Abstract
Limited access for high-quality biologics due to cost of treatment constitutes an unmet medical need in the United States (US) and other regions of the world. The term “biosimilar” is used to designate a follow-on biologic that meets extremely high standards for comparability or similarity to the originator biologic drug that is approved for use in the same indications. Use of biosimilar products has already decreased the cost of treatment in many regions of the world, and now a regulatory pathway for approval of these products has been established in the US. The Food and Drug Administration (FDA) led the world with the regulatory concept of comparability, and the European Medicines Agency (EMA) was the first to apply this to biosimilars. Patents on the more complex biologics, especially monoclonal antibodies, are now beginning to expire and biosimilar versions of these important medicines are in development. The new Biologics Price Competition and Innovation Act allows the FDA to approve biosimilars, but it also allows the FDA to lead on the formal designation of interchangeability of biosimilars with their reference products. The FDA’s approval of biosimilars is critical to facilitating patient access to high-quality biologic medicines, and will allow society to afford the truly innovative molecules currently in the global biopharmaceutical industry’s pipeline.
Introduction to the Opportunity Represented by Biosimilars for ALL Stakeholders
Biologics are medicines made in or isolated from living systems. They increasingly use recombinant DNA technology, although many important biologics continue to be naturally sourced, such as certain childhood vaccines and blood products. The first recombinant product for human use, human insulin (Humulin®), was approved in the US in 1982. The number of approved recombinant protein therapeutics, including monoclonal antibodies (mAbs), has expanded considerably, not least because these complex products can be created to bind unique targets.
Global sales of biologics were $93 billion in 2009, and these sales are expected to continue to grow at least twice as fast as those of small molecules. Of this cohort of highly successful biologic products, mAbs are the largest and fastest growing segment. The substantial clinical utility and commercial success of these products has convinced large pharmaceutical companies to engage in the research and development (R&D) of biologics. Approximately 30% of the pharmaceutical and biotechnology industry R&D pipeline is composed of biologics, and nearly a third of these are mAb-based.Citation1 It is anticipated that by 2016, ten of the top-selling 20 drugs will be biologics; of these, seven (Humira®, Avastin®, Rituxan®, Herceptin®, Remicade®, Prolia® and Lucentis®) are mAbs and one (Enbrel®) is a fusion protein containing antibody components.
However, the growing success of these biologics has been inevitably paralleled by the increasing challenge to the ability of health care systems worldwide to pay for them. The products are highly effective, life-altering therapies, but they have a high unit cost and are often used for chronic conditions that require on-going treatment. It is estimated that the average daily treatment cost for a small molecule brand drug is around $1 per day (with small molecule generic drug costing cents per day), whereas that of a branded biopharmaceutical is $22 per day. It was widely reported in So et al.Citation2 that a breast cancer patient's average cost for Herceptin® (trastuzumab) is $37,000, the treatment costs for rheumatoid arthritis or Crohn disease with Humira® (adalimumab) is $50,000 per year, and the cost to a Gaucher disease patient for treatment with Cerezyme® (imiglucerase) can be $200,000 per year for the rest of their life. The top six biologics already consume 43% of the drug budget for Medicare Part B,Citation2 and if the current trend in increasing usage continues, expenditure will inevitably also increase. This situation is unsustainable and may lead to limited access whereby only the more severely affected patients are treated with these agents when disability might be prevented or limited with earlier intervention. Access for even the current patients treated with biologics can become restricted due to costs.
The development of high-quality, but lower cost, biologics when patents expire addresses the need to improve access to patients who can benefit from treatment. In 2004, legislation enacted in Europe created a pathwayCitation3 for the approval of biosimilar products that has been adapted more broadly. The EMA used their authority to approve subsequent versions of biologic products that explicitly refer to a previously approved biologic, based on the comparability approach, by approving Omnitrope® (somatropin) as the first biosimilar product available in Europe in 2006.Citation4
With nearly three decades of experience with marketed biotech medicines in the US, patents on a number of these recombinant biologics are beginning to expire. It is estimated that off-patent sales will represent approximately 40% of the anticipated global sales in 2015 and by 2020, over $100 billion in biologic sales will be off patent.
Until 2010, the FDA lacked the legal authority to approve biosimilar medicines in the manner that had occurred in Europe. This deficit was addressed with the passage of healthcare reform legislation that included the BPCIA. As of March 2010, FDA has had the authority to review and approve biosimilar products that are judged to be “highly similar” to the originator reference product. Although the terms “highly similar” and comparability are used by FDA for different purposes, the science underpinning the approaches is identical. FDA uses the term “comparability” only when referring to the comparison of product attributes of a biologic when the manufacturing process is altered. This can result in changes in the biologic molecule. The sponsor must show that the pre-change and post-change products are “comparable.” FDA uses the term “highly similar” when referring to comparisons of an originator reference product and a biosimilar, although the methods and analytical tools used in both exercises are identical.
The Science-Based Regulatory Concept of “Comparability” is Fundamental for All Biologics
With the commercialization and rapid uptake of biologic drugs, it became apparent in the 1990s that companies would have to scale up their manufacturing process or transfer their processes to other manufacturing sites to meet demand. These changes in manufacturing can induce changes in the product attributes that can have an impact on clinical safety and efficacy. To address this issue, the FDA and other regulatory agencies developed a scientifically valid approach to evaluate these products pre- and post-manufacturing change. In doing so, the FDA originated the term “comparability” for head-to-head product comparisons of products, as described in their 1996 Guidance “Demonstration of Comparability of Human Biological Products, including Therapeutic Biotechnology-derived Products.”Citation5 If a product is judged to be comparable pre- and post-manufacturing change, then the products resulting from the process before and after the change are interchangeable. Comparability, as first conceived, was a soundly-grounded scientific concept and neither statutory history nor relative complexity of the product factored into the equation. The authority to assess and designate pre- and post-manufacturing change biologics as interchangeable was assumed by the FDA under the pre-BPCIA Public Health Service Act (PHSA), and did not entail legislation.
Comparability was supported by industry, both specifically through the trade organization Pharmaceutical Research and Manufacturers of America (PhRMA), and also more generally by individual companies. The concept of comparability has been used extensively by sponsors of licensed biologic products. It is data-driven and the burden remains with the sponsor to collect the necessary data to convince the FDA of the validity of their chosen approach, just as is the case with the initial biologic approval. These comparability “exercises” rarely entailed clinical trials, and they never required switching studies between pre- and post-change product. It has never been suggested that the use of comparability puts patients' well-being at risk, even though its use has not been entirely without incident.Citation6,Citation7
In Europe, the comparability concept was also of great value for all stakeholders, but the European use of the term “comparable” was also extended to products from different sponsors as per a 2003 guideline.Citation8 The EMA (known then as the European Medicines Evaluation Agency) also participated in the development and implementation of the International Conference on Harmonization (ICH) Tripartite Comparability Guideline,Citation9 even though they continue to maintain their own EU Comparability Guideline. The former only applies to a sponsor making a manufacturing change to their own product. Nonetheless, with minimal extrapolation the cross-sponsor use of comparability became the basis for the EU approach to biosimilars and informed their definition of biosimilarity.Citation10 With the extensive experience of regulators and sponsors in highly regulated markets, comparability is the universal standard for judging interchangeability of pre- and post-manufacturing changes of any biologic. With the development of the biosimilar approval pathway, the scientific approach underlying interchangeability can be broadly applied to an originator product undergoing a manufacturing change or a biosimilar at initial approval or a biosimilar undergoing a manufacturing change. Comparability is already being used for post-approval manufacturing changes to biosimilars in Europe and elsewhere.
An Overview of Requirements for Biosimilars Development
Europe has led in the approval of biosimilars because EMA was the first regulatory authority to be given the mandate to review and approve biologic products that explicitly referred to a previously approved product and because patents on key products expired in Europe sooner than in the US. Due to successful experiences in the EU, use of the EU approach to biosimilars regulation in the US has been advocated,Citation11,Citation12 and the European approach was endorsed by a wide variety of the stakeholders presenting at the recent FDA public hearing on this topic.Citation13 This meeting sought public input on the implementation by the FDA of the new abbreviated biosimilar pathway created in the BPCIA.Citation14 However, it is unlikely, despite the many recent efforts at harmonizing the regulations in the highly regulated markets, that the FDA will adopt the EU biosimilars guidelines () even though it could save substantial time and resources and allow FDA to concentrate on defining a guideline for interchangeability, which is an issue not addressed in Europe as a regulatory matter.
The highly-regulated markets have specific requirements for biosimilars regarding the comparative studies that a biosimilar sponsor must conduct to prove biosimilarity to their chosen originator reference biologic. We have estimated that the costs of developing a biosimilar product for highly-regulated markets such as Europe or the US ranges from $75–250 million, which will limit the number of successful companies in this space.
Developing markets also have compelling unmet medical needs for targeted biologics, which has fostered local regulatory processes with reduced requirements, if any, for head-to-head studies. Such “non-highly-regulated” jurisdictions approve what we term “alternative biologics.” These jurisdictions address the unmet medical need by allowing a follow-on biologic for the same target as the originator molecule to be approved without a demonstration of biosimilarity. Such products may be adequate for the purpose, but they are not biosimilars, i.e., they are not close enough to the originator molecule to be judged biosimilar in analytical or clinical testing. Some use these “alternative” biologics to cast doubt on the clinical utility of true biosimilars. Schellekens evaluated several alternative recombinant erythropoietins manufactured in Asia and demonstrated with isoelectric focusing gels that they were not comparable with the originator product.Citation15 This is unlike approved biosimilars, which are highly similar to the originator reference as shown by Brockmeyer and Seidl who, also using isoelectric focusing gels, compared biosimilar erythropoietin to the originator product.Citation16 Alternative biologics are not biosimilar and have not been approved as biosimilar, and they should be clearly distinguished from approved biosimilars in discussions about the new biosimilars pathway implemented in the US. The FDA is expected to approve biosimilars to the same standards as the marketed reference biologics, given that the same standards of safety, purity and potency apply. Questions about other biologics in other jurisdictions that apply lower standards are not relevant to the biosimilars debate in the US or other highly-regulated markets, but will likely continue to be invoked by some to confuse or frighten patients or providers.
Likewise, Europe has expressed confidence in the quality and consistency of its biologic products, including biosimilars. At the 2008 European Generics Medicines Association Biosimilars Symposium, Nicolas Rossignol, former administrator of the European Commission Pharmaceuticals Unit, said that “biosimilar products approved by the European Commission in accordance with EMEA guidelines should not be subject to unfounded questions regarding their safety” and “a biosimilar product is as safe and efficacious as any other product authorized by the European Commission in the EU.”Citation17
In order to develop a biosimilar, a sponsor must necessarily ensure quality, plus safety and efficacy (EU) or safety, purity and potency (US), and it must be biosimilar to the reference product. However, in order to be viable as a business, a sponsor must also ensure their development is efficient by receiving product approval in a timely manner and with lower costs than those required for development of a novel agent. This combination will allow competitive pricing, improved access and an appropriate return on investment. Thus, the key to biosimilar development is the demonstration of a high degree of molecular similarity, as well as adequate safety and efficacy data to achieve commercial viability (represented by a broad product label).
If an originator biologic product has multiple indications, extrapolation to additional indications for a biosimilar, based on solid biosimilar data in a sensitive indication, may be granted in the EU. This is appropriate where comparability is shown, clinical data supports equivalent safety and efficacy to the originator molecule in at least one indication, and a common mechanism of action can be presumed for the clinical effect.
Defining the “Goal Posts”: Product Attribute Distribution of Originator Product
In the US, the BPCIA is unique in allowing the FDA to designate a biosimilar as interchangeable with its reference product [as stated in the statute, “the biological product (biosimilar) may be substituted for the reference product without the intervention of the health care provider who prescribed the reference product”]. An application for interchangeability can be concurrent or subsequent to the application for a biosimilar product under 351(k) of the BPCIA.
As discussed previously, originator biologic products undergo changes over time, e.g., when the manufacturing process is modified by scaling up or transferred to alternate facilities. Such changes in the originator product attributes provide a distribution of these attributes that should be considered acceptable because the products were subsequently administered to patients successfully. The limits to this distribution of product attributes are what we call “goal posts,” and these can be used to evaluate the acceptability of a biosimilar. If the product attributes fall within the variability of the originator molecule after manufacturing change, then the biosimilar should be considered “highly similar.” The foundational principle and goal for the development of biosimilars at Sandoz has been to achieve a high degree of similarity to the originator reference product such that our biosimilar is as similar to the originator product as the originator product is to itself considering the variability induced by manufacturing changes. As an example of establishing these goal posts, plots one specific glycan structure in a commercial mAb followed in many batches over a four year period. Both US and EU commercial formulations were analyzed over time at Sandoz using state of the art technologies for this specific glycan structure. There is very little variability between batches over time when using the same manufacturing process. However, a major shift in this product attribute does occur with a manufacturing change wherein the relative amount of this structure drops from approximately 50–30%. This change was first seen with European product, but eventually the same product attribute was seen with US commercial product. This figure confirms several points: (1) batch to batch variability is small during routine manufacturing, (2) major shifts in product attributes may occur as a result of manufacturing changes, (3) in this case EU and US products are “highly similar” and are probably manufactured in the same plant, (4) the variance induced by manufacturing changes establishes “goal posts” that a biosimilar company can employ for establishing comparability and (5) it is apparent that originators are essentially manufacturing biosimilars of their original product.Citation18
Once “goal posts” are established for a originator reference product, an iterative process is used in developing a biosimilar where the product is fully evaluated by physicochemical and biological characterization compared to the originator product. When product attributes fall outside established “goal posts,” various process steps are modified to produce product attributes that are within the variability of the originator product (). This process is repeated until highly similar product attributes are achieved. The degree of similarity that is established is based on analytical characterization that is run in parallel with iterative process development activities.
Depending on the extent that product attributes of the biosimilar overlap with those of the originator product, preclinical and clinical development can be abbreviated compared to developing a novel biologic. An abbreviated development program is justified when the biosimilar product attributes are highly similar to the originator product. As a historical perspective, when evaluating manufacturing changes, FDA has rarely required sponsors to repeat preclinical or clinical studies,Citation19,Citation20 but for biosimilars such studies are currently expected in both the EU and US. The authority is given to the FDA in BPCIA to waive preclinical and clinical studies, yet it is clear that they will be conservative and not waive such requirements in the near future.
Once a biosimilar candidate is shown to be “highly similar” (also a BPCIA statutory requirement), at the analytical level and through biological characterization, the subsequent pre-clinical and clinical studies can be streamlined as a scientific matter. However, if any of the product attributes of the proposed biosimilar deviate from those of the reference product, then scientific justification of presumed safety/efficacy impact is required or additional pre-clinical and clinical data demonstrating similarity, are needed, just as is the case for the use of comparability in the context of a manufacturing change. Sponsors' use of the new US biosimilars pathway will depend on the degree to which FDA is driven by this scientific justification for an abbreviated clinical development program, as opposed to their endorsement of a precautionary approach that requires many of the typical development steps of a the traditional novel biologic 351(a) biologics license application.
To achieve highly similar product attributes, the “goal posts” chosen by the biosimilar sponsor, which represent variability of originator product attributes for their design space,Citation21 become key to the development of a 351(k) biosimilar. The “goal posts” for biosimilarity are established through an examination of the reference product attributes by the biosimilar sponsor by obtaining commercial reference product and analyzing product characteristics, and not through access to the originators data (). The “goal posts” approach is dependent upon analysis of multiple batches of reference product manufactured in the recent past, but also, to the extent possible, over an extended period of time since its initial approval, including batches pre- and post-manufacturing change. Hence, sample collection and evaluation of commercial drug of the chosen originator reference product is a critical part of the planning for the development of a biosimilar. “Goal posts” of originator product attributes are only broadened if the reference product sponsor changes their product's specifications after manufacturing changes even if such changes occur toward the end of their patent life in anticipation of life cycle management or to block biosimilar competition.
Most biologics that will become reference products for future biosimilar development have undergone comparability exercises over their lifetimes during scale-up activities to meet demand. Even though these manufacturing changes are tightly regulated, they are still associated with observable changes in the product attributes over time. However, by remaining on the market with the same label during these times, regulators are indicating that each of these pre- and post-manufacturing change products is interchangeable with itself. This regulatory scientific approach can be used to evaluate and approve biosimilar candidates with attributes falling between the goal posts of reference products that would be considered highly similar to the originator (“highly similar” is the term used in BPCIA and ICH Q5E). If judged highly similar and shown to produce equivalent clinical responses after switching with the originator, the sponsor should receive approval as interchangeable with the reference product ( and ). Such an approach by sponsors and regulators is consistent with all prior regulatory decisions regarding major manufacturing changes of originator products.
Like the use of comparability to make manufacturing changes, the type and extent of preclinical and clinical studies will be governed by how well the biosimilar sponsor can keep their product within the bounds defined by the “goal posts” of the reference product. It is not trivial to achieve overlapping product attributes, and any differences that are not within the “goal posts” will have to be defended or demonstrated not to alter the clinical outcomes of the product. Thus, preclinical and clinical study designs will be tailored to the comprehensive comparability data generated analytically.
While the US is generally behind the EU with regard to biosimilars, two products are worth mentioning as examples of FDA experience using the science appropriately within their prior regulatory authority. Most biologics are regulated under the PHSA. For historical reasons, a few biologics are regulated under the Federal Food, Drug and Cosmetic Act, and therefore the Hatch Waxman generic pathways are available for these products, which include somatropin and enoxaparin. In May 2006, Sandoz achieved an approval for Omnitrope® (somatropin) based on a demonstration of “high similarity”Citation22/comparability to Pfizer's Genotropin® (somatropin), and in July 2010, the approval of enoxaparin as a fully substitutable generic to Sanofi-Aventis' Lovenox® (enoxaparin). While the statutes within the BPCI and Hatch Waxman legislation differ, many of the same scientific and regulatory considerations apply, not least because the latter represents a naturally-sourced complex mixture that was appropriately characterized as part of the application and no clinical trials were required. In reviewing the enoxaparin file, FDA established five criteria to determine “sameness” of Sandoz enoxaparin to Lovenox® (). These included physiochemical properties, source material, the sugar building blocks, biologic assays and in-vitro pharmacodynamic profile. Each of these criteria had to be comparable for FDA to rule that the two products were the “same”. In this case, the analytical characterization, along with other information, provided sufficient data for FDA to approve the product. Again, enoxaparin was not approved as a biosimilar under the new BPCIA pathway, but the scientific approach used by FDA is equally applicable.
The Need for a “Global” Biosimilar Pathway
Biosimilars are now recognized around the world as safe and effective medicines, and the World Health Organization has also generated guidelinesCitation23 to assist the less highly regulated markets in their consideration of applications from biosimilar sponsors. The standards are largely derived from those demonstrated to be successfully applied in the EU, and other regulated markets such as Australia, Canada and Japan.
The need for access to high quality, safe and effective biologics is global, as is the regulatory science, and increasingly, the biomanufacturing experiences. However, healthcare priorities and resources, as reflected in infrastructure and payment for medicines, vary between individual regions and countries, intellectual properties rights differ, and the regulatory policy by which medicines first reach the market can be extremely specific. For potential sponsors of all biologics, including biosimilars, considerations must include the research and development costs of the regulatorily-required studies. Regional regulatory authorities now generally require biosimilar sponsors to compare their biosimilar to the reference product approved in the local region. That is, when performing studies in Europe, EMA generally requires use of a European-labeled reference product as a comparator, while a US-labeled reference product would be necessary as a comparator for approval in the US. Thus these policies currently require essentially a duplicative clinical development program, which vastly increases development costs. To date, each law in each country has defaulted to the need for a reference product previously approved by that regulatory authority. This is the case even if the biologic drug used as a reference is made by a single facility for worldwide use, and the only difference in the product in each country is the label. Presently, it is the label on the product that defines its regulatory status, and not what is in the tube. Thus, there is a need for scientifically-appropriate bridging to be accepted across jurisdictions. The data showing that, for example, an EU reference product and a US reference product are indistinguishable should obviate the need for a repetition of critical studies. If originators can prepare global data sets and dossiers, but biosimilar sponsors cannot, this will have a major impact on the supply of the respective products, and the consequent access by patients. Scientific principles suggest that using a single global reference product is reasonable, provided that it is proven comparable to the same product labeled in other major regions.
Interchangeability or Substitution
The one explicit authority granted to the FDA, and not available to EMA, is that they can designate a biosimilar as interchangeable with its reference originator product. Interchangeability, as defined in the BPCIA, allows a pharmacist to substitute an interchangeable biosimilar with the originator drug without the intervention of the original prescribing physician. Such a designation would enhance adoption of biosimilars and more rapidly enable costs savings and increased access, just as is the case today with traditional generic drugs. FDA can lead the world in establishing criteria for an interchangeable designation, but even this, as discussed above, should be covered by the concept of comparability, as initiated by the FDA in 1996, adopted by ICH in 2005 and converted for biosimilar use in the EU in 2004. Namely, a product for which comparability is demonstrated is interchangeable with its comparator. This comparator may be from the same sponsor or a different sponsor, but the comparison is always based on data and a scientific rationale.
Conclusion
Biosimilars can have a major impact on the affordability and availability of important biologic medicines in all markets. Approval of biosimilars will facilitate patient access, and lower costs, thus making health care dollars available for the next generations of originator medicines. Such was the case in the US in 1984 when the Hatch Waxman Act allowed sponsors to refer to the previously approved product on which the patents had expired as part of their abbreviated new drug application for generic small molecule drugs. With passage of BPCIA, we believe the same can be encouraged to occur for biologics.
The quality of the biosimilars and the originator biologics to which they refer will be the same if the FDA applies consistent, science-based and data-driven standards equally to all products. The US is already the leading market for biotechnology-based products. Having led the world with the development of comparability in 1996 through guidance alone, we believe that the FDA has the expertise and experience needed, and is ideally suited, to review and approve biosimilar applications now. The FDA can encourage biosimilar applications today by expressing confidence in the science, as well as in their own reviewers experience and expertise. Their support of a scientifically-justified and appropriate reduction in the data burdens for biosimilars through the FDA's recognition of quality characterization and support of abbreviated clinical development programs would create great confidence amongst sponsors, and no changes in risks for consumers. Such an approach occurs today, under FDA oversight, during the use of comparability for manufacturing changes for already licensed biopharmaceuticals of all sorts. Comparability applies to complex products such as mAbs just as it does to other smaller and simpler biologics. By engaging today in advocating and implementing the highly similar standard for mAb biosimilars and working with EU on their published draft guidelines, the FDA can rejoin EMA in leading the oversight of all biologics for a global patient need. The FDA engagement in the creation of the new regulatory approaches for interchangeable biologics, originator and biosimilar, based on their original insights with comparability, gives the FDA additional opportunity for leadership.
Note
This paper is based on presentations given by Mark McCamish on November 30, 2010 at the 6th European Antibody Congress in Geneva, Switzerland;Citation24 on December 8, 2010 at IBC's 8th Annual International Conference on Antibody Therapeutics held in San Diego, CA and also on the presentation and submission made by the Novartis Group of companies to the FDA Docket on Approval Pathway for Biosimilar and Interchangeable Biological Products.Citation18
Abbreviations
| ANDA | = | Abbreviated New Drug Application |
| BLA | = | Biologics License Application |
| BPCIA | = | Biologics Price Competition and Innovation Act |
| EMA | = | European Medicines Agency |
| Hatch Waxman | = | Hatch Waxman Act |
| FFDCA | = | Federal Food Drug and Cosmetic Act |
| mAbs | = | monoclonal antibodies |
| NDA | = | New Drug Application (FFDCA) |
| NME | = | new molecular entity |
| PHSA | = | Public Health Service Act |
Appendix
Definitions.
Biosimilar, PHSA 351(k) application (US) or “similar biological medicinal products” (EU); BPCIA, Biologics Price Competition and Innovation Act, the biosimilars provisions of the US health care reform legislation enacted on March 23, 2010); EMA, European Medicines Agency, formerly known as the European Medicines Evaluation agency (EMEA); Hatch Waxman act, Drug Price Competition and Patent Term Restoration act of 1984, also known as Hatch Waxman; FFDCA, Federal Food, Drug and Cosmetic act, the statute under which most drugs have been approved in the US; PHSA, Public Health Service Act, the statute under which most biologics have been licensed (approved) in the US.
Figures and Tables
Figure 1 European biosimilar guidelines. The EMA began with an overarching guideline on biosimilars and then general guidelines, before issuing product class specific data requirements. The EU Guidelines that have been finalized are indicated in blue. A draft guideline for mAbs is currently available for public comment.Citation25
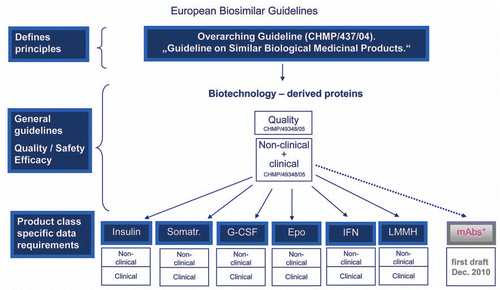
Figure 2 Analyzing complex product attributes over time. The bG2 glycan structure was quantified by Sandoz in many batches of commercial product distributed by the originator in the EU (light blue) and the US (dark blue). Expiry date of the product batches is listed on the x-axis and relative amount of product attribute enrichment is listed on the y-axis. Pre-shift quality refers to the content of the attribute prior to a manufacturing change and post-shift quality after the manufacturing change.
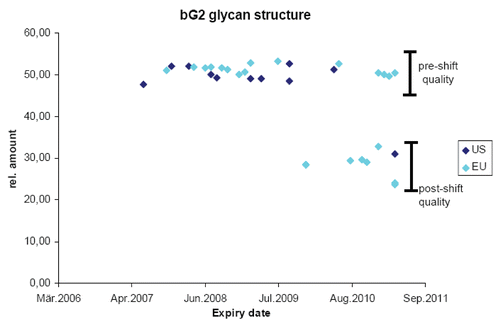
Figure 3 Biosimilar development process. The development of a biosimilar relies on creation of a design space based on analysis of the reference product and then iterative development of a biosimilar to fit the chosen specifications. There is no access to, nor need for, originator data at any point in this process. The early process development is essential, and later development cannot compensate for this initial generation of a “highly similar” candidate product. As the complexity of the reference product increases, the initial development becomes more challenging, and the likelihood that multiple iterations will be needed increases.
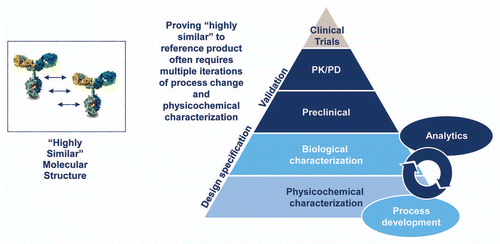
Figure 4 Biosimilarity goal posts. The “goal posts” of biosimilarity are established by the biosimilar sponsor by their analysis of the distribution of product attributes present in the reference product pre- and post-manufacturing change. They then use these to select the design space for their biosimilar candidate. While the complete quality range may be quite broad for the life time of the reference product, the biosimilar sponsor will select a tighter range of control for their biosimilar product.
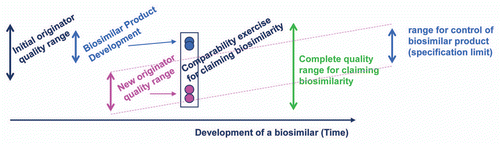
Figure 5 The one US marketed generic biologic: enoxaparin. Generic enoxaparin was approved by FDA on July 23, 2010. Although not a “biosimilar” insofar as it was not approved through the new biosimilar 351(k) regulatory pathway, scientifically it is a fully interchangeable generic biologic and many of the FDA considerations anticipated for biosimilars s apply.

Notes
For explanation of abbreviations, see Appendix.
References
- Pharmaceutical Research and Manufacturers of America. Biotechnology Medicines in Development Report 2008; www.phrma.org/sites/phrma.org/files/attachments/Biotech%202008.pdf
- So AD, Katz SL. Biologics Boondoggle New York Times 2010; March 8
- DIRECTIVE 2004/27/EC OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL of 31 March 2004 amending Directive 2001/83/EC on the Community code relating to medicinal products for human use eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2004:136:0034:0057:EN:PDF
- European Public Assessment Report EMEA/H/C/607. Omnitrope (somatropin) www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Summary_for_the_public/human/000607/WC500043689.pdf
- Demonstration of Comparability of Human Biological Products, Including Therapeutic Biotechnology-derived Products, Center for Biologics Evaluation and Research (CBER), Center for Drug Evaluation and Research (CDER) April 1996 www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm122879.htm
- Casadevall N. Immune-response and adverse reactions: PRCA case example http://www.ema.europa.eu/docs/en_GB/document_library/Presentation/2009/11/WC500011064.pdf
- Usdin S. Myozyme's zig zags. BioCentury 2008; 16:1 - 6
- EU Guideline on Comparability of Medicinal Products containing Biotechnology-derived Proteins as Active Substances: Quality issues (CPMP December 2003) www.emea.europa.eu/pdfs/human/bwp/320700en.pdf
- ICH Q5E: Comparability of Biotechnological/Biological Products Subject to Changes in Their Manufacturing Process. EU: Adopted by CMPM, December 1, 2004, CPMP/ICH/5721/03, date for coming into operation: June 2005; MHLW: Adopted 26 April 2005, PFSB/ELD Notification No. 0426001; FDA: Published in the Federal Register, Vol. 70, No. 125, June 30, 2005; 37861-37862 www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q5E/Step4/Q5E_Guideline.pdf
- Directive 2004/27/EC of the European Parliament and of The Council of 31 March 2004 amending Directive 2001/83/EC on the Community code relating to medicinal products for human use http://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2004:136:0034:0057:EN:PDF
- Senate HELP Full Committee Hearing “Follow-On Biologics” March 8, 2007 http://help.senate.gov/hearings/hearing/?id=0d7fdce8-b5cd-5a56-8930-31dfb58e7c11
- FDA Public Hearing on Approval Pathway for Biosimilar and Interchangeable Biological Products, 11/3/2010 Transcript Document ID: FDA-2010-N-0477-012 www.regulations.gov/search/Regs/home.html#documentDetail?R=0900006480bafe46
- Federal register notice Food and Drug Administration [Docket No. FDA-2010-N-0477] Approval Pathway for Biosimilar and Interchangeable Biological Products; Public Hearing; Request for Comments http://edocket.access.gpo.gov/2010/pdf/2010-24853.pdf
- Biologics Price Competition and Innovation Act (BPCIA) provisions of the Patient Protection and Affordable Care Act (PPACA) http://frwebgate.access.gpo.gov/cgi-bin/getdoc.cgi?dbname=111_cong_bills&docid=f:h3590enr.txt.pdf
- Schellekens H. Biosimilar epoetins: How similar are they?. Eur J Hosp Pharm 2004; 10:243 - 247
- Brockmeyer C, Seidl A. Binocrit: assessment of quality, safety and efficacy of biopharmaceuticals. Eur J Hosp Pharm Prac 2009; 15:34 - 40
- European Generics Medicines Association. EGA Conference Highlights: 6th European Generics Medicines Association Biosimilars Symposium 2008 www.egagenerics.com/doc/ega_highlights_2008_02.pdf
- Novartis submission to FDA-2010-N-0477 docket on Notice: Approval Pathway for Biosimilar and Interchangeable Biological Products; Hearing www.regulations.gov/#!documentDetail;D=FDA-2010-N-0477-064.1
- Assessing the Impact of a Safe and Equitable Biosimilar Policy in the United States. Statement of Janet Woodcock MD, Deputy Commissioner, Chief Medical Officer, Food and Drug Administration before the Subcommittee on Health, House Committee on Energy and Commerce May 2, 2007 www.fda.gov/NewsEvents/Testimony/ucm154017.htm
- Follow-on Protein Products. Statement of Janet Woodcock MD, Deputy Commissioner, Chief Medical Officer Food and Drug Administration before the House Committee on Oversight and Government Reform, March 26, 2007 www.fda.gov/NewsEvents/Testimony/ucm154070.htm
- FDA Guidance for Industry. Q8, Q9 and Q10 Questions and Answers www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM210822.pdf
- FDA response to the Citizen petitions of Pfizer, BIO and Genentech. Re: Dockets Nos. 2004P-0231/CP1 and SUP1, 2003P-0176/CP1 and EMC1, 2004P-0171/CP1 and 2004N-0355 www.fda.gov/ohrms/dockets/dockets/04P0231/04P-0231-pdn0001.pdf
- WHO Expert Committee on Biological Standardization, Geneva. Guidelines on evaluation of similar biotherapeutic products (SBPs) 2009; October 9 to 23 www.who.int/biologicals/areas/biological_therapeutics/BIOTHERAPEUTICS_FOR_WEB_22APRIL2010.pdf
- Beck A, Wurch T, Reichert JM. 6th European Antibody Congress 2010: November 29–December 1, 2010, Geneva, Switzerland. mAbs 2011; 3:111 - 134
- European Medicines Agency. Biosimilar guidelines www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/general/general_content_000408.jsp&murl=menus/regulations/regulations.jsp&mid=WC0b01ac058002958c