Abstract
TWEAK, a TNF family ligand with pleiotropic cellular functions, was originally described as capable of inducing tumor cell death in vitro. TWEAK functions by binding its receptor, Fn14, which is up-regulated on many human solid tumors. Herein, we show that intratumoral administration of TWEAK, delivered either by an adenoviral vector or in an immunoglobulin Fc-fusion form, results in significant inhibition of tumor growth in a breast xenograft model. To exploit the TWEAK-Fn14 pathway as a therapeutic target in oncology, we developed an anti-Fn14 agonistic antibody, BIIB036. Studies described herein show that BIIB036 binds specifically to Fn14 but not other members of the TNF receptor family, induces Fn14 signaling, and promotes tumor cell apoptosis in vitro. In vivo, BIIB036 effectively inhibits growth of tumors in multiple xenograft models, including colon (WiDr), breast (MDA-MB-231), and gastric (NCI-N87) tumors, regardless of tumor cell growth inhibition response observed to BIIB036 in vitro. The anti-tumor activity in these cell lines is not TNF-dependent. Increasing the antigen-binding valency of BIB036 significantly enhances its anti-tumor effect, suggesting the contribution of higher order cross-linking of the Fn14 receptor. Full Fc effector function is required for maximal activity of BIIB036 in vivo, likely due to the cross-linking effect and/or ADCC mediated tumor killing activity. Taken together, the anti-tumor properties of BIIB036 validate Fn14 as a promising target in oncology and demonstrate its potential therapeutic utility in multiple solid tumor indications.
Introduction
The tumor necrosis factor (TNF) superfamily represents an attractive opportunity for therapeutic targeting in cancer because of its tumor cell killing activity. A number of TNF family members, including TNF and Fas/Apo1, have been evaluated in clinical studies, but toxicities related to systemic exposure have severely limited their development as cancer therapies, although alternative strategies for targeted or local delivery are still being pursued.Citation1 More recently, targeting of other TNF family members, including TNF-related apoptosis inducing ligand (TRAIL/Apo2L) and CD40, have emerged as promising therapeutic approaches.Citation1,Citation2 Notably, recombinant soluble TRAIL and agonist antibodies to the TRAIL receptors, TRAIL-R1 (death receptor (DR)4) and TRAIL-R2 (DR5), which exhibited impressive efficacy in tumor xenograft models, are currently undergoing early clinical testing with encouraging results regarding safety and tolerability.Citation3
TNF-like weak inducer of apoptosis (TWEAK) and its receptor, FGF-inducible molecule 14 (Fn14), are members of the TNF superfamily. Like TNF, TWEAK is a type II transmembrane protein which forms homotrimers that can function as soluble cytokine upon cleavage from the cell surface. TWEAK is a pleiotropic factor with a broad range of biological capabilities, such as pro-inflammatory activity and promotion of angiogenesis, migration, invasion and survival.Citation4 TWEAK was initially described and named for its ability to weakly induce HT29 tumor cell killing in vitro,Citation5 typically requiring co-incubation with sensitization agents such as IFNγ.Citation6
While Fn14 is generally expressed at relatively low levels on normal tissues, elevated Fn14 expression is observed in settings of tissue injury and regeneration,Citation7–Citation10 and, notably, in tumors including breast, pancreatic, esophageal and glioma.Citation7,Citation11–Citation15 In the largest survey to-date examining 1,655 tumor samples across 22 solid tumor subtypes by immunohistochemistry, Fn14 expression was detected in the majority of tumor types, including pancreatic cancer (60%), non-small cell lung cancer (55%), bone metastases (54%) and liver metastases in colorectal cancer (50%).Citation16 A significant correlation between increased Fn14 expression and higher tumor grade or poor prognosis has been documented in glioma, breast cancer and esophageal cancer.Citation14,Citation15,Citation17
Upon engaging TWEAK, the intracellular domain of Fn14 recruits TNF receptor associated factor (TRAF) molecules and induces signaling through nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB) and mitogen-activated protein kinases pathways.Citation18,Citation19 NFκB pathway stimulation by TWEAK/Fn14 has been documented in numerous contexts,Citation4 with evidence of stimulation of both canonical and non-canonical signaling.Citation18,Citation19 Although the scavenger receptor CD163 has been proposed to be an alternative receptor for TWEAK,Citation20 the biological implications of this interaction are unknown. Notably, tumor cell death induced by TWEAK is thought to be solely mediated through Fn14.Citation21
Unlike many other TNF family receptors with death-inducing activity, Fn14 does not contain a death domain, and consequently the mechanism by which TWEAK induces cell death is not well understood. In fact, there appear to be multiple mechanisms by which TWEAK can induce tumor cell death, and in some cases cell death induced by TWEAK may be mediated through other pathways. For example, TWEAK-induced cell death of some tumor cell lines, such as Kym-1, SKOV-3 and OVCAR, is TNF-dependent and involves recruitment of TRAF2 and cIAP-1 degradation.Citation22–Citation24 On the other hand, TWEAK-induced cell death of tumor cells such as HSC3, HT-29 and KATO-III is independent of TNF.Citation22 Similarly, TWEAK-induced cell death can be caspasedependent or caspase-independent, with features of both apoptosis and cathepsin-B dependent necrosis.Citation21,Citation25
To date, the anti-tumor activity of TWEAK has been exclusively demonstrated in vitro. Herein, we show that in vivo administration of TWEAK is efficacious in inhibiting tumor growth in a xenograft model. To exploit the tumor cell death inducing capacity of the TWEAK-Fn14 pathway, we developed an anti-Fn14 agonistic antibody, BIIB036, which mimics the signaling activity of TWEAK and can stimulate tumor cell apoptosis. BIIB036 exhibits potent anti-tumor activity in multiple xenograft models, despite its variable tumor cell inhibition activity in vitro. Interestingly, tumor cell killing in these cell lines does not appear to be TNF-dependent. The anti-tumor activity of BIIB036 is enhanced by increasing antigen-binding valency, indicating a requirement for Fn14 receptor cross-linking to maximize activity. BIIB036 can also induce antibody-dependent cell-mediated cytotoxicity (ADCC) in vitro. Consistent with these observations, we found that full Fc effector function is required for maximal activity of BIIB036 in xenograft models, likely reflecting the contribution of Fc receptors in mediating Fn14 cross-linking or ADCC activity.
Results
Administration of TWEAK is efficacious in a breast tumor xenograft model.
TWEAK has been shown to induce tumor cell death in vitro.Citation5,Citation22,Citation23 To determine whether in vivo delivery of TWEAK can inhibit tumor growth, adeno-TWEAK (or control adeno-GFP) was directly injected into MDA-MB-231 human breast tumors grown in nude mice. As shown in , the average tumor volumes were statistically different (p < 0.007) between the adeno-TWEAK treated groups and control (adeno-GFP) from day 23 through the end of the study (day 76). At day 76, tumors from the adeno-TWEAK treated mice had an 82% reduction in volume compared with tumors from the adeno-GFP treated group. shows the actual tumor weights determined upon dissection at termination of the study, confirming a significant reduction in tumor mass in the adeno-TWEAK treated group (). Notably, upon dissection at day 76, two of the seven mice in the adeno-TWEAK treated groups had no discernable tumor mass. Upon histological examination, the adeno-TWEAK treated tumors exhibited reduced density of tumor cells and decreased levels of proliferation as evident by H&E and Ki-67 staining, respectively (). Consistent with this, increased levels of apoptotic cells as assessed by TUNEL staining was observed in the adeno-TWEAK treated group (). Thus, intratumoral adeno-TWEAK administration was efficacious in reducing tumor growth in a xenograft model.
To verify that TWEAK is indeed responsible for the observed anti-tumor effect, we also tested whether administration of soluble Fc-TWEAK, in the absence of the viral delivery system, would be efficacious in the xenograft model. MDA-MB-231 tumor bearing mice were treated bi-weekly with intratumoral administration of Fc-TWEAK (or vehicle control). shows that the average tumor volume in the Fc-TWEAK treated mice was statistically smaller (p < 0.02) compared to tumors in control treated mice from day 54 through termination of the study (day 96). shows that on average there was an 86% reduction in actual tumor weight upon dissection at termination of the study (). Taken together, these results demonstrate that TWEAK can inhibit tumor growth in vivo, suggesting a potential therapeutic opportunity to exploit the pathway for the treatment of cancer.
Development of an agonistic anti-Fn14 antibody, BIIB036.
The studies described above provided evidence of the in vivo tumor cell killing capacity of TWEAK. However, as a therapeutic agent, an adenoviral delivery system was not considered a viable option. Fc-TWEAK was also not a feasible approach given its complex oligomeric structure resulting from the dimerizing capacity of the Fc domain coupled with the trimerizing region of TWEAK (Hsu Y-M, unpublished results). Since Fn14 is the sole receptor for TWEAK with biological relevance and is responsible for mediating tumor cell killing,Citation21 we chose to develop an agonistic anti-Fn14 monoclonal antibody as a candidate therapeutic agent to target the pathway.
The murine antibody mBIIB036 (P4A8)Citation14 was selected from a panel of anti-Fn14 antibodies that was generated in Fn14 knockout mice.Citation8 We demonstrated that mBIIB036 binds specifically to Fn14, but not to other members of the TNF receptor family (Sup. Fig. 1). mBIIB036 was subsequently humanized by CDR graftingCitation29 to generate a drug candidate, BIIB036, with a human IgG1 backbone. The binding affinity of BIIB036 to human monomeric Fn14 as measured by Biacore is 1.7 nM (Sup. Fig. 2), a value indistinguishable from that calculated for the murine version of the antibody (mBIIB036). To evaluate whether BIIB036 is capable of binding Fn14 expressed on the cell surface, FACS analysis was performed. shows that BIIB036 binds to 293E cells transiently transfected with full length human Fn14, but not to 293E cells transfected with empty vector. In addition, shows similar binding affinities of BIIB036 to 293E cells transfected with mouse, rat or cynomolgus monkey Fn14 (). We also characterized the binding site of BIIB036 by competitive binding of TWEAK versus BIIB036 to cell surface Fn14 in the WiDr colon carcinoma cell line. shows that binding of TWEAK to Fn14 can be blocked by BIIB036 in a dose-dependent fashion. Likewise, binding of BIIB036 can be blocked by TWEAK (data not shown). Thus, BIIB036 binds Fn14 with high affinity and specificity, binds Fn14 across species, and is capable of reciprocally competing with TWEAK for binding to Fn14.
Agonistic activity was a key requirement for the anti-Fn14 antibody and a basis upon which BIIB036 was selected from the panel of antibodies. We first tested BIIB036 agonistic activity in a chemokine release assay. shows that mBIIB036 is capable of inducing IL-8 release in A375 cells, albeit significantly less potently compared with Fc-TWEAK. Similar results were observed with measurement of MCP-1 and IP-10 release in A375 cells (data not shown). In addition, we evaluated signaling through the non-canonical NFκB pathway, as has previously been described for TWEAK,Citation18,Citation19 by monitoring p100 cleavage to p52. We found that treatment of WiDr or MDA-MB-231 tumor cells with BIIB036 resulted in the appearance of p52 (), similar to the effect observed upon treatment with Fc-TWEAK (). Canonical NFκB pathway signaling was also observed in response to BIIB036 treatment (data not shown). Taken together, the data indicated that BIIB036 exhibits agonistic activity, including induction of chemokine release and NFκB signaling, analogous to the effects of TWEAK.
BIIB036 can induce tumor cell death in vitro.
Next, the ability of BIIB036 to inhibit tumor growth in vitro was examined. BIIB036 and Fc-TWEAK were evaluated in an MTT assay on a panel of 38 human tumor cell lines (breast, colon, gastric, pancreatic, ovarian, non-small cell lung, head/neck, melanoma and renal). A range of sensitivity was observed in the various cell lines. Approximately 50% of the cell lines showed growth inhibition in response to BIIB036. All of the BIIB036-sensitive cell lines also exhibited growth inhibition in response to Fc-TWEAK. However, cell lines were typically more sensitive to Fc-TWEAK compared with BIIB036, with approximately half of the BIIB036-insensitive lines showing sensitivity to TWEAK. Moreover, Fc-TWEAK was typically more potent than BIIB036 in inhibiting tumor growth in cell lines that were sensitive to both agents. Of note, there was no correlation between sensitivity to tumor cell killing and the relative levels of Fn14 expression (Amatucci A and Michaelson JS, unpublished results), similar to the case of TRAIL receptor expression not correlating with sensitivity to anti-TRAIL antibodies.Citation2
WiDr was identified as one of the most highly sensitive cell lines in the MTT assays. shows that both BIIB036 and Fc-TWEAK can inhibit WiDr cell growth, although BIIB036 is significantly less potent than Fc-TWEAK. To confirm that the inhibition of tumor growth observed in the MTT assay was an apoptotic effect, a TUNEL assay was performed. shows that BIIB036 and Fc-TWEAK treated cells are positive for TUNEL staining. This effect was specific, as the positive staining was abrogated when WiDr cells were treated with BIIB036 or Fc-TWEAK in the presence of soluble Fn14 (data not shown). Compared with WiDr cells, many other cell lines were less sensitive to Fn14 pathway stimulation. NCI-N87 gastric tumor cells (), exhibited modest tumor cell growth inhibition in response Fc-TWEAK, but the cells were not sensitive to BIIB036. MDA-MB-231 breast cancer cells were similarly modestly sensitive to Fc-TWEAK, but not growth inhibited in response to BIIB036 (). Notably, all three cell lines (WiDr, NCI-N87 and MDA-MB-231) showed relatively similar levels of Fn14 cell surface expression as measured by flow cytometry (data not shown). Taken together, our results indicate that the degree of sensitivity to Fn14 pathway stimulation varies among tumor cell lines, and typically cells are more sensitive to TWEAK compared with BIIB036.
Multimerization of BIIB036 enhances anti-tumor activity in vitro.
Because Fc-TWEAK consistently exhibited more potent tumor inhibition compared to BIIB036, we investigated whether increasing the Fn14-binding valency might enhance the activity of BIIB036. We generated a multimeric version of BIIB036 (BIIB036-multimer) by complexing the antibody with soluble protein A; the multimer was purified by size exclusion chromatography to remove uncomplexed components (Sup. Fig. 3). The tumor inhibition activity of BIIB036-multimer was compared to that of BIIB036 and Fc-TWEAK in a panel of 24 cell lines. In the majority of cell lines tested, BIIB036-multimer exhibited enhanced inhibition of tumor cell growth relative to BIIB036; in fact, BIIB036-multimer showed activity in >90% of the cell lines that were sensitive to Fc-TWEAK. Although more potent than BIIB036, BIIB036-multimer nevertheless exhibited less potent anti-tumor activity compared with Fc-TWEAK. The enhanced anti-tumor activity of BIIB036-multimer relative to BIIB036 in WiDr cells is shown in . In the NCI-N87 cell line, which was insensitive to BIIB036, BIIB036-multimer was modestly active, exhibiting slightly less potent activity compared with Fc-TWEAK (). In the MDA-MB-231 cell line, where BIIB036 also had no anti-tumor activity, BIIB036-multimer was only minimally active ().
The increased activity of BIIB036-multimer suggested that in an in vivo context, interaction of the BIIB036 Fc domain with an array of Fc receptors on the cell surface of immune cells might lead to oligomeric structures and result in enhanced anti-tumor activity. To more closely mimic Fc engagement by Fc receptors, we evaluated BIIB036 activity in the presence of a cross-linking secondary antibody. As shown in , WiDr tumor growth inhibition by BIIB036 is increased under these conditions. Notably the enhanced activity of the cross-linked BIIB036 is comparable to that of BIIB036-multimer. In contrast, no activity was observed upon cross-linking of a control Ig. Of note, no tumor inhibition was observed in response to a monomeric Fab fragment derived from BIIB036 (data not shown). Thus, oligomeric BIIB036, generated either by secondary antibody-mediated cross-linking or by complexing via protein A, exhibited enhanced anti-tumor activity in vitro.
Anti-tumor activity is TNF-independent in most cell lines.
As it has been reported that tumor cell killing induced by TWEAK in various cell lines (e.g., Kym-1, SKOV-3, OVCAR) is mediated by TNF,Citation23,Citation24,Citation30 we evaluated whether cell killing induced by TWEAK, BIIB036-multimer or BIIB036 is TNF dependent in a panel of 12 tumor cell lines. TNF dependence was determined by performing the MTT assay in the presence or absence of the anti-TNF antibody, adalimumab (500 pM), which was previously shown to effectively inhibit TNF at concentrations up to 200 pM (data not shown). Interestingly, no diminution in tumor cell inhibition was observed when the MTT assay was performed in the presence of adalimumab in the majority (9/12) of cell lines tested, suggesting that TNF is not required for TWEAK-induced killing in these cell lines. An example of a TNF-independent cell line, WiDr, shows growth inhibition induced by Fc-TWEAK, BIIB036-multimer or BIIB036 is comparable in the presence or absence of adalimumab (). NCI-N87 and MDA-MB-231 were also TNF-independent cell lines (data not shown). In contrast, adalimumab was effective in inhibiting TWEAK-mediated killing in SKOV-3 ovarian tumor cells (), a cell line previously reported to be TNF-dependent.Citation24 We could detect TNF in supernatants and cell extracts of SKOV-3 cells treated with Fc-TWEAK (data not shown), consistent with the previously reported effect of TWEAK in this cell line.Citation24 However, TNF was not detectable in supernatants or cell extracts prepared from other cell lines treated with Fc-TWEAK (data not shown). Interestingly, while TWEAK-induced growth inhibition in Panc-1 cells was TNF-dependent in the MTT assay, soluble TNF was not detectable in supernatants or cell extracts prepared from Panc-1 cells treated with TWEAK (data not shown). Taken together, our findings demonstrate that TWEAK-mediated cell killing is TNF independent in a majority of cell lines tested, including WiDr, NCI-N87 and MDA-MB-231.
BIIB036 exhibits ADCC activity in vitro.
Given that the Fc portion of BIIB036 is a human IgG1, we reasoned that, in addition to effecting cross-linking, the Fc portion of BIIB036 might also contribute to anti-tumor activity by inducing ADCC or CDC. To test this hypothesis, we generated variants of BIIB036 with aglycosylated Fc ends to ablate effector function,Citation31 including aglycosylated hIgG1 (hIgG1agly) with reduced effector function and aglycosylated IgG4 variant (hIgG4Pagly) with no effector function. To assess ADCC activity in vitro, a chromium release assay was performed in the presence of NK effector cells. As shown in and B, ADCC activity was observed in MDA-MB-231 and WiDr tumor cells treated with BIIB036. In contrast, the Fc crippled variants (hIgG1agly and hIgG4Pagly) did not exhibit activity in these assays, as expected ( and B). We also performed CDC assays. No CDC activity was observed with BIIB036 in multiple cell lines tested including WiDr and MDA-MB-231 (data not shown). Thus, an in vitro evaluation of Fc effector function revealed potential ADCC, but not CDC activity of BIIB036.
BIIB036 is efficacious in multiple xenograft models.
Having established that BIIB036 can inhibit tumor cell growth in vitro, we next tested whether it was efficacious in tumor xenograft models in vivo. We first chose to evaluate BIIB036 in the WiDr colon carcinoma model, since WiDr was among the most sensitive cell lines in vitro. Mice bearing WiDr tumors with an average volume of 250 mm3 were treated intraperitoneally (IP) with BIIB036 at doses ranging from 0.9–12.8 mg/kg, or an irrelevant human IgG antibody (12.8 mg/kg), once a week for six weeks. As shown in , BIIB036 exhibited significant tumor growth inhibition at all doses examined (p < 0.001 compared to control from days 25–61). A dose response was observed, with maximal efficacy achieved at 6.4 and 12.8 mg/kg. Maximal tumor inhibition in the 6.4 mg/kg group relative to control IgG treated mice was 94% on day 61. Notably, at doses of 3.2 mg/kg and higher, BIIB036 appeared to induce tumor regression. We also monitored body weights over the course of the study, and BIIB036 was well tolerated with no reduction in body weight observed in any of the treatment groups ().
To understand whether the growth inhibition of BIIB036 was associated with induction of apoptosis in vivo, tumor samples were taken 24 and 48 h after BIIB036 treatment (6.4 mg/kg) and evaluated for caspase-3 cleavage. Substantial staining of cleaved caspase-3, indicative of caspase-3 activation, at both time-points was evident (). We continued to follow the levels of cleaved caspase-3 over time and found a return to baseline by day 7 post-treatment (). Following a second dose of BIIB036, another wave of caspase cleavage was observed, with significant induction at the 24 and 48 h time points (data not shown).
We next evaluated BIIB036 in the NCI-N87 gastric carcinoma xenograft model where in vitro results indicated a lack of sensitivity to the antibody, but modest sensitivity to BIIB036-multimer. Although NCI-N87 tumor cells were not sensitive to BIIB036, the in vitro sensitivity to BIIB036-multimer suggested that the potential interaction of BIIB036 with Fc receptors on immune effector cells might facilitate generation of multimeric forms of BIIB036 thereby promoting anti-tumor activity in vivo. Established NCI-N87 tumor (250 mm3) bearing mice were treated with BIIB036 (at 3.2, 6.4 or 12.8 mg/kg intraperitoneally) or an irrelevant human IgG antibody (12.8 mg/kg) on a weekly dosing schedule for 6 weeks. BIIB036 treatment significantly inhibits tumor growth compared with control antibody (p < 0.01 compared to control from days 21–54), with no dose-dependency at the doses evaluated (). Maximal tumor inhibition in the 6.4 mg/kg dose group compared with the control group was 77% on day 54. Thus, despite the reduced activity observed in vitro in NCI-N87 cells, BIIB036 exhibited potent anti-tumor efficacy in the NCI-N87 xenograft model.
Finally, we tested the efficacy of BIIB036 in a xenograft model using a breast tumor cell line, MDA-MB-231, which lacked in vitro sensitivity to BIIB036 and was only marginally sensitive to BIIB036-multimer. MDA-MB-231 tumor-bearing mice were treated once weekly for six weeks with BIIB036 at 6.4, 12.8 or 25.6 mg/kg or an irrelevant human IgG antibody (25.6 mg/kg).As shown in , significant tumor inhibition was observed at all doses tested. In the 6.4 mg/kg group, maximal tumor inhibition compared to the control group was 74% on day 70. Notably, caspase-3 cleavage was observed in the BIIB036-treated tumors (data not shown). Thus, significant tumor inhibition was observed following BIIB036 treatment despite the lack of anti-tumor activity observed in MDA-MB-231 cells in vitro.
Since MDA-MB-231 cells grown subcutaneously in the flank region metastasize to the lymph nodes, we had the opportunity to assess whether BIIB036 might affect the incidence or size of metastases. Treatment with BIIB036 reduced the incidence of gross metastases to the axillary lymph nodes compared with control Ig (40 versus 100%, respectively; ). The metastatic incidence was confirmed by a similar reduction in micrometastastic lesions, as determined by histological staining with cytokeratin (). In addition, shows that the average size of the metastatic lesion was dramatically reduced in the BIIB036-treated mice compared with the control group (5.5 mm3 versus 709.4 mm3, respectively, p < 0.03). Furthermore, the extent of metastases in the lymph nodes, as quantified by cytokeratin staining, was significantly reduced (7% versus 68.2%, p = 0.02; ). Similar results showing a diminution in incidence and size of metastases were observed in mice treated with BIIB036 at doses ranging from 6.4–25.6 mg/kg (data not shown). In addition, a reduction in mitotic events was also recorded in BIIB036-treated mice (data not shown). Taken together, BIIB036 exhibited strong anti-tumor activity in vivo in multiple xenograft models, despite varying levels of sensitivity in vitro, and also demonstrated anti-metastatic activity in a lymph node metastasis model.
Fc effector function is required for maximal antitumor activity of BIIB036.
Based on our observations that BIIB036 multimerization can enhance anti-tumor activity in vitro and that BIIB036 can induce ADCC in vitro, and given the activity of BIIB036 in xenograft models grown from relatively insensitive cell lines, we posited that the Fc domain likely contributes to BIIB036 activity in vivo. To test this, we compared the in vivo anti-tumor activity of BIIB036 (hIgG1) to the Fc crippled versions (hIgG1agly and hIgG4Pagly).
shows that in the MDA-MB-231 xenograft model, BIIB036 (hIgG1) exhibited 75% tumor growth inhibition compared with the control Ig (p < 0.0001 on day 62), whereas the reduced effector hIgG1agly version exhibited only 57% inhibition of tumor growth (p < 0.001 compared to control Ig on day 62). Moreover, the hIgG4Pagly version, which completely lacks effector function, had further reduced tumor growth inhibitory activity of 29% compared to control (p < 0.05 on day 62). Similar results were observed at other doses tested, including 6.4 and 25.6 mg/kg (data not shown). shows that in the WiDr model, the effectorless hIgG4Pagly version similarly exhibited significantly reduced tumor inhibitory activity (48%, p < 0.05 compared to control on day 56) compared with BIIB036 (91%, p < 0.001 compared with control on day 56). Similar results were obtained in the WiDr model when the different Fc versions of BIIB036 were compared at other doses, such as 3.2 and 6.4 mg/kg (data not shown). Thus, results from xenograft models clearly indicate that the Fc domain of the antibody contributes significantly to the growth inhibitory activity of BIIB036 in vivo.
Discussion
The data presented herein validate the TWEAK/Fn14 pathway as a target in oncology. We show that administration of TWEAK in vivo is efficacious in inhibiting tumor growth in a xenograft model. To therapeutically target the pathway, we developed a humanized agonistic anti-Fn14 antibody, BIIB036, which mimics many activities of TWEAK, including the ability to inhibit tumor growth. Importantly, BIIB036 is highly efficacious in inhibiting tumor growth in multiple xenograft models irrespective of its relative activity in vitro in the various cell lines. We found that the anti-tumor activity of BIIB036 can be enhanced by multimerization of the antibody, suggesting an effect of higher order cross-linking of Fn14 molecules on the cell surface. Indeed, we observed that maximal activity in vivo is achieved only with full effector function of the antibody, suggesting the role of Fc receptors in scaffolding the antibody or ADCC activity in vivo.
BIIB036 was developed as an anti-Fn14 agonistic antibody to mimic the activity of the natural ligand, TWEAK. Indeed, BIIB036 can induce chemokine release, activate NFκB pathway signaling, and, importantly, stimulate tumor cell death. However, BIIB036, the most potent agonist in our panel of anti-Fn14 antibodies, is significantly less potent than TWEAK. Notably, other anti-Fn14 antibodies that have been described also show reduced potency relative to TWEAK.Citation16,Citation32 The decreased potency might be a function of the dimeric interaction of an antibody with the Fn14 receptor in contrast to TWEAK which presumably engages in a trimer-trimer interaction with Fn14. It is also possible that a particular geometrical configuration of Fn14 engagement is unique to TWEAK. Nevertheless, as a therapeutic, BIIB036 is highly efficacious in multiple xenograft models. Moreover, as a monoclonal antibody, BIIB036 is a better drug candidate than soluble TWEAK. The adenoviral system, while delivering abundant quantities of soluble TWEAK, is not feasible for human use. Fc-TWEAK, consisting of a dimerizing Ig Fc domain and a trimerizing extra-cellular domain of TWEAK, can potentially fold into several oligomeric forms as observed by size exclusion chromatography (Hsu Y-M, unpublished observations). The minimal structure of Fc-TWEAK is expected to be a hexamer consisting of two TWEAK trimers and three dimeric Ig Fc domains. This complexity and the presence of heterogeneous populations of Fc-TWEAK, make it a poor drug candidate. Recombinant soluble TWEAK would also not be a desirable drug candidate due to an extremely short half-life in vivo (Burkly LC, data not shown). Finally, BIIB036 may be a safer alternative because TWEAK administration in vivo has been associated with significant weight loss in mice (reviewed in ref. Citation33 and Michaelson JS unpublished observations), whereas no weight loss was observed in response to BIIB036 treatment even with weekly administration at a high dose of 25.6 mg/kg.
Fn14 is emerging as a therapeutic target for the treatment of cancer, joining other TNF family member receptors, such as the CD40 and TRAIL receptors, that are currently under investigation. Similar to the robust anti-tumor activity we observed in xenograft models in response to TWEAK or BII036 treatment, efficacy was observed with recombinant soluble TRAIL and agonistic antibodies to the TRAIL receptors, (TRAIL-R1 (DR4) and TRAIL-R2 (DR5)) in numerous tumor xenograft models.Citation2 Ongoing clinical trials with soluble TRAIL and anti-TRAIL receptor antibodies have thus far demonstrated acceptable safety and tolerability in humans,Citation3 thereby providing precedence for safety with systemic administration of TNF family agonists in man. However, one notable distinction between Fn14 and TRAIL receptors is that Fn14 does not contain a death domain which is responsible for DR4- and DR5-mediated assembly of the death inducing signaling complex (DISC).Citation1 The differential mechanism of inducing tumor cell death, along with the distinct pattern of receptor expression in human tumors, may allow for a differentiated opportunity for Fn14 agonists in the clinic.
In the absence of a death domain in the cytoplasmic region of Fn14, it is less clear how stimulation of the Fn14 pathway leads to death of tumor cells. We showed that, like TWEAK, BIIB036 is capable of inducing NFκB pathway activation. An early report suggested that TWEAK-induced killing is TNF-dependent in Kym-1 cells.Citation23 However, Nakayama et al.Citation22 reported that TWEAK-induced death of HT-29, HS-C or KATO-III cells does not exhibit TNF dependence, so it appears that not all Fn14-mediated cell death is TNF-dependent. Our analysis showed that tumor cell killing induced by Fn14 agonists is not TNF dependent in a majority of tumor cell lines evaluated. Specifically, we showed that only three (SKOV-3, MCF7 and Panc-1) of 12 cell lines were TNF dependent. Interestingly, Vince et al. showed that TWEAK killing is TNF dependent in the three cell lines (Kym-1, SKOV-3 and OVCAR) that they tested, and TNF was detectable in the supernatants and cell extracts of TWEAK-treated cells. However, in our evaluation of six cell lines, we could only detect TNF in supernatants or cell extracts from SKOV-3-treated cells, suggesting that the cell lines described in Vince et al. may not be representative. Studies in several other cell types have also documented that TWEAK signaling is independent of TNF, including pro-inflammatory responses in synoviocytes, dermal fibroblasts and keratinocytes.Citation34,Citation35 Taken together, the results suggest that TWEAK signaling in general, and TWEAK-induced tumor cell killing in particular, may be predominantly independent of TNF.
We observed robust anti-tumor efficacy with BIIB036 in multiple xenograft models regardless of relative activity in vitro. Notably, in the NCI-N87 and MDA-MB-231 cell lines, BIIB036 exhibited negligible anti-tumor activity in MTT assays, yet in vivo a 75% reduction in tumor growth was observed. This lack of correlation between in vitro and in vivo sensitivity highlights the limitations of making predictions based on cell-based assays alone. Stromal cells, tumor architecture and host-tumor interactions likely contribute to the anti-tumor response in vivo. Indeed, one explanation for the enhanced activity that we observed in vivo with BIIB036 is the contribution of Fc effector function. Fc receptor expressing immune effector cells can bind the Fc portion of BIIB036 and may effectively induce ADCC, as we showed in the in vitro reconstituted ADCC assay. Moreover, binding of BIIB036 by Fc receptor expressing cells may result in effective multimerization of BIIB036 leading to higher order cross-linking of Fn14 molecules and thereby an enhanced anti-tumor effect. Indeed, we show that multimerization through protein A or secondary antibody cross-linking of BIIB036 resulted in significantly enhanced activity in MTT assays and signaling studies (Amatucci A and Michaelson JS, unpublished observations). This is consistent with a recent reportCitation36 showing that Fcγ receptors are capable of driving antibody-dependent signaling in target cells in the context of agonistic DR5 and CD40 antibodies, indicating that Fcγ receptor-dependent mechanisms contribute to the activity of antibodies directed against other TNF family member receptors as well. Our in vivo studies, demonstrating that effectorless versions of BIIB036 exhibited reduced activity in xenograft models, confirmed the contribution of Fc effector function to BIIB036 activity. Residual tumor inhibition was nevertheless observed with the fully effectorless versions of BIIB036 in xenograft models, suggesting that not all of the activity is Fc-mediated. Our data suggest that this is due to signaling through Fn14 on tumor cells (Michaelson JS, unpublished observations), although, given species cross-reactivity of BIIB036 to mouse Fn14, a direct effect of BIIB036 binding to non-tumor cells cannot be ruled out.
It is important to note that a monoclonal antibody targeting Fn14, PDL192 (Facet Biotech/Abbott), is currently undergoing evaluation in a Phase 1 clinical study. One difference between PDL192 and BIIB036 is that the binding affinity, as determined by Biacore, is five times greater for BIIB036 compared to that of PDL192.Citation16 While it is possible that there may not be a direct correlation between binding affinity and activity, nevertheless there could be implications in terms of relative competition for Fn14 binding to TWEAK. Like BIIB036, PDL192 exhibited anti-tumor activity in multiple xenograft models.Citation16 BIIB036 was administered at significantly lower doses and on a less frequent dosing schedule compared with PDL192, suggesting that BIIB036 may be a more potent anti-tumor agent. It is also likely that BIIB036 and PDL192 have different binding epitopes on Fn14. Indeed, a notable feature of BIIB036 is an ability to bind Fn14 across species, with equivalent affinity to rodent, monkey and human Fn14. Perhaps this is due to the fact that BIIB036 was selected from a panel of anti-Fn14 antibodies that were generated in Fn14-deficient mice, which would favor cross-reactivity to rodent Fn14. The species cross-reactivity is critical for drug development because it provides a path forward for informative toxicology studies in rodents and non-human primates.
In considering BIIB036 as a therapeutic for the treatment of cancer, it is important to note that high expression of Fn14 has been reported in multiple tumor types, including breast, pancreatic, glioma and non-small cell lung.Citation7,Citation11–Citation16 Likewise, our xenograft studies, in which we observed robust efficacy, also spanned a range of tumor types, including colon, breast and gastric. Moreover, upregulated Fn14 expression was specifically observed in liver and bone metastases,Citation16 and indeed BIIB036 demonstrated impressive anti-metastatic activity in the MDA-MB-231 model. In several tumor types, including glioma, breast and esophageal, a significant correlation between Fn14 expression and tumor grade and/or prognosis has been noted.Citation13–Citation15,Citation17,Citation37 The upregulated expression of Fn14 in tumors may be due to some of the other features of the TWEAK/Fn14 pathway, such as the ability to promote migration, invasion and survival.Citation38 Interestingly, although these tumor promoting features, as well as a capacity to induce angiogenesis,Citation9,Citation10,Citation39–Citation42 have been attributed to TWEAK, it is important to emphasize that no enhancement in tumor growth was observed following BIIB036 treatment in any xenograft models tested to date. Thus, the fact that Fn14 is expressed in a wide variety of cancer types, and particularly in more aggressive tumors, together with our observation of in vivo anti-tumor efficacy in multiple tumor types, suggest that BIIB036 could be efficacious in the treatment of a broad range of tumors, and specifically in patients with advanced disease who are most in need of effective therapy.
Materials and Methods
Cell lines and reagents.
WiDr cells (NCI) were grown in MEM Earle's supplemented with 10% FBS, 2 mM L-glutamine, 1x non-essential amino acids, 0.5 mM sodium pyruvate. MDA-MB-231 cells (NCI) were grown in MEM Earle's or RPMI-1640 supplemented with 10% FBS. NCI-N87 cells (ATCC) were grown in RPMI-1640 medium with 10% FBS.
BIIB036-multimer was made by combining 5.58 mg BIIB036 with 0.35 mg Protein A (Pierce) in PBS overnight at 4°C, followed by purification on a Superdex 200 column (GE Healthcare) by AKTA FPLC (GE Healthcare).
The human IgG4 heavy chain was stabilized by mutation of hinge Kabat residue 228 from Ser to Pro (hIgG4P).Citation26 The hIgG1agly and hIgG4Pagly heavy chains were made by mutation of the CH2's N-linked glycosylation site Asn-Ser-Thr (Kabat residues 297–299): residue 299 from Thr to Ala. Site-directed mutagenesis was performed using Stratagene's Quikchange mutagenesis kit following the manufacturer's recommended protocols. Mutations were confirmed by sequencing the hBIIB036 heavy chain cDNA insert in the relevant expression vectors.
The control antibody (control Ig) used in these studies was IDEC151.Citation27
FACS binding studies.
Full-length Fn14 cDNAs (human, cynomolgus monkey, rat and mouse) were engineered to remove extraneous 5′ and 3′ UTRs and add an identical optimized Kozak sequence, then were subcloned into pNE001, a fully sequence-confirmed pUC-based EBV expression vector derived from the Invitrogen expression vector pCEP4, in which heterologous gene expression is controlled by a CMV-IE promoter and an SV40 polyadenylation signal, but lacking the EBNA gene and the hygromycin resistance gene. The Fn14 expression vectors were co-transfected into 293E cells at a 1:1 molar ratio with an EBV expression vector carrying an EGFP reporter. Cells were transfected using Qiagen's Effectene reagent, following the manufacturer's recommended protocol. Cells were used in FACS at 2 days post-transfection, staining with a dilution titration series of BIIB036 (detected with PE-conjugated goat anti-human IgG secondary antibody) and gating on green EGFP-positive living cells.
The FACS competition experiment was performed by titrating the antibodies (starting at 100 nM) followed by the addition of equal volume of a static concentration of recombinant soluble TWEAK (40 ng/mL). This combination was incubated with WiDr cells. Detection was with a PE labeled goat anti-mouse-Fc (JacksonImmuno#115-116-071). The geometric mean was determined on ungated cells and plotted.
Chemokine release assays.
A375 (2 × 104) human melanoma cells were grown in 96-well plates in the presence of varying concentrations of Fc-TWEAK,Citation12 mBIIB036 or control Ig. After 36 h, the supernatants were collected and IL-8 release was measured by ELISA using the human IL-8 DuoSet ELISA kit (R&D Systems).
Signaling studies.
Western blots were performed according to standard procedures. The following antibodies were used: NFκB-p52/p100 (Cell Signaling #4882) and GAPDH (Ambion #AM4300).
Tumor cell killing assays.
(3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide (MTT) assays were performing by seeding cells in a 96-well plate in media containing 80 U/ml human IFNγ (Biogen Idec) along with a serial dilution of Fc-TWEAK,Citation12 BIIB036-multimer, BIIB036 or control Ig in triplicate. In some wells, the anti-TNF inhibitor adalimumab (Abbott) was added at a concentration of 500 pM. Cells were incubated for 3–4 days and developed using Cell Titer 96 Aqueous One Solution Cell Proliferation Assay (Promega). The percentage survival was determined as follows: % survival = (OD of treated wells/average OD of the untreated wells) × 100. All samples were run in triplicate, and data is presented with standard deviation.
For cross-linking studies, 16 µg/mL goat anti-human IgG F(ab')2 (Jackson Immuno#109-006-098) was combined with an equal volume of 12 µg/mL BIIB036 followed by nine serial 1:3 dilutions of this mixture in a 96-well plate. This titration resulted in a 2× solution of a 1:4 (molar) crosslinker:BIIB036 ratio, which was added to cells 1:1 by volume as above.
For terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assays, cells were grown in 10-centimeter plates and treated with Fc-TWEAKCitation12 (50 ng/ml) or BIIB036 (500 ng/ml) in the presence of IFNγ for 48 h. The assay was performed using the In-Situ Cell Death Detection Kit, Fluorescein-TUNEL (Roche) according to the manufacturer's instructions.
ADCC assays.
An enriched population of natural killer (NK) cells was purified from human peripheral blood monocytes (PBMC) using biotin-conjugated antibodies and microbeads cocktails (Miltenyi Biotec NK Cell Isolation Kit, Auburn, CA). The NK cells were grown overnight at 3 × 106/mL in RPMI with 10% Fetal Bovine Serum (Hyclone) and 20 units/mL of recombinant human interleukin-2 (IL-2) (Invitrogen). Target cells (WiDr or MDA-MB-231) were labeled with 150 µCi of 51Cr (Amersham Pharmacia Biotech) per 1 × 106 cells for 1 h at 37°C. The cells were washed four times, and then incubated in triplicate in 96-well plates in the presence of test antibodies at varying concentrations and effector (NK) cells at a 5:1 effector to target ratio for 4 h at 37°C. Target cells incubated with complete medium was used to determine spontaneous release, and target cells incubated with 0.5% Triton X 100 (Sigma Aldrich Corp.) was used to determine maximum release. 51Cr released in the culture supernatant was measured by a gamma counter (ISODATA). The cytotoxicity was expressed as the percentage of specific lysis and calculated as follows: 1 − [51Cr release of test samples − spontaneous 51Cr release/maximum 51Cr release − spontaneous 51Cr release] × 100.
Tumor studies.
All in vivo procedures were performed in accordance with Biogen Idec Institutional Animal Care and Use Committee guidelines. All mice (Charles River Laboratories, Inc.) were 8–12 weeks old at the time of cell implantation. MDA-MB-231 breast cancer cells (5 × 106 cells per mouse) or NCI-N87 gastric cancer cells (2 × 106 cells per mouse) in 200 µl media containing 50% Matrigel™ Matrix (BD Biosciences) were implanted subcutaneously into the right flank of female CB17 SCID mice (CB17-Prkdcscid/NCrCrl). WiDr colon carcinoma cells (5 × 106 cells per mouse) in 200 µl media were inoculated into the female nu/nu mice (Crl:NU-Foxn1nu). Tumors were size-matched across groups prior to initiation of treatment.
For the adenoviral delivery study, a single intratumoral injection of adeno-TWEAK,Citation9 or the control adeno-GFP, (1 × 1011 viral particles) was administered to the mice (n = 7 mice per group) at day 9, when tumors had reached approximately 90 mm3. At the conclusion of the study (day 76 post-implantation), the tumors were weighed and then formalin-fixed and embedded in paraffin for histological analysis. For the Fc-TWEAK study, bi-weekly intratumoral injections of 100 µg of human Fc-TWEAKCitation12 or vehicle control (PBS) (n = 8 mice per group) was begun at day 19 post-implantation. At the conclusion of the study (day 99 post-implantation), the tumors were weighed.
All antibodies (BIIB036; control Ig; hIgG1agly and hIgG4Pagly versions of BIIB036) were administered via intraperitoneal injection, once a week for 6 weeks. When average tumor volume reached ∼220 mm3, mice were assigned to groups (n = 10 per group for WiDr and NCI-N87, n = 9 per group for MDA-MB-231) and antibody treatments were initiated.
During the course of the xenograft studies, tumors were measured twice a week using digital calipers and body weights were recorded. Tumor volume was calculated using the formula: L × W2/2 = mm3, where L is the length and W the tumor width. One way repeated measures ANOVA followed by Dunett's post test was used to evaluate differences between test and control groups across the study. Student's t-test (two-tail) was used to determine statistical significance of differences in mean tumor volume of test groups compared to control groups on individual measurement days.
Histological analysis.
Paraffin embedded tumor sections were alternatively stained with hematoxylin and eosin (H&E); anti-Ki-67 antibody to detect proliferating cells (mouse anti-human Ki-67); or stained with the TUNEL assay (ApopTag Peroxidase In Situ Apoptosis Detection Kit, Intergen Co.) to detect apoptotic cells.
Tumor sections (3 µm) were stained with a rabbit anti-cleaved Caspase 3 monoclonal antibody (Cell Signaling, Danvers, MA) or control rabbit IgG (Vector Lab) on a Ventana Discovery XT Instrument. The amount of cleaved Caspase 3 staining was quantified using Aperio's Spectrum Software Image Analysis algorithm for nuclear stain.
For the metastasis study, at terminal sacrifice (40 days post final dosing of antibody), axillary lymph nodes of mice were examined grossly for the presence of metastatic lesions and the volume of the lymph nodes was measured using calipers. Nodes from a representative set of mice in each group were harvested and placed in 10% neutral buffered formalin. The samples were processed and stained with anti-pan cytokeratin (Ventana) for histopathologic evaluation to determine presence of epithelial cells, i.e., tumor micro-metastases. The lymph nodes were examined for cytokeratin staining at 100x magnification. The lymph nodes were measured in situ and the volume calculated using the same formula as for tumor volume, L × W2/2 = mm3. Left and right axillary lymph node volumes of each animal were combined. The mean extent of micrometastases was scored as the percentage of the lymph node that stained positive with cytokeratin. Lymph nodes were scored blindly. The mean percentages were calculated for each group, and the p value was calculated by Wilcoxon signed rank test.
Conflict of Interest
The authors acknowledge a conflict of interest as employees and stock holders of Biogen Idec.
Figures and Tables
Figure 1 Adeno-TWEAK or Fc-TWEAK treatment is efficacious in tumor xenograft model. (A) Efficacy of adeno-TWEAK treatment in MDA-MB-231 xenograft model is shown. MDA-MB231 cells were grown subcutaneously in nude mice and a single dose of adeno-TWEAK or adeno-GFP was administered intratumorally at day 9 post-implantation. Tumor volume is plotted over time, indicating marked reduction in tumor size of adeno-TWEAK treated mice. Statistical significance (p < 0.007) was achieved at each time point beginning at day 23 and continuing through the end of the study (day 76). (B) Actual tumor weights were determined upon dissection at termination of the study (day 76) as shown, indicating significantly reduced tumor mass in the adeno-TWEAK treated group compared with the adeno-GFP group. Shown are averages with standard deviation. Statistical significance (t-test) is shown. (C) H&E staining of tumors at termination of the study shows reduced tumor tissue in the adeno-TWEAK treated group compared with the adeno-GFP treated group. Ki-67 staining reveals reduced proliferation in tumors from the adeno-TWEAK treated mice compared with adeno-GFP treated group. TUNEL staining demonstrates increased levels of apoptotic cells in tumors from the adeno-TWEAK treated mice compared with the adeno-GFP treated group. (D) Efficacy of Fc-TWEAK treatment in MDA-MB-231 xenograft model is shown. Nude mice bearing MDA-MB-231 tumors were given biweekly intratumoral administration of Fc-TWEAK or vehicle control beginning at day 19 post-tumor cell implantation. Tumor volume is plotted over time, indicating a marked reduction in tumor size of Fc-TWEAK treated mice. Statistical significance was achieved (p < 0.02) at each time point beginning at day 54 and continuing through termination of the study (day 99). (E) Comparison of tumor weights as determined upon dissection at termination of the study (day 99) is shown, indicating significantly reduced weights in the Fc-TWEAK treated group. Shown are averages with standard deviation. Statistical significance (t-test) is shown.
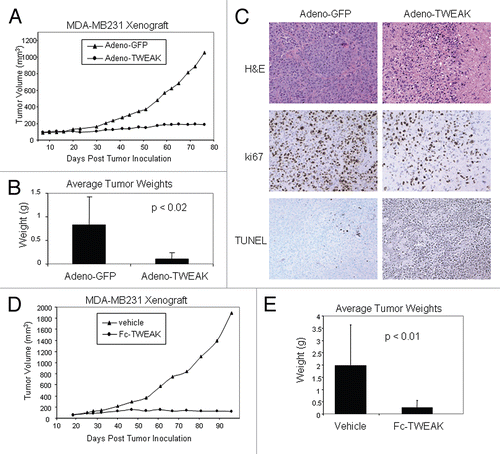
Figure 2 BIIB036 binds Fn14 and blocks TWEAK binding to Fn14. (A) Direct FACS binding assay of BIIB036 to 293E cells transiently transfected with full-length Fn14 from various species is shown. An empty vector (“control”) was included as a negative control in this experiment. BIIB036 exhibits similar binding to human, cynomolgous monkey, rat and mouse Fn14. The geometric mean fluorescence intensity (MFI) is plotted. (B) FACS competition assay of BIIB036 binding to Fn14-expressing WiDr cells in the presence of TWEAK is shown. WiDr cells were incubated with mFc-TWEAK in the presence of varying concentrations of BIIB036, human Ig control, or recombinant soluble TWEAK. Binding of mFc-TWEAK to the cells was detected with a labeled anti-mouse Ig antibody. mFc-TWEAK binding was inhibited at increasing concentrations of BIIB036 (or recombinant TWEAK). The geometric mean fluorescence intensity (MFI) is plotted.
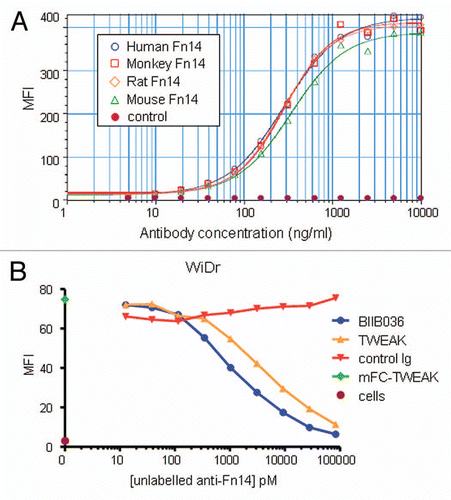
Figure 3 BIIB036 exhibits agonistic activity. (A) IL-8 release assay in A375 cells is shown. Supernatants from A375 cells incubated for 36 h with mBIIB036, but not control Ig, showed increased levels of IL-8, as measured by ELISA, in a dose-dependent fashion. Fc-TWEAK was included as a positive control in this experiment. (B) Protein extracts were prepared from WiDr and MDA-MB-231 cells incubated with BIIB036 (1 µg/ml) or Fc-TWEAK (100 ng/ml) for 24 h. Western blotting with an anti-NFκB-p52/p100 antibody detects p100 and the p52 cleavage product. p52 is apparent in BIIB036-treated WiDr and MDA-MB231 cells similar to the induction observed in response to Fc-TWEAK.
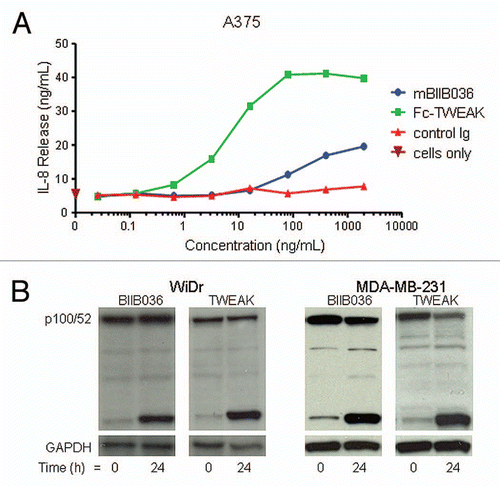
Figure 4 BIIB036 can induce tumor cell killing in vitro. (A) MTT assay was performed with WiDr cells treated with a range of doses of Fc-TWEAK, BIIB036 or human Ig control antibody in the presence of IFNγ for four days. Results are graphed as percent survival relative to control IFNγ treated WiDr cells. (B) A TUNEL assay was performed with WiDr cells treated with BIIB036 for 48 h in the presence of IFNγ. TUNEL-positive cells are observed in BIIB036-treated but not control cells. Fc-TWEAK was included in this experiment as a positive control. (C) MTT assay was performed with NCI-N87 cells treated with a range of doses of Fc-TWEAK, BIIB036-multimer, BIIB036 or Ig control in the presence of IFNγ for 3 days. Results are graphed as percent survival relative to control IFNγ treated NCI-N87 cells. (D) MTT assay was performed with MDA-MB-231 cells treated with a range of doses of Fc-TWEAK, BIIB036-multimer, BIIB036 or Ig control in the presence of IFNγ for four days. Results are graphed as percent survival relative to control IFNγ treated MDA-MB-231 cells.
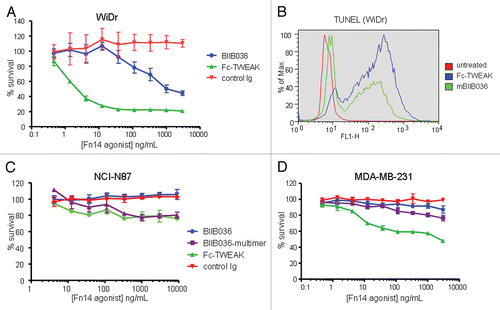
Figure 5 Multimerization of BIIB036 results in enhanced anti-tumor activity. MTT assay was performed with WiDr cells in the presence of IFNγ treated with a range of doses of BIIB036 or control Ig, in the presence or absence of a secondary cross-linking antibody, or with BIIB036-multimer, for 4 days. Results are graphed as percent survival relative to control IFNγ treated WiDr cells.
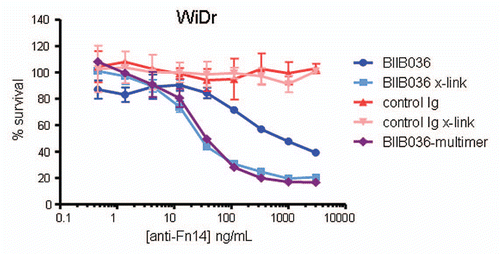
Figure 6 Cell killing in multiple tumor cell lines is TNF independent. MTT assays in (A) WiDr cells and (B) SKOV-3 cells were performed in the presence or absence of the anti-TNF inhibitor, Adalimumab (500 pM). Cell survival is plotted as a function of concentration of Fc-TWEAK, BIIB036-multimer or BIIB036. Only in SKOV-3 cells, but not in WiDr cells, is tumor cell killing inhibited in the presence of Adalimumab.
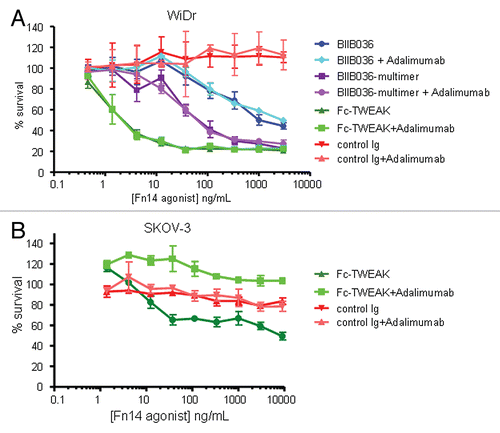
Figure 7 BIIB036 exhibits ADCC activity in vitro. ADCC assays were performed in (A) MDA-MB-231 and (B) WiDr cells with a 5:1 ratio of NK to target tumor cells in the presence of increasing concentrations of antibody. BIIB036 (hIgG1) was tested and compared to other Fc versions of the antibody (hIgG1agly or hIgG4Pagly) or control human Ig. Percent lysis is shown. Only BIIB036 exhibited activity in this assay.
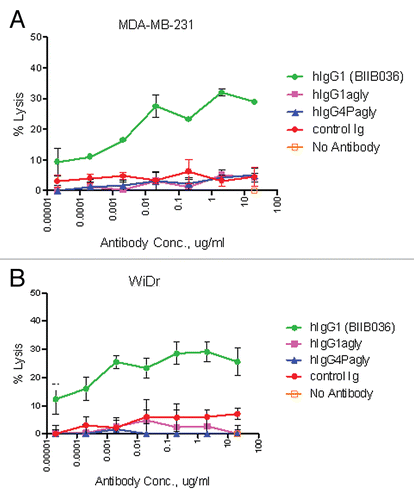
Figure 8 BIIB036 inhibits tumor growth and induces apoptosis in WiDr colon tumor xenograft model. (A) Efficacy of BIIB036 treatment in WiDr colon carcinoma xenograft model is shown. Tumor volume as a function of time in WiDr xenograft model is plotted. Nude mice implanted with WiDr tumor cells were treated with varying doses of BIIB036 (12.8, 6.4, 3.2, 1.8 or 0.9 mg/kg) or human Ig control (12.8 mg/kg) on a weekly basis for 6 weeks starting when the tumors were approximately 250 mm3. Data are mean ± SEM of 10 mice per group. Dose-responsive anti-tumor activity is observed, with significant efficacy evident at all doses relative to control group (p value < 0.001 on day 61 for dose groups 3.2 mg/kg and higher). (B) Percent change in body weight of each group of mice from the WiDr xenograft experiment in (A) is plotted as a function of time. No reduction in body weight was observed in any of the treatment groups. (C) WiDr tumors harvested from mice treated with mBIIB036 (6.4 mg/kg) were sectioned and stained for cleaved caspase-3. Increased cleaved caspase-3 staining is observed at 24 and 48 h post-BIIB036 treatment. (D) Graphical presentation of cleaved caspase-3 staining in WiDr tumors from mice at 1–7 days following treatment with mBIIB036 (6.4 mg/kg). The percent of nuclei staining positive for cleaved caspase-3 is plotted. Average values with standard deviation for n = 5 mice per group are shown.
Figure 9 BIIB036 inhibits tumor growth in NCI-N87 gastric and MDA-MB-231 breast tumor xenograft models and reduces lymph node metastases. (A) Efficacy of BIIB036 treatment in NCI-N87 gastric xenograft model is shown. Nude mice implanted with NCI-N87 tumor cells were treated with varying doses of BIIB036 (3.2, 6.4 and 12.8 mg/kg) or human Ig control (12.8 mg/kg) on a weekly basis for 6 weeks starting when the tumors were approximately 250 mm3. Data are mean ± SEM of ten mice per group. Statistically significant robust anti-tumor activity is observed at all doses relative to control Ig over the entire dosing period (p < 0.0001). (B) Efficacy of BIIB036 treatment in MDA-MB-231 breast xenograft model is shown. Nude mice implanted with MDA-MB-231 tumor cells were treated with varying doses of BIIB036 (6.4, 12.8 and 25.6 mg/kg) or human Ig control (25.6 mg/kg) on a weekly basis for 6 weeks starting when the tumors were approximately 250 mm3. Robust anti-tumor activity compared to the control is observed at all doses (p value < 0.000001 for all BIIB036 groups relative to control group at day 58, one week following the final dose). For all dose groups, statistically significant anti-tumor efficacy was maintained through the entire dosing period and beyond (p < 0.0001 compared to control from days 16 to 70). (C) Metastatic tumor burden in the lymph nodes is shown. Each bar represents the mean combined volume of right and left axillary lymph nodes. A dramatic reduction in metastatic volume is observed in BIIB036-treated mice compared with mice treated with human Ig control (p < 0.03, t-test).
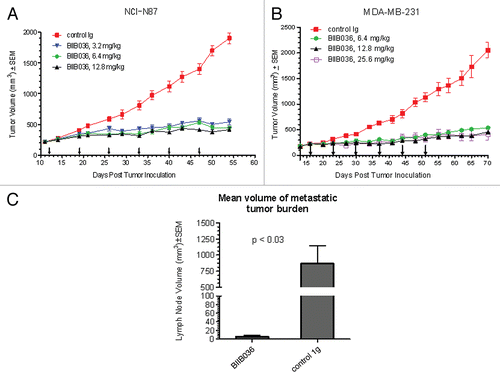
Figure 10 BIIB036 exhibits superior anti-tumor activity in vivo relative to versions of the antibody with reduced effector function. (A) MDA-MB-231 and (B) WiDr tumor xenograft model were used to compare the efficacy of different Fc versions of BIIB036. BIIB036 (hIgG1) exhibited superior efficacy compared with the other Fc versions of the antibody (hIgG1agly and hIgG4Pagly) when dosed weekly for a 6 week period. Control human Ig (dosed at 12.8 mg/kg in the WiDr study and 25.6 mg/kg in the MDA-MB-231 study) was included as a negative control.
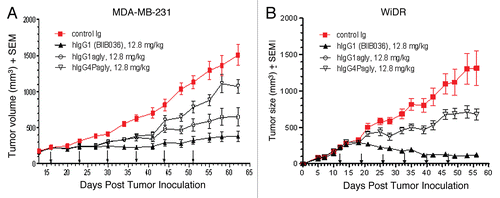
Table 1 Effect of BIIB036 on metastasis in MDA-MB-231 breast tumor model
Additional material
Download Zip (1.7 MB)Acknowledgments
The authors acknowledge Bob Dunstan for caspase-3 and cytokeratin staining and scoring, Dian Olson for contribution to cell viability assays, and Michael Corbley for his many insights.
References
- Daniel D, Wilson NS. Tumor necrosis factor: renaissance as a cancer therapeutic?. Curr Cancer Drug Targets 2008; 8:124 - 131
- Buchsbaum DJ, Zhou T, Lobuglio AF. TRAIL receptor-targeted therapy. Future Onc 2006; 2:493 - 508
- Wiezorek J, Holland P, Graves J. Death receptor agonists as a targeted therapy for cancer. Clin Cancer Res 2010; 16:1701 - 1708
- Burkly LC, Michaelson JS, Hahm K, Jakubowski A, Zheng TS. TWEAKing tissue remodeling by a multifunctional cytokine: role of TWEAK/Fn14 pathway in health and disease. Cytokine 2007; 40:1 - 16
- Chicheportiche Y, Bourdon PR, Xu H, Hsu YM, Scott H, Hession C, et al. TWEAK, A new secreted ligand in the tumor necrosis factor family that weakly induces apoptosis. J Biol Chem 1997; 272:32401 - 32410
- Wiley SR, Winkles JA. TWEAK, a member of the TNF superfamily, is a multifunctional cytokine that binds the TweakR/Fn14 receptor. Cytokine Growth Factor Rev 2003; 14:241 - 249
- Feng SL, Guo Y, Factor VM, Thorgeirsson SS, Bell DW, Testa JR, et al. The Fn14 immediate-early response gene is induced during liver regeneration and highly expressed in both human and murine hepatocellular carcinomas. A J Pathol 2000; 156:1253 - 1261
- Girgenrath M, Weng S, Kostek CA, Browning B, Wang M, Brown SA, et al. TWEAK, via its receptor Fn14, is a novel regulator of mesenchymal progenitor cells and skeletal muscle regeneration. EMBO J 2006; 25:5826 - 5839
- Jakubowski A, Ambrose C, Parr M, Lincecum JM, Wang MZ, Zheng TS, et al. TWEAK induces liver progenitor cell proliferation. J Clin Invest 2005; 115:2330 - 2340
- Wiley SR, Cassiano L, Lofton T, Davis-Smith T, Winkles JA, Lindner V, et al. A novel TNF receptor family member binds TWEAK and is implicated in angiogenesis. Immunity 2001; 15:837 - 846
- Han H, Bearss DJ, Browne LW, Calaluce R, Nagle RB, Von Hoff DD. Identification of differentially expressed genes in pancreatic cancer cells using cDNA microarray.[ erratum appears in Cancer Res 2002; 62:4532]. Cancer Res 2002; 62:2890 - 2896
- Michaelson JS, Cho S, Browning B, Zheng TS, Lincecum JM, Wang MZ, et al. Tweak induces mammary epithelial branching morphogenesis. Oncogene 2005; 24:2613 - 2624
- Tran NL, McDonough WS, Donohue PJ, Winkles JA, Berens TJ, Ross KR, et al. The human Fn14 receptor gene is upregulated in migrating glioma cells in vitro and overexpressed in advanced glial tumors. A J Pathol 2003; 162:1313 - 1321
- Tran NL, McDonough WS, Savitch BA, Fortin SP, Winkles JA, Symons M, et al. Increased fibroblast growth factor-inducible 14 expression levels promote glioma cell invasion via Rac1 and nuclear factor{kappa} B and correlate with poor patient outcome. Cancer Res 2006; 66:9535 - 9542
- Wang S, Zhan M, Yin J, Abraham JM, Mori Y, Sato F, Xu Y, et al. Transcriptional profiling suggests that Barrett's metaplasia is an early intermediate stage in esophageal adenocarcinogenesis. Oncogene 2006; 25:3346 - 3356
- Culp PA, Choi D, Zhang Y, Yin J, Seto P, Ybarra SE, et al. Antibodies to TWEAK receptor inhibit human tumor growth through dual mechanisms. Clin Cancer Res 2010; 16:497 - 508
- Willis AL, Tran NL, Chatigny JM, Charlton N, Vu H, Brown SA, et al. The fibroblast growth factor-inducible 14 receptor is highly expressed in HER2-positive breast tumors and regulates breast cancer cell invasive capacity. Mol Cancer Res 2008; 6:725 - 734
- Brown SA, Richards CM, Hanscom HN, Feng SL, Winkles JA. The Fn14 cytoplasmic tail binds tumournecrosis-factor-receptor-associated factors 1, 2, 3 and 5 and mediates nuclear factorkappaB activation. Biochem J 2003; 371:395 - 403
- Saitoh T, Nakayama M, Nakano H, Yagita H, Yamamoto N, Yamaoka S. TWEAK induces NFκB2 p100 processing and long lasting NFκB activation. J Biol Chem 2003; 278:36005 - 36012
- Bover LC, Cardo-Vila M, Kuniyasu A, Sun J, Rangel R, Takeya M, et al. A previously unrecognized protein-protein interaction between TWEAK and CD163: potential biological implications. J Immunol 2007; 178:8183 - 8194
- Nakayama M, Harada N, Okumura K, Yagita H. Characterization of murine TWEAK and its receptor (Fn14) by monoclonal antibodies. Biochem Biophys Res Comm 2003; 306:819 - 825
- Nakayama M, Ishidoh K, Kayagaki N, Kojima Y, Yamaguchi N, Nakano H, et al. Multiple pathways of TWEAK-induced cell death. J Immunol 2002; 168:734 - 743
- Schneider P, Schwenzer R, Haas E, Muhlenbeck F, Schubert G, Scheurich P. TWEAK can induce cell death via endogenous TNF and TNF receptor 1. Eur J Immunol 1999; 29:1785 - 1792
- Vince JE, Chau D, Callus B, Wong WW, Hawkins CJ, Schneider P, et al. TWEAK-FN14 signaling induces lysosomal degradation of a cIAP1-TRAF2 complex to sensitize tumor cells to TNFalpha. J Cell Biol 2008; 182:171 - 184
- Wilson CA, Browning JL. Death of HT29 adenocarcinoma cells induced by TNF family receptor activation is caspase-independent and displays features of both apoptosis and necrosis. Cell Death Diff 2002; 9:1321 - 1333
- Angal S, King DJ, Bodmer MW, Turner A, Lawson AD, Roberts G, et al. A single amino acid substitution abolishes the heterogeneity of chimeric mouse/human (IgG4) antibody. Mol Immunol 1993; 30:105 - 108
- Dong J, Demarest SJ, Sereno A, Tamraz S, Langley E, Doern A, et al. Combination of two insulin-like growth factor-I receptor inhibitory antibodies targeting distinct epitopes leads to an enhanced antitumor response. Mol Cancer Ther 2010; 9:2593 - 2604
- Holler N, Tardivel A, Kovacsovics-Bankowski M, Hertig S, Gaide O, Martinon F, et al. Two adjacent trimeric Fas ligands are required for Fas signaling and formation of a death-inducing signaling complex. Mol Cell Biol 2003; 23:1428 - 1440
- Co MS, Avdalovic NM, Caron PC, Avdalovic MV, Scheinberg DA, Queen C. Chimeric and humanized antibodies with specificity for the CD33 antigen. J Immunol 1992; 148:1149 - 1154
- Wicovsky A, Salzmann S, Roos C, Ehrenschwender M, Rosenthal T, Siegmund D, et al. TNF-like weak inducer of apoptosis inhibits proinflammatory TNF receptor-1 signaling. Cell Death Differ 2009; 16:1445 - 1459
- Sarmay G, Lund J, Rozsnyay Z, Gergely J, Jefferis R. Mapping and comparison of the interaction sites on the Fc region of IgG responsible for triggering antibody dependent cellular cytotoxicity (ADCC) through different types of human Fc gamma receptor. Mol Immunol 1992; 29:633 - 639
- Nakayama M, Ishidoh K, Kojima Y, Harada N, Kominami E, Okumura K, et al. Fibroblast growth factor-inducible 14 mediates multiple pathways of TWEAK-induced cell death. J Immunol 2003; 170:341 - 348
- Dogra C, Changotra H, Wedhas N, Qin X, Wergedal JE, Kumar A. TNF-related weak inducer of apoptosis (TWEAK) is a potent skeletal muscle-wasting cytokine. FASEB J 2007; 21:1857 - 1869
- Chicheportiche Y, Chicheportiche R, Sizing I, Thompson J, Benjamin CB, Ambrose C, et al. Proinflammatory activity of TWEAK on human dermal fibroblasts and synoviocytes: blocking and enhancing effects of anti-TWEAK monoclonal antibodies. Arthritis Res 2002; 4:126 - 133
- Zimmermann M, Koreck A, Meyer N, Basinski T, Meiler F, Simone B, et al. TNF-like weak inducer of apoptosis (TWEAK) and TNFalpha cooperate in the induction of keratinocyte apoptosis. J Allergy Clin Immunol 2011; 127:200 - 207
- Wilson NS, Yang B, Yang A, Loeser S, Marsters S, Lawrence D, et al. An Fcgamma receptor-dependent mechanism drives antibody-mediated target-receptor signaling in cancer cells. Cancer Cell 2011; 19:101 - 113
- Watts GS, Tran NL, Berens ME, Bhattacharyya AK, Nelson MA, Montgomery EA, et al. Identification of Fn14/TWEAK receptor as a potential therapeutic target in esophageal adenocarcinoma. Int J Cancer 2007; 121:2132 - 2139
- Michaelson JS, Burkly LC. Therapeutic targeting of TWEAK/Fnl4 in cancer: exploiting the intrinsic tumor cell killing capacity of the pathway. Results Probl Cell Differ 2009; 49:145 - 160
- Lynch CN, Wang YC, Lund JK, Chen YW, Leal JA, Wiley SR. TWEAK induces angiogenesis and proliferation of endothelial cells. J Biol Chem 1999; 274:8455 - 8459
- Donohue PJ, Richards CM, Brown SA, Hanscom HN, Buschman J, Thangada S, et al. TWEAK is an endothelial cell growth and chemotactic factor that also potentiates FGF-2 and VEGF-A mitogenic activity. Arterioscler Thromb Vasc Biol 2003; 23:594 - 600
- Ho DH, Vu H, Brown SA, Donohue PJ, Hanscom HN, Winkles JA. Soluble tumor necrosis factor-like weak inducer of apoptosis overexpression in HEK293 cells promotes tumor growth and angiogenesis in athymic nude mice. Cancer Res 2004; 64:8968 - 8972
- Kawakita T, Shiraki K, Yamanaka Y, Yamaguchi Y, Saitou Y, Enokimura N, et al. Functional expression of TWEAK in human colonic adenocarcinoma cells. Int J Oncol 2005; 26:87 - 93