T cell-mediated lysis of malignant human B cells. Int J Cancer 1998; 77:763 - 72; http://dx.doi.org/10.1002/(SICI)1097-0215(19980831)77:5<763::AID-IJC16>3.0.CO;2-2; PMID: 9688311
- Stork R, Müller D, Kontermann RE. A novel tri-functional antibody fusion protein with improved pharmacokinetic properties generated by fusing a bispecific single-chain diabody with an albumin-binding domain from streptococcal protein G. Protein Eng Des Sel 2007; 20:569 - 76; http://dx.doi.org/10.1093/protein/gzm061; PMID: 17982179
- Seimetz D, Lindhofer H, Bokemeyer C. Development and approval of the trifunctional antibody catumaxomab (anti-EpCAM x anti-CD3) as a targeted cancer immunotherapy. Cancer Treat Rev 2010; 36:458 - 67; http://dx.doi.org/10.1016/j.ctrv.2010.03.001; PMID: 20347527
- Sheridan C. Amgen swallows Micromet to BiTE into ALL market. Nat Biotechnol 2012; 30:300 - 1; http://dx.doi.org/10.1038/nbt0412-300c; PMID: 22491268
- Frankel SR, Baeuerle PA. Targeting T cells to tumor cells using bispecific antibodies. Curr Opin Chem Biol 2013; 17:385 - 92; http://dx.doi.org/10.1016/j.cbpa.2013.03.029; PMID: 23623807
- Winkler U, Jensen M, Manzke O, Schulz H, Diehl V, Engert A. Cytokine-release syndrome in patients with B-cell chronic lymphocytic leukemia and high lymphocyte counts after treatment with an anti-CD20 monoclonal antibody (rituximab, IDEC-C2B8). Blood 1999; 94:2217 - 24; PMID: 10498591
- Wing MG, Moreau T, Greenwood J, Smith RM, Hale G, Isaacs J, Waldmann H, Lachmann PJ, Compston A. Mechanism of first-dose cytokine-release syndrome by CAMPATH 1-H: involvement of CD16 (FcgammaRIII) and CD11a/CD18 (LFA-1) on NK cells. J Clin Invest 1996; 98:2819 - 26; http://dx.doi.org/10.1172/JCI119110; PMID: 8981930
- Chatenoud L, Ferran C, Reuter A, Legendre C, Gevaert Y, Kreis H, Franchimont P, Bach JF. Systemic reaction to the anti-T-cell monoclonal antibody OKT3 in relation to serum levels of tumor necrosis factor and interferon-gamma [corrected]. N Engl J Med 1989; 320:1420 - 1; http://dx.doi.org/10.1056/NEJM198905253202117; PMID: 2785642
- Suntharalingam G, Perry MR, Ward S, Brett SJ, Castello-Cortes A, Brunner MD, Panoskaltsis N. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N Engl J Med 2006; 355:1018 - 28; http://dx.doi.org/10.1056/NEJMoa063842; PMID: 16908486
- Klinger M, Brandl C, Zugmaier G, Hijazi Y, Bargou RC, Topp MS, Gökbuget N, Neumann S, Goebeler M, Viardot A, et al. Immunopharmacologic response of patients with B-lineage acute lymphoblastic leukemia to continuous infusion of T cell-engaging CD19/CD3-bispecific BiTE antibody blinatumomab. Blood 2012; 119:6226 - 33; http://dx.doi.org/10.1182/blood-2012-01-400515; PMID: 22592608
- Kiewe P, Hasmüller S, Kahlert S, Heinrigs M, Rack B, Marmé A, Korfel A, Jäger M, Lindhofer H, Sommer H, et al. Phase I trial of the trifunctional anti-HER2 x anti-CD3 antibody ertumaxomab in metastatic breast cancer. Clin Cancer Res 2006; 12:3085 - 91; http://dx.doi.org/10.1158/1078-0432.CCR-05-2436; PMID: 16707606
- Heiss MM, Ströhlein MA, Jäger M, Kimmig R, Burges A, Schoberth A, Jauch KW, Schildberg FW, Lindhofer H. Immunotherapy of malignant ascites with trifunctional antibodies. Int J Cancer 2005; 117:435 - 43; http://dx.doi.org/10.1002/ijc.21165; PMID: 15906359
- Chang CH, Rossi EA, Goldenberg DM. The dock and lock method: a novel platform technology for building multivalent, multifunctional structures of defined composition with retained bioactivity. Clin Cancer Res 2007; 13:5586s - 91s; http://dx.doi.org/10.1158/1078-0432.CCR-07-1217; PMID: 17875793
- Rossi EA, Goldenberg DM, Chang CH. The dock-and-lock method combines recombinant engineering with site-specific covalent conjugation to generate multifunctional structures. Bioconjug Chem 2012; 23:309 - 23; http://dx.doi.org/10.1021/bc2004999; PMID: 22168393
- Rossi EA, Goldenberg DM, Cardillo TM, McBride WJ, Sharkey RM, Chang CH. Stably tethered multifunctional structures of defined composition made by the dock and lock method for use in cancer targeting. Proc Natl Acad Sci U S A 2006; 103:6841 - 6; http://dx.doi.org/10.1073/pnas.0600982103; PMID: 16636283
- Rossi EA, Chang CH, Cardillo TM, Goldenberg DM. Optimization of multivalent bispecific antibodies and immunocytokines with improved in vivo properties. Bioconjug Chem 2013; 24:63 - 71; http://dx.doi.org/10.1021/bc300488f; PMID: 23116517
- Rossi DL, Rossi EA, Goldenberg DM, Chang CH. A new mammalian host cell with enhanced survival enables completely serum-free development of high-level protein production cell lines. Biotechnol Prog 2011; 27:766 - 75; http://dx.doi.org/10.1002/btpr.584; PMID: 21473000
- Hultin LE, Hausner MA, Hultin PM, Giorgi JV. CD20 (pan-B cell) antigen is expressed at a low level on a subpopulation of human T lymphocytes. Cytometry 1993; 14:196 - 204; http://dx.doi.org/10.1002/cyto.990140212; PMID: 7679964
- Chang CH, Wang Y, Trisal P, Li R, Rossi DL, Nair A, Gupta P, Losman M, Cardillo TM, Rossi EA, et al. Evaluation of a novel hexavalent humanized anti-IGF-1R antibody and its bivalent parental IgG in diverse cancer cell lines. PLoS One 2012; 7:e44235; http://dx.doi.org/10.1371/journal.pone.0044235; PMID: 22952934
- Gupta P, Goldenberg DM, Rossi EA, Cardillo TM, Byrd JC, Muthusamy N, Furman RR, Chang CH. Dual-targeting immunotherapy of lymphoma: potent cytotoxicity of anti-CD20/CD74 bispecific antibodies in mantle cell and other lymphomas. Blood 2012; 119:3767 - 78; http://dx.doi.org/10.1182/blood-2011-09-381988; PMID: 22271448
- Rossi EA, Goldenberg DM, Cardillo TM, Stein R, Wang Y, Chang CH. Novel designs of multivalent anti-CD20 humanized antibodies as improved lymphoma therapeutics. Cancer Res 2008; 68:8384 - 92; http://dx.doi.org/10.1158/0008-5472.CAN-08-2033; PMID: 18922911
- Rossi EA, Goldenberg DM, Cardillo TM, Stein R, Chang CH. Hexavalent bispecific antibodies represent a new class of anticancer therapeutics: 1. Properties of anti-CD20/CD22 antibodies in lymphoma. Blood 2009; 113:6161 - 71; http://dx.doi.org/10.1182/blood-2008-10-187138; PMID: 19372261
- Gold DV, Goldenberg DM, Karacay H, Rossi EA, Chang CH, Cardillo TM, McBride WJ, Sharkey RM. A novel bispecific, trivalent antibody construct for targeting pancreatic carcinoma. Cancer Res 2008; 68:4819 - 26; http://dx.doi.org/10.1158/0008-5472.CAN-08-0232; PMID: 18559529
- Sharkey RM, van Rij CM, Karacay H, Rossi EA, Frielink C, Regino C, Cardillo TM, McBride WJ, Chang CH, Boerman OC, et al. A new Tri-Fab bispecific antibody for pretargeting Trop-2-expressing epithelial cancers. J Nucl Med 2012; 53:1625 - 32; http://dx.doi.org/10.2967/jnumed.112.104364; PMID: 22952342
- Rossi EA, Goldenberg DM, Cardillo TM, Stein R, Chang CH. CD20-targeted tetrameric interferon-alpha, a novel and potent immunocytokine for the therapy of B-cell lymphomas. Blood 2009; 114:3864 - 71; http://dx.doi.org/10.1182/blood-2009-06-228890; PMID: 19710501
- Rossi EA, Rossi DL, Stein R, Goldenberg DM, Chang CH. A bispecific antibody-IFNalpha2b immunocytokine targeting CD20 and HLA-DR is highly toxic to human lymphoma and multiple myeloma cells. Cancer Res 2010; 70:7600 - 9; http://dx.doi.org/10.1158/0008-5472.CAN-10-2126; PMID: 20876805
- Rossi EA, Rossi DL, Cardillo TM, Stein R, Goldenberg DM, Chang CH. Preclinical studies on targeted delivery of multiple IFN?2b to HLA-DR in diverse hematologic cancers. Blood 2011; 118:1877 - 84; http://dx.doi.org/10.1182/blood-2011-03-343145; PMID: 21680794
- Chang CH, Rossi EA, Cardillo TM, Nordstrom DL, McBride WJ, Goldenberg DM. A new method to produce monoPEGylated dimeric cytokines shown with human interferon-?2b. Bioconjug Chem 2009; 20:1899 - 907; http://dx.doi.org/10.1021/bc9001773; PMID: 19736932
- Chang CH, Hinkula J, Loo M, Falkeborn T, Li R, Cardillo TM, Rossi EA, Goldenberg DM, Wahren B. A novel class of anti-HIV agents with multiple copies of enfuvirtide enhances inhibition of viral replication and cellular transmission in vitro. PLoS One 2012; 7:e41235; http://dx.doi.org/10.1371/journal.pone.0041235; PMID: 22844444
- Bargou R, Leo E, Zugmaier G, Klinger M, Goebeler M, Knop S, Noppeney R, Viardot A, Hess G, Schuler M, et al. Tumor regression in cancer patients by very low doses of a T cell-engaging antibody. Science 2008; 321:974 - 7; http://dx.doi.org/10.1126/science.1158545; PMID: 18703743
- Topp MS, Kufer P, Gökbuget N, Goebeler M, Klinger M, Neumann S, Horst HA, Raff T, Viardot A, Schmid M, et al. Targeted therapy with the T-cell-engaging antibody blinatumomab of chemotherapy-refractory minimal residual disease in B-lineage acute lymphoblastic leukemia patients results in high response rate and prolonged leukemia-free survival. J Clin Oncol 2011; 29:2493 - 8; http://dx.doi.org/10.1200/JCO.2010.32.7270; PMID: 21576633
- Bluemel C, Hausmann S, Fluhr P, Sriskandarajah M, Stallcup WB, Baeuerle PA, Kufer P. Epitope distance to the target cell membrane and antigen size determine the potency of T cell-mediated lysis by BiTE antibodies specific for a large melanoma surface antigen. Cancer Immunol Immunother 2010; 59:1197 - 209; http://dx.doi.org/10.1007/s00262-010-0844-y; PMID: 20309546
- Dreier T, Baeuerle PA, Fichtner I, Grün M, Schlereth B, Lorenczewski G, Kufer P, Lutterbüse R, Riethmüller G, Gjorstrup P, et al. T cell costimulus-independent and very efficacious inhibition of tumor growth in mice bearing subcutaneous or leukemic human B cell lymphoma xenografts by a CD19-/CD3- bispecific single-chain antibody construct. J Immunol 2003; 170:4397 - 402; PMID: 12682277
- Schlereth B, Fichtner I, Lorenczewski G, Kleindienst P, Brischwein K, da Silva A, Kufer P, Lutterbuese R, Junghahn I, Kasimir-Bauer S, et al. Eradication of tumors from a human colon cancer cell line and from ovarian cancer metastases in immunodeficient mice by a single-chain Ep-CAM-/CD3-bispecific antibody construct. Cancer Res 2005; 65:2882 - 9; http://dx.doi.org/10.1158/0008-5472.CAN-04-2637; PMID: 15805290