
Abstract
Background: Development of functional monoclonal antibodies against intractable GPCR targets.
Results: Identification of structured peptides mimicking the ligand binding site, their use in panning to enrich for a population of binders, and the subsequent challenge of this population with receptor overexpressing cells leads to functional monoclonal antibodies.
Conclusion: The combination of techniques provides a successful strategic approach for the development of functional monoclonal antibodies against CXCR2 in a relatively small campaign.
Significance: The presented combination of techniques might be applicable for other, notoriously difficult, GPCR targets.
Summary: The CXC chemokine receptor-2 (CXCR2) is a member of the large ‘family A’ of G-protein-coupled-receptors and is overexpressed in various types of cancer cells. CXCR2 is activated by binding of a number of ligands, including interleukin 8 (IL-8) and growth-related protein α (Gro-α). Monoclonal antibodies capable of blocking the ligand-receptor interaction are therefore of therapeutic interest; however, the development of biological active antibodies against highly structured GPCR proteins is challenging. Here we present a combination of techniques that improve the discovery of functional monoclonal antibodies against the native CXCR2 receptor.
The IL-8 binding site of CXCR2 was identified by screening peptide libraries with the IL-8 ligand, and then reconstructed as soluble synthetic peptides. These peptides were used as antigens to probe an antibody fragment phage display library to obtain subpopulations binding to the IL-8 binding site of CXCR2. Further enrichment of the phage population was achieved by an additional selection round with CXCR2 overexpressing cells as a different antigen source. The scFvs from the CXCR2 specific phage clones were sequenced and converted into monoclonal antibodies. The obtained antibodies bound specifically to CXCR2 expressing cells and inhibited the IL-8 and Gro-α induced ß-arrestin recruitment with IC50 values of 0.3 and 0.2 nM, respectively, and were significantly more potent than the murine monoclonal antibodies (18 and 19 nM, respectively) obtained by the classical hybridoma technique, elicited with the same peptide antigen. According to epitope mapping studies, the antibody efficacy is largely defined by N-terminal epitopes comprising the IL-8 and Gro-α binding sites. The presented strategic combination of in vitro techniques, including the use of different antigen sources, is a powerful alternative for the development of functional monoclonal antibodies by the classical hybridoma technique, and might be applicable to other GPCR targets.
Abbreviations
| ABTS | = | 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulphonic acid) |
| BSA | = | bovine serum albumin |
| CLIPS™ | = | Chemical LInkage of Peptides onto Scaffolds |
| ECL | = | extracellular loop |
| EDTA | = | ethylenediaminetetraacetic acid |
| ELISA | = | enzyme-linked immunoabsorbent assay |
| Fmoc | = | fluorenylmethyloxycarbonyl |
| GPCR | = | G-protein coupled receptor |
| Gro-α | = | growth-related protein α |
| IL-8 | = | interleukin 8 |
| IPTG | = | isopropyl β-D-1-thiogalactopyranoside |
| MFI | = | mean fluorescence intensity |
| PBS | = | phosphate buffer saline |
| PCR | = | polymerase chain reaction |
| PEG | = | polyethyleneglycol |
| scFv | = | single-chain variable fragment |
| TES | = | 2-[[1,3-dihydroxy-2-(hydroxymethyl)propan-2-yl]amino]ethanesulfonic acid |
| TRIS | = | tris(hydroxymethyl)aminomethane |
Introduction
G-protein-coupled receptors (GPCRs) are the target of ∼50–60% of all current drug discoveries.Citation1,2 Members of this large superfamily of proteins account for 2–4% of all genes encoded by the human genome.Citation3,4 Through sequence analysis, many receptors have been identified, but the full range of their ligands, their function and the effects, especially on disease states, caused by modulating that function remains unclear. GPCRs have been grouped into 4 families named Family A or the rhodopsin family, Family B or the Secretin and Adhesion family (sometimes classified as 2 separate familiesCitation3), Family C or metabotropic Glutamate family, and Family D the Frizzled family. The Rhodopsin family (family A) is the largest, and it has the most diverse set of ligands, including amines and purines, small peptides such as neuropeptides, and small proteins such as chemokines.
Family A member CXC chemokine receptor-2 (CXCR2), or interleukin 8 receptor, β (IL8RB), is involved in a number of diseases, including cancers,Citation5-9 inflammatory diseases,Citation10-12 and Alzheimer's disease.Citation13 It has the GPCR-characteristic 7 transmembrane structure with 3 internal and 3 external loops and an extracellular N-terminus domain. The promiscuous receptor is activated by a number different ligands, including interleukin 8 (IL-8) and growth-related protein α (Gro-α), both known for their high affinity binding for CXCR2.Citation14 When the ligand-binding site is blocked, signal transduction will be inhibited; therefore, it is an interesting target for the development of therapeutic antibodies.
GPCRs generally have an extracellular N terminus that is exposed, together with the 3 extracellular loops, to the environment where their ligands act.Citation15 It has long been recognized that antibodies could provide a means of specifically altering the interaction between GPCRs and their ligands, and thus provide a general means of validating a given GPCR as a useful pharmaceutical target or even be useful as therapeutics in their own right. However, the generation of such functional antibodies has proven to be a challenging task.
There are several issues when trying to raise functional antibodies to GPCRs, all of which relate to the nature of GPCRs themselves. Only the N-terminus and 3 short extracellular loops are exposed; this is particularly a problem for the largest family, Family A, which only has a relatively short N-terminal region. GPCRs are expressed in low levels, which means purification is difficult, and, if whole cells are considered as antigens, other proteins will dominate the immune response or antibody library selection techniques. Even if large purification is achieved, GPCRs can be unstable outside the membrane and may not be in a biologically relevant conformation. Genetic immunization whereby the gene encoding the GPCR of interest is used to transform the host animal's own cells seeks to circumvent these problems, but no attractive alternative has been found to be universally applicable.Citation16
Peptides have long been used as inexpensive and effective surrogates for full-length proteins in antibody generation. However, in the case of GPCRs with their highly structured nature and discontinuous epitopes, linear peptides can be poor imitators of the protein in its natural state. While antibodies raised against linear peptides can often recognize GPCRs in denaturing assays, such as western blot, functional antibodies generated in this manner are rare. More sophisticated peptide approaches have sought to try and mimic the GPCR extracellular loops, and some success has been obtained.Citation17, 18 However, apart from synthetic issues this approach has its limitations. Firstly, the structure of GPCRs is largely based on homology models,Citation15 and thus the precise structure cannot be known. Secondly, the relevance of the crystal structure to the structure in vivo is a matter of conjecture. Finally, the interaction of the ligand with the surface of the GPCR can be multipoint, relying on contacts with more than one loop or with loops and the N terminus.Citation19 We have sought to address these issues by a combination of techniques (), firstly generation of a large array of structured peptides encompassing a variety of constraint positions and juxtapositions of the component parts of the GPCR external surface. This array was then probed with a labeled version of a GPCR's ligand, in order to determine which constructs best mimicked the natural protein. Based upon this data and in combination with published structural data available for other members of the GPCR family A,Citation20-24 antigens were designed and synthesized. These antigens were used to generate antibodies by conventional immunization and for the selection of peptide specific antibodies from 3 naïve antibody phage display libraries, before conducting a further round of selection against cells expressing the GPCR of interest. For this study we selected human CXCR2 (hCXCR2) and subsequently tested the resulting antibodies for their ability to modulate the effect of 2 of its ligands: IL-8 and growth-related protein α (Gro-α).
Figure 1. Strategy for monoclonal antibody development by naïve antibody phage display library panning and B cell hybridoma technique.
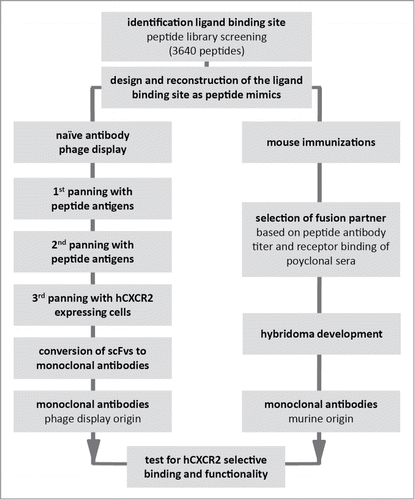
Results
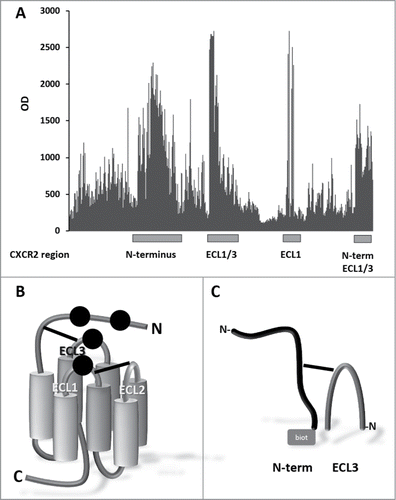
Identification of the IL-8 binding sites on CXCR2 and reconstruction as synthetic peptides. The IL-8 ligand binding site of hCXCR2 was identified by screening peptide libraries based on the extracellular domains of the receptor. Four major hCXCR2 regions were identified as strong IL-8 binding sequences, including regions of the N-terminus and extracellular loops (ECL)1 and combinations of ECL1/ECL3 and N-terminus/ECL1/ECL3 (). This suggests a discontinuous binding site on the CXCR2 receptor for IL-8 binding, with contact regions at the N-terminus, ECL1 and ECL3 (). This, in combination with the available structural data of similar GPCR receptors,Citation20-24 was used to synthesize peptides mimicking 2 CXCR2 domains involved with the IL-8 binding, comprising the N-terminal sequence DSFEDFWKGEDLSNYSYSSTLPPFLLDAAPCEPESLEINK combined with ECL3 sequence DTLMRTQVIQETCERRNHIDR, tethered by either a disulfide bridge (antigen A) or a CLIPS™ moiety (antigen B) (). Additional immunogens were successfully synthesized including combinations of Nterm-ECL1-ECL2 constructs and single extracellular domains ECL1 and ECL2 (linear and cyclized). These antigens were also used for mice immunizations and phage display library panning. Although high antigen specific polyclonal antibody titers were elicited in mice, binding to CXCR2 expressing cells was absent (data not shown). It was decided to select the most promising mice for hybridization. Also, these antigens did not bind, or bound very marginally, to the scFv phage display libraries (data not shown). Further monoclonal development was therefore not opportune for these antigens.
Figure 2. Identification of IL-8 binding sites on CXCR2. (A) Binding of IL-8 to array of > 3,600 different peptide constructs. The peptide arrays were challenged with biotinylated IL-8 and detected with streptavidin-HRP. (B) Cartoon of CXCR2. Seven transmembrane domains (cylinders) are connected by 3 intra cellular loops (not labeled) and 3 extracellular loops (ECL1, ECL2 and ECL3) with 2 disulfide bonds (solid lines), between ECL3 and N-terminus, and between ECL1 and ECL2, respectively. Black dots represent the inferred binding positions of IL-8. (C) Peptide reconstruction of the extracellular regions of hCXCR2. N-terminus (DSFEDFWKGEDLSNYSYSSTLPPFLLDAAPCEPESLEINK) (black structure) and ECL3 (DTLMRTQVIQETCERRNHIDR) (dark gray arch) are connected by a disulfide bond (antigen A) or a CLIPS™ moiety (antigen B). The biotin moiety (gray square), for antibody phage display library panning, is located at the C-terminal side of the N-terminus.
Monoclonal antibody development
Isolation of scFvs from antibody phage display libraries. Two populations of phage scFvs were selected for each of the peptide antigens from each of 3 naïve scFv antibody libraries (BMV, CS, DP47). The first population arose from 2 rounds of biopanning against the peptide antigen, and the second population arose from 2 rounds of biopanning against the peptide antigen and a third round against whole cells known to express hCXCR2 protein in their surface membrane. It is known that using a peptide as a surrogate for the natural protein will also produce many irrelevant antibodies that recognize the peptide, but not the natural protein. The panning against cells was designed to eliminate these and enrich for antibodies which recognized the natural protein.
All isolates were tested by enzyme-linked immunosorbent assay (ELISA) for their ability to specifically bind the peptide antigen used in their isolation. All unique anti-peptide positive scFvs from both panning strategies (2 panning rounds (peptide/peptide) or 3 panning rounds (peptide/peptide/cells)) were assessed for their ability to bind recombinant hCXCR2 HEK cells (positive), but not parental HEK cells (negative).
Anti-peptide antibodies were isolated against each antigen after 2 rounds of panning (). A similar or reduced number of unique anti-peptide scFvs were isolated after a third round of panning with hCXCR2 bearing cells. However, when the ability of the isolated anti-peptide antibodies to recognize native hCXCR2 was tested, it was clear that the third panning round with a different antigen source (cells) significantly increased the percentage of hCXCR2 binding scFvs. The enrichment after a third cell panning round was increased 12-fold in the group initially panned with the CLIPS™ moiety containing antigen (B) and 2-fold in the thiol-bridged antigen (A) group (). Together, these data were used to select scFvs to be converted to whole rabbit IgG1 antibodies.
Table 1A. scFvs isolated from panning naïve antibody phage display libraries
Generation of murine monoclonal antibodies. In parallel to the antibody phage display library panning technique, monoclonal antibodies were developed by the traditional B-cell hybridoma approach. For this strategy, mice were immunized with the same antigen ‘A’ as was used for the phage display panning, but without the biotin moiety. The antigen was able to elicit high polyclonal antibody titers, and showed cross reactive binding to hCXCR2 overexpressing cells (). Two mice were selected for hybridoma development based on the combination of the presence of high antibody titers against their antigen and binding to the native hCXCR2 receptor. Fifteen peptide-specific hybridomas were produced, of which 7 were shown to specifically bind recombinant hCXCR2 HEK cells ().
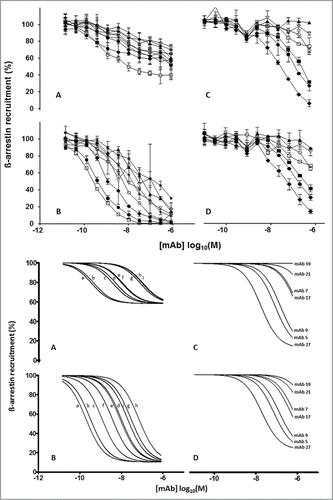
Characterization of antibodies from both antibody phage display libraries and mouse monoclonals. The IgGs (converted from scFvs) and murine monoclonal antibodies were tested for their ability to inhibit the ß-arrestin stimulating effects of IL-8 and Gro-α in a functional Tango™ GPCR Assay System (Invitrogen). Eight IgGs (converted from scFvs) from the peptide/peptide/cells panning strategy and one from the peptide/peptide panning (IgG 4081.73) were effective inhibitors of IL-8 and Gro-α ligand binding to the hCXCR2 receptor (). Gro-α induced ß-arrestin recruitment was inhibited almost completely (90%) while the maximum inhibition of IL-8 induced ß-arrestin recruitment was limited to 40%. This large difference in maximum IL-8 and Gro-α ligand inhibition was absent with the murine IgGs obtained by the classical hybridoma technique (). When the ligand inhibition is expressed as IC50 values (), the recombinant IgGs (converted from scFvs) were more effective inhibitors than IgGs obtained by the classical hybridoma technique as is demonstrated by factor of 50 and 100 differences in IC50 values for IL-8 and Gro-α, respectively.
Table 1B. Antibodies from mouse hybridomas, elicited with antigen A
Figure 3. Top Panel: Inhibition of IL-8 (A and C) and Gro-α (B and D) induced ß-arrestin recruitment in hCXCR2-HEK cells (Tango™ GPCR Assay System, Invitrogen) by IgGs converted from scFvs (A and B): 4138.51 (open square), 4081.73 (solid circle), 4138.73 (filled diamond), 4138.06 (asterisk), 4138.27 (open triangle), 4138.39 (open circle), 4138.14 (solid triangle) 4138.54 (solid star), and 4138.40 (open diamond). Murine IgGs (C and D): mAb27 (solid diamond), mAb5 (solid circle), mAb9 (solid square), mAb17 (open square), mAb7 (open circle), mAb21 (open diamond) and mAb19 (solid triangle). Each curve is based on 2 independent measurements except for panel A which is based on 4 independent measurements. The error bars represent the standard deviations. Bottom panel: Using GraphPad Prism software (Software MacKiev) the top values of each dataset were constrained at 100% and the bottom values of each dataset were shared. In all panels the Hill slopes were constrained at -1.000. IgGs converted from scFvs (A and B): 4138.51 (a), 4081.73 (b), 4138.73 (c), 4138.06 (d), 4138.27 (e), 4138.39 (f), 4138.14 (g) 4138.54 (h), and 4138.40 (i). Murine IgGs (C and D): mAb27, mAb5, mAb9, mAb17, mAb7, mAb21 and mAb19. Each curve is based on 2 independent measurements except for panel A which is based on 4 independent measurements.
Specificity of antibody phage display library derived and murine monoclonal antibodies. On average the murine mAbs show a higher affinity for CXCR2 expressing cells than the phage mAbs when tested at 1 μg/ml, but at lower concentrations (0.01 and 0.1 μg/ml) there is no difference (). Not all CXCR2 binding antibodies were able to significantly inhibit the IL-8 or Gro-α induced signal transduction of the receptor (). We speculated whether this may be caused by a different binding profile. In ELISA binding studies, the functional recombinant IgGs (converted from scFvs) bound prominently to the N-terminal part of the CXCR2 N-terminus (), which was confirmed by epitope mapping showing the core epitope to center around the 11FEDFWKCitation16 region (). Non-functional recombinant IgGs (converted from scFvs) did bind to the same N-terminal regions as the functional ones, but flow cytometry showed that they did not cross react with the native CXCR2 protein expressed on the cell surface (). In contrast to the recombinant IgGs from scFvs, all the functional murine monoclonal antibodies were specific for peptides representing the C-terminal part of the CXCR2 N-terminus (Nterm-C: SYSSTLPPFLLDAAPAEPESLEINK), but did not bind to either the N-terminal part of the CXCR2 N-terminus (Nterm-N: MEDFNMESDSFEDFWKGEDLSNYSYSS) (), or any of the 3 ECL domains presented as linear and cyclic versions (, footnote 1). A more detailed analysis of the binding site by epitope mapping showed the dominant epitope to center around residuesCitation31 PFLLDCitation35 and a much weaker second epitope at positions11 FEDFWKGEDLCitation20, which was not detected by ELISA (). The very weak and non-functional murine monoclonal antibodies bound the single Citation31PFLLDCitation35 epitope ().
Table 2. Binding to native CXCR2 and functional testing of recombinant IgGs (converted from scFvs) and murine monoclonal antibodies
Table 3. Epitope mapping of recombinant IgGs (converted from scFv) and murine monoclonal antibodies
Discussion
GPCRs are a vast group of membrane-bound protein structures important for many functions.Citation25 The CXC chemokine receptor-2 (CXCR2), a member of the large Family A type GPCRs, is involved in many ailments, including cancers and inflammatory diseases, and it is therefore an important target for drug discovery.Citation1,5-9,16,26-28 A diversity of therapeutics is used to inhibit the receptor signaling function of a number of GPCRs, e.g., small molecules,Citation29,30 peptide analogs of the natural ligands,Citation31,32 or monoclonal antibodies;Citation16 however, no monoclonal antibody therapeutics targeting CXCR2 have been clinically developed so far.
An important characteristic of monoclonal antibodies is their high target specificity, which makes them very attractive therapeutics capable of steric interference with the ligand binding site of the receptor. However, the generation of anti-GPCR antibodies is hampered by the nature of the target. The only exposed epitopes are the extracellular N-terminus and 3 ECLs. Whole cells and cell membrane fragments can be used as antigens,Citation33,34 but will elicit undesired immune responses against other surface proteins or intracellular domains. To circumvent the undesired immune responses, sophisticated peptide mimics of the extracellular domains can be used as antigens. This seems a straight-forward approach, but simply binding an antigen does not necessarily mean that an antibody will have appropriate functionality. We therefore first determined the ligand-binding site of IL-8, this being the most logical site for steric hindrance by an antibody. The CXCR2 binding sites of IL-8 and Gro-α are known from literatureCitation19 and were used to validate the generic application of our method for the identification of ligand binding sites.
In our studies, IL-8 binds to multiple sites of the extracellular domains of the receptor with contacts spanning over the N-terminus, ECL1, and ECL3 (), which agrees with reported interaction sites of IL-8 and Gro-α in site-directed mutagenesis studies of CXCR2.Citation19 Furthermore, emerging evidence suggests the participation of the N-terminus in receptor activation of rhodopsin like (family A) GPCRs.Citation35-37 With the intention to develop a strategy that might be applied to GPCRs in general, the design of the experiments was focused on the combinations of the N-terminus with an ECL domain. Several antigen designs have been synthesized; however, the single N-terminus was omitted. This was based on the fact that in earlier experiments on other GPCRs (e.g., CXCR7 or CCR7), the N-terminus as antigen alone did not result in any neutralizing antibodies (data not shown). An exception was found in mouse anti human CXCR2 monoclonal 6C6, which inhibits CXCR2 mediated responses after binding to the N-terminus, this monoclonal antibody was elicited against whole cells.Citation38 However, for a number of other GPCRs, it was confirmed that an immune response against the N-terminus alone resulted in binding antibodies, but these were without neutralizing capability.Citation16 Using this data and published structural information of other members of the GPCR Family A,Citation20-24 2 peptides likely to be representative for the N-terminus and ECL3 domain of the GPCR's external surface were designed and synthesized. These were used to isolate antibodies from 3 naïve scFv libraries and to generate hybridomas from immunized mice. Both approaches produced functional antibodies. Both also produced anti-peptide antibodies that failed to bind the native receptor. However, we showed that introducing a third round of panning with CXCR2 expressing cells strongly increased the number of functional antibodies: 8 out of 40 (20%) of the hCXCR2 binding scFvs resulted in functional IgGs, in contrast to just 1 out of 12 (8%) after 2 peptide panning rounds (). The first 2 panning rounds with peptides will enrich the phage population for the specific extracellular domains of the GPCR and the third panning round selects for phages capable of binding to the native receptor presented in its natural matrix, the cell membrane. Alternative panning round sequences might be considered, e.g., cells-peptide-(peptide): the first round with cells to enrich the phage population for the target (GPCR), and subsequently one or 2 rounds with peptides to enrich for the target. This panning sequence may be less efficient, as the first round might yield a high number of cell surface specific sites (e.g., other receptors etc.), but not GPCR specific phages per sé.
Monoclonal antibody binding to the native receptor, e.g., CXCR2, alone is not enough to be functional in ligand-binding inhibition. A difference in ß-arrestin recruitment inhibition efficacy of the IgGs converted from scFvs was observed between Gro-α, which was inhibited completely, and IL-8 (max. 40%; ). In ELISA binding studies with peptides representing N-terminal and the 3 ECL domains and epitope mapping studies, all functional monoclonal antibodies recognized N-terminal but not ECL epitopes (). A plausible explanation for this difference in efficacy may originate from the ligand-specific binding residues: for IL-8 the critical residues are located on the N-terminus (Asp-9, Glu-12) as well as ECL1 (Lys-108 and Lys-120), while Gro-α specific residues are located on the N-terminus only (Glu-7, Asp-9 and Glu-12).Citation19 The functionality of IgGs converted from scFvs is defined by binding to the N-terminal FEDFWK epitope, largely blocking the Gro-α binding site and partially blocking the N-terminal part of the IL-8 binding site. This mechanism might be different for the murine monoclonal antibodies because the maximum inhibition of the IL-8 and Gro-α induced ß-arrestin recruitment is of a similar order of magnitude (). The functionality of the murine monoclonal antibodies 27, 5 and 9 might be defined by a discontinuous epitope comprising the N-terminal dominant PFLLD binding site and the much weaker FEDFWKGEDL binding site that seems to contribute to the ligand inhibition (). Part of the explanation might be the fact that the residues for ligand binding in the native protein are blocked in a similar fashion as with the IgGs converted from scFvs. Although the FEDFWKGEDL region seems to be necessary to inhibit the ligand binding, the major binding energy of the murine monoclonal antibody discontinuous epitope is provided by the dominant PFLLD site. The monoclonal antibody may bind first to the PDLL epitope and subsequently to the FEDFK epitope (). Murine mAb 21 binds to the C-terminal part of the N-terminus in the epitope mapping study, but, in contrast, it binds to the N-terminal part in the ELISA results (). To a lesser extent, a similar difference can be found for mAb17. It might be an effect of specific conformational differences of the peptide antigens used in the epitope mapping and the coated peptide antigens in ELISA. If this speculation is true, it hints to specific characteristics of these monoclonal antibodies, which are not interesting for ligand inhibition ().
A complicating factor in general is the redundancy in ligand binding. Many GPCRs can bind a large number of different ligands, all with their specific signal-triggering pathway. As shown in this study, delicate differences are found for the phage and murine monoclonal antibodies in ligand-binding inhibition (e.g., IL-8 and Gro-α), a feature that may be an advantage for a potential therapeutic antibody, e.g., if elimination of one specific type of signal transduction might be required. Another factor involved in ligand binding might be post-translational modifications of the extra-cellular domains. These modifications are known to influence the actions of a number of chemokine receptors (e.g., CCR5, CXCR4, but were not reported for CXCR2 to this date.Citation39-41 In conclusion, the production of a relatively high number of functional monoclonal antibodies against a notoriously difficult target such as a GPCR shows the potential of combining identification and reconstruction of the ligand binding site and alternating antibody phage display library pannings, considering the relatively small campaign. Furthermore, this study suggests that it would be fairly straightforward to apply this approach to other GPCR targets. The presented strategic combination of in vitro techniques, including different antigen sources, is a powerful alternative for the development of functional monoclonal antibodies by the classical hybridoma technique. Additionally, further experiments are needed to evaluate the therapeutic value of the obtained monoclonal antibodies.
Materials and Methods
Peptide array synthesis and screening. The extracellular domains of human CXCR2 were synthesized as peptide arraysCitation42 of 3640 peptides comprising the N-terminus (MEDFNMESDSFEDFWKGEDLSNYSYSSTLPPFLLDAAPCEPESLEINK, ECL1 (ASKVNGWIFGTFLCK), ECL2 (RRTVYSSNVSPACYEDMGNNTANWR) and ECL3 (DTLMRTQVIQETCERRNHIDR), using all overlapping linear 7- and 15-mers and all overlapping looped 17-mers in a Cys-X15-Cys-X15-Cys format and all overlapping looped 7-mers of the combined N-terminus-ECL1, -ECL2, and -ECL3 constructs in a X7-Cys-Cys-X7-Cys-X7-Cys-X7-Cys format. Conformational loops are created by connecting the cysteine residues with a T3 CLIPS™ scaffold.Citation43
Identification of IL-8 binding site. The peptide arrays were first pre-incubated with blocking solution, and, after washing, incubated with his-tagged human IL-8 (overnight at 4°C) in 2 different test conditions: i) 1 μg/ml diluted in strong blocking solution (4% horse serum, 5% ovalbumin (w/v) in PBS/1% Tween); ii) 0.05 μg/ml diluted in mild blocking buffer, PBS/1% Tween. Detection was done with anti-his antibody and horse radish peroxidase labeled Rabbit-anti-Mouse antibody (Birmingham, AL, USA) according to previously described protocols.Citation42
Epitope mapping of monoclonal antibodies was performed according to previously published protocols.Citation42,43 In short: The binding of antibody to each peptide is tested in a PEPSCAN-based ELISA. The peptide arrays are incubated with a primary antibody solution, for example consisting of 1 μg/ml diluted in blocking solution (4% horse serum, 5% ovalbumin (w/v) in PBS/1% Tween). After washing, the peptides are incubated with a 1000-fold dilution of antibody peroxidase conjugate for one hour at 25° C. After washing, the peroxidase substrate solution (0.5 mg/ml 2,2’-azino-di-3-ethylbenzthiazoline sulfonate (ABTS) and 0.006% H2O2 in 0.05 M citrate buffer pH 4) was added. After one hour incubation at room temperature, the color development was measured. The color development was quantified with a charge coupled device (CCD) – camera and an image processing system
Peptide synthesis. CXCR2 domains comprising the N-terminus (DSFEDFWKGEDLSNYSYSSTLPPFLLDAAPCEPESLEINK), and ECL3 (DTLMRTQVIQETCERRNHIDR), were reconstructed as peptide mimics and synthesized by standard Fmoc chemistry.Citation44 Disulfide bridges and CLIPS™ chemistryCitation45 were used to mimic the 3-dimensional structure of the of the extracellular domains of CXCR2. The peptides were synthesized with a biotin moiety for antibody phage display library panning and to coat microtiter plates for antibody titer measurements by ELISA. The purity and identity of the peptides were analyzed by UPLC (C18 column, Waters Corporate, Milford, MA, USA) and electrospray ionization mass spectrometry detection (Aqcuity UPLC-MS, Waters Corporate, Milford, MA, USA).
Phage Display peptide panning. Three phage display libraries were used: the Bone Marrow Vaughan (BMV) library, a nonimmunized library of 1.4 × 1010 scFv fragments,Citation46 a combined spleen (CS) library of 1.29 × 101 scFv fragmentsCitation47 and the DP47 library, a naive human library of 1 × 1010 scFv fragments.Citation48 All of these libraries are phagemid-based and require rescue with helper phage M13K07 for the production of scFv-bearing phage. The BMV, CS and DP47 libraries are available from Medimmune Cambridge (formerly Cambridge Antibody Technology). Washed streptavidin beads (M280, Dynal) were charged with the biotinylated antigens at 1 μg/100 μl beads, before being blocked with 2% dried milk powder (Marvel, Cadbury) in PBS (137 mM NaCl, 2.7 mM KCl, 4.3 mM Na2HPO4, 1.47 mM KH2PO4, adjust to a final pH of 7.4.)
Before use, the charged beads were washed 3 times in PBS. Aliquots of each of the 3 scFv phage libraries (BMV, CSand the DP47) were blocked in 2% milk powder in PBS before being exposed to the peptide-charged streptavidin beads. After mixing for 1 hour, the beads were washed 10 times in PBS-Tween, and then twice in PBS alone. Bound phage were release by 10-minute exposure to 100 mM triethylamine, (TEA; Sigma), which was immediately neutralized with 1 M TRIS-HCl pH7.4. After mixing for 1 hour, the beads were precipitated and the supernatant was treated with 10 μg/ml trypsin 30 minutes at 37°C and used to infect TG1 cells. These were grown overnight on 2TY-plates containing ampicillin and glucose. The following morning the bacteria were used to seed a liquid culture that was subsequently infected with the helper phage M13KO7 trpR.Citation49 After overnight growth at 30°C, the bacteria were pelleted and the supernatant used to perform subsequent rounds of panning.
Fingerprinting and sequencing isolates from antibody phage display libraries. To assess the diversity among positive isolates, fingerprinting was initially performed, whereby the gene encoding the scFv was amplified by PCR and the product digested with the high frequency cutter BStNI. These digests were then separated on a 3% agarose gel and the patterns obtained from each compared. Subsequently, all isolates of interest were fully sequenced, using Applied Biosystems Big Dye v 3.1 cycle-sequencing chemistry and an Applied Biosystems 3730xl capillary electrophoresis system. All scFvs that had different amino acid sequences were defined as unique.
Cell panning. Six-well dishes (pre-coated with poly-lysine) were seeded 2 ml/well with 5 × 104 cells/ml with HEK293T cells stably expressing human CXCR2 before growing overnight at 37°C 4% CO2. Dishes were cooled to 4°C on a rocking platform, removing media and replacing with 1 ml of ice-cold fresh growth media for 1 hour. 200 μl of each of the purified phage (from 2 rounds of peptide panning) were blocked by the addition of 800 μl of 4% milk powder (Marvel, Cadbury, UK), 1% BSA (v/v) in PBS for 1 hour at 4°C before adding to the media in each well and panning for 1 hour at 4°C with gentle rocking. Each of the wells was washed 6 times over 30 minutes by removing the media/buffer and adding 2 ml of ice cold PBS. 500 μl of 100 mM TEA was added for 10 minutes to each well in order to elute bound phage. The eluate was taken from the well and added to 0.25 ml of Tris pH 8.0 to neutralize before treating with 10 μg/ml trypsin 30 minutes at 37°C.
Hybridoma Generation. Hybridomas were generated by Antibody Applications (Macclesfield, UK), using traditional PEG fusion technology.
Primary screen of isolates from antibody Phage Display Library. In order to screen the selected phage populations after 2 or 3 rounds of panning, isolated phages were used to infect a culture of HB2151, which was diluted and plated to obtain single colonies. 96 colonies per library per antigen were grown in 2TY broth (10 g Bacto-tryptone/L, 10 g yeast extract/L, 5 g NaCl/L, pH 7.0) supplemented with 100 μg/mL ampicillin. ScFv production was induced by adding 1 mM IPTG. The following day, the bacteria were precipitated and treated with TES (20% Sucrose, 50 mM TRIS, 1 mM EDTA, pH 8.0) followed by 10 mM MgS04. After centrifugation, the supernatant was used in a variety of screens as described below.
Maxisorp immunoassay plates (Nunc, Roskilde, Denmark) were coated with 100 μl per well of biotinylated BSA at 2 μg/ml in 50 mM carbonate buffer, pH 8.5 for 2 hours at 37°C. After washing 3 times with PBS-Tween (0.1%), 100 μl of streptavidin at 10 μg/ml in PBS was added to each well for 1 hour at room temperature before blocking by the addition of 200 ml per well of 2% milk powder (Marvel, Cadbury, UK) in PBS. After washing 100 μl biotinylated antigen was added at 1 μg/ml in 2% Marvel in PBS for 1 hour. Coated, blocked plates were washed 3 times with PBS-Tween (0.1%) and supernates diluted in PBS with 2% milk powder, were applied to the assay plates. After an hour at room temperature, plates were washed 3 times with PBS-Tween (0.1%) and a 10000-fold dilution of rabbit anti-cmyc HRP conjugate (Ab19312, Abcam, Cambridge, UK) secondary antibody was added to detect bound scFv. Plates were incubated for a further hour at room temperature and washed 3 times with PBS-Tween (0.1%) prior to developing by the addition of 3,3’,5,5’-tetramethylbenzidine (TMB) substrate (Sigma, St. Louis, Mo, USA). Absorbances were read at 370 nm on a Molecular Devices VERSAmax plate reader.
Assessing binding of isolates from antibody Phage Display Library to hCXCR2 expressing cells. In order to assess the binding to hCXCR2 expressing HEK cells, supernates from scFv clones (prepared exactly as described above) were titrated before adding to a premixed solution: mouse anti-6His Serotec MCA1396, at a final concentration of 1 μg/ml, and Alexafluor 647 goat anti-mouse (A21236), at a final concentration of 2 μg/ml, for scFv supernates in FMAT buffer (0.1% BSA in PBS) in 96 or 384 well FMAT 8100HTS plates. HEK cells stably expressing hCXCR2 or a parental cell line which had negligible expression of huCXCR2 were resuspended in FMAT buffer at 1 × 105 cells/ml before adding to relevant wells. The plate was incubated for 2 hours in the dark at room temperature before reading on an FMAT 8200 cellular detection system (Applied Biosystems).
Mouse Immunization and ELISA screening. Female Balb/c mice of 6–8 weeks were immunized with 100 μg antigen at day 0 and at 3 and 6 weeks, and with 50 μg antigen at 9 weeks, all in the presence of an adjuvant. The vaccine was inoculated subcutaneously (s.c.) at 2 locations at both flanks or in the neck and 2 locations intramuscular (i.m) in the hind thigh muscle. Blood samples were taken at 0 (pre-immune serum), 6, 9 and 12 weeks. At week 23, 5 d prior to harvesting the B cells from the spleen for hybridoma generation, the mice were boosted i.p. with 25 μg antigen doses in the absence of adjuvant. All animal procedures were performed in the facilities of CVI, Lelystad, The Netherlands, part of the Animal Science Group (ASG) of Wageningen, and were performed in accordance with the provisions of the European Convention for the protection of vertebrate animals used for experimental and other scientific purposes (86/609 EG) and with the Act on Experimental Animals of the Netherlands by which the use of experimental animals is granted to ASG (license DLO dd. 17 febr. 2010. Licencenr. 40100). After the experiments were approved by the Animal Welfare Committee of ASG prior to execution of the experiments and executed in accordance to the guidelines of the Code of practice for the immunization of laboratory animals, amended second version August 2000; Keuringsdienst van Waren.
The polyclonal sera were evaluated for the antigen specific antibody titer development by an indirect ELISA. Nunc Maxisorp 96-wells microtiter plates (Nunc; Thermo Fisher Scientific, Denmark) were pre-coated with 100 μl avidin (4 μg/ml in 50 mM bicarbonate pH 9.5 buffer) and incubated overnight at 37° C. Subsequently, the microtiter plates were washed 4 times with 0.05% (w/v) Tween80 in water and pre-blocked (1 h, room temperature) with 100 μl 4% horse serum and 1% Tween in PBS, and subsequently washed 4 times. Per well, 100 μl 0.5 μg/ml biotinylated peptide solution was added, incubated for 90 minutes at 37°C and washed 4 times. Sera dilutions in incubation buffer (4% Horse serum, 0.48 mol/l NaCl, 5.5 mmol/l Na2HPO4.H2O, 1.1 mmol/l NaH2PO4.H2O, 1% Tween80) were added to the wells and incubated one hour at 25°C. After 4 washes, 100 μl of horse radish peroxidase labeled Rabbit-anti-Mouse antibody (Birmingham, AL, USA), 1000-fold diluted in incubation buffer, was added to the wells. After one hour incubation at 25°C, the plates were washed and 150 μl freshly prepared ABTS substrate solution was added. After 45 minutes incubation at room temperature, the absorbance at 418 nm was measured with a spectrophotometer (SpectraMax M5, Molecular Devices, Sunnyvale, CA, USA). The titers were defined as the serum dilution that had an OD equivalent to 4 times the background OD, and the values are expressed as 10log(dilution).
Flow cytometry. Cells were cultured to 70–80% confluence, detached with cell dissociation buffer (Invitrogen, Carlsbad, CA, USA) and resuspended (106 cells/ml) in FACS buffer (PBS + 5%FCS + 1 mM EDTA + 0.04 %(m/v) sodium azide). Sera, hybridoma supernates or monoclonal antibodies were incubated for 30 min at 4°C with cells (45 μl) in 96-well plates unless stated otherwise. After washing with FACS buffer, bound antibodies were stained with Fcγ fragment specific goat anti- mouse IgG conjugated to R-Phycoerythrin (R-PE) or goat anti-human IgG (H+L) conjugated to R-PE (Jackson ImmunoResearch Europe Ltd, Suffolk, UK). Cells were washed again and analyzed on a FACScalibur flow cytometer (Becton Dickinson, San Jose, CA, USA).
Conversion of scFv to whole IgG. All scFvs with unique amino acid sequences that had been selected based upon their performance in a variety of screens (described above) were converted into IgG format by amplifying the variable heavy and variable light chains separately by PCR, and then cloned into a whole heavy chain and or a whole light chain vector, respectively, by ligation-independent cloning (Infusion, Clontech). The heavy and light chain vectors were then co-expressed in a transient CHO expression system.
Functional assays. The potency of antibodies to inhibit the activation of hCXCR2 by either IL-8 or Gro-α hCXCR2 was measured using the TANGO ß-arrestin GPCR assay system (Invitrogen). Briefly, 30 μl per well of U2OS hCXCR2-bla cells (10000 cells/well) were added to a 384-well Greiner PDL plate before incubating overnight at 37°C in a CO2 incubator. Selected IgGs were added to the plate before incubating for 30 minutes at 37°C in a CO2 incubator (11 point, 3-fold dilution series in assay media: FreeStyleTM Expression Medium, Invitrogen). IL-8 (7 nM) or Gro-α (15 nM) was added to the plate followed by 5 hours incubation at 37°C in a CO2 incubator. Detection mix was prepared (LiveBLAzerTM FRET B/G Substrate, Invitrogen) before adding 8 μl/well, incubating at room temp for 2 hours and reading on an Envision plate reader (Ex 400, Em460/Em535).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This paper is dedicated to Jerry Slootstra.
We would like to thank Danielle Mitchell, David Fisher, Magdalena Florkowska, Mariska Ruiter, Drohpati Timmers and Mariët Zekveld for their contributions to the experiments.
References
- Dorsam RT, Gutkind JS. G-protein-coupled receptors and cancer. Nat Rev Cancer 2007; 7:79-94; PMID:17251915; http://dx.doi.org/10.1038/nrc2069
- Pierce KL, Premont RT, Lefkowitz RJ. Seven-transmembrane receptors. Nat Rev Mol Cell Biol 2002; 3:639-50; PMID:12209124; http://dx.doi.org/10.1038/nrm908
- Fredriksson R, Lagerstrom MC, Lundin LG, Schioth HB. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol Pharmacol 2003; 63:1256-72; PMID:12761335; http://dx.doi.org/10.1124/mol.63.6.1256
- Vassilatis DK, Hohmann JG, Zeng H, Li F, Ranchalis JE, Mortrud MT, Brown A, Rodriguez SS, Weller JR, Wright AC, et al. The G protein-coupled receptor repertoires of human and mouse. Proc Natl Acad Sci U S A 2003; 100:4903-8; PMID:12679517; http://dx.doi.org/10.1073/pnas.0230374100
- Keane MP, Belperio JA, Xue YY, Burdick MD, Strieter RM. Depletion of CXCR2 inhibits tumor growth and angiogenesis in a murine model of lung cancer. J Immunol 2004; 172:2853-60; PMID:14978086; http://dx.doi.org/10.4049/jimmunol.172.5.2853
- Murphy C, McGurk M, Pettigrew J, Santinelli A, Mazzucchelli R, Johnston PG, Montironi R, Waugh DJ. Nonapical and cytoplasmic expression of interleukin-8, CXCR1, and CXCR2 correlates with cell proliferation and microvessel density in prostate cancer. Clin Cancer Res 2005; 11:4117-27; PMID:15930347; http://dx.doi.org/10.1158/1078-0432.CCR-04-1518
- Wang B, Hendricks DT, Wamunyokoli F, Parker MI. A growth-related oncogene/CXC chemokine receptor 2 autocrine loop contributes to cellular proliferation in esophageal cancer. Cancer Res 2006; 66:3071-7; PMID:16540656; http://dx.doi.org/10.1158/0008-5472.CAN-05-2871
- Wislez M, Fujimoto N, Izzo JG, Hanna AE, Cody DD, Langley RR, Tang H, Burdick MD, Sato M, Minna JD, et al. High expression of ligands for chemokine receptor CXCR2 in alveolar epithelial neoplasia induced by oncogenic kras. Cancer Res 2006; 66:4198-207; PMID:16618742; http://dx.doi.org/10.1158/0008-5472.CAN-05-3842
- Yang G, Rosen DG, Liu G, Yang F, Guo X, Xiao X, Xue F, Mercado-Uribe I, Huang J, Lin SH, et al. CXCR2 promotes ovarian cancer growth through dysregulated cell cycle, diminished apoptosis, and enhanced angiogenesis. Clin Cancer Res 2010; 16:3875-86; PMID:20505188; http://dx.doi.org/10.1158/1078-0432.CCR-10-0483
- Belperio JA, Keane MP, Burdick MD, Londhe V, Xue YY, Li K, Phillips RJ, Strieter RM. Critical role for CXCR2 and CXCR2 ligands during the pathogenesis of ventilator-induced lung injury. J Clin Invest 2002; 110:1703-16; PMID:12464676; http://dx.doi.org/10.1172/JCI0215849
- Olszyna DP, Florquin S, Sewnath M, Branger J, Speelman P, van Deventer SJ, Strieter RM, van der Poll T. CXC chemokine receptor 2 contributes to host defense in murine urinary tract infection. J Infect Dis 2001; 184:301-7; PMID:11443555
- Weathington NM, van Houwelingen AH, Noerager BD, Jackson PL, Kraneveld AD, Galin FS, Folkerts G, Nijkamp FP, Blalock JE. A novel peptide CXCR ligand derived from extracellular matrix degradation during airway inflammation. Nat Med 2006; 12:317-23; PMID:16474398; http://dx.doi.org/10.1038/nm1361
- Bakshi P, Margenthaler E, Reed J, Crawford F, Mullan M. Depletion of CXCR2 inhibits gamma-secretase activity and amyloid-beta production in a murine model of Alzheimer's disease. Cytokine 2011; 53:163-9; PMID:21084199; http://dx.doi.org/10.1016/j.cyto.2010.10.008
- Cerretti DP, Kozlosky CJ, Vanden Bos T, Nelson N, Gearing DP, Beckmann MP. Molecular characterization of receptors for human interleukin-8, GRO/melanoma growth-stimulatory activity and neutrophil activating peptide-2. Mol Immunol 1993; 30:359-67; PMID:8384312; http://dx.doi.org/10.1016/0161-5890(93)90065-J
- Lagerstrom MC, Schioth HB. Structural diversity of G protein-coupled receptors and significance for drug discovery. Nat Rev Drug Discov 2008; 7:339-57; PMID:18382464; http://dx.doi.org/10.1038/nrd2518
- Hutchings CJ, Koglin M, Marshall FH. Therapeutic antibodies directed at G protein-coupled receptors. MAbs 2010; 2:594-606; PMID:20864805; http://dx.doi.org/10.4161/mabs.2.6.13420
- Misumi S, Nakayama D, Kusaba M, Iiboshi T, Mukai R, Tachibana K, Nakasone T, Umeda M, Shibata H, Endo M, et al. Effects of immunization with CCR5-based cycloimmunogen on simian/HIVSF162P3 challenge. J Immunol 2006; 176:463-71; PMID:16365439; http://dx.doi.org/10.4049/jimmunol.176.1.463
- Zhang Y, Pool C, Sadler K, Yan HP, Edl J, Wang X, Boyd JG, Tam JP. Selection of active ScFv to G-protein-coupled receptor CCR5 using surface antigen-mimicking peptides. Biochemistry 2004; 43:12575-84; PMID:15449947; http://dx.doi.org/10.1021/bi0492152
- Katancik JA, Sharma A, de Nardin E. Interleukin 8, neutrophil-activating peptide-2 and GRO-alpha bind to and elicit cell activation via specific and different amino acid residues of CXCR2. Cytokine 2000; 12:1480-8; PMID:11023662; http://dx.doi.org/10.1006/cyto.2000.0742
- Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI, Kobilka BK, et al. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science 2007; 318:1258-65; PMID:17962520; http://dx.doi.org/10.1126/science.1150577
- Gautier A, Mott HR, Bostock MJ, Kirkpatrick JP, Nietlispach D. Structure determination of the seven-helix transmembrane receptor sensory rhodopsin II by solution NMR spectroscopy. Nat Struct Mol Biol 2010; 17:768-74; PMID:20512150
- Granier S, Manglik A, Kruse AC, Kobilka TS, Thian FS, Weis WI, Kobilka BK. Structure of the delta-opioid receptor bound to naltrindole. Nature 2012; 485:400-4; PMID:22596164; http://dx.doi.org/10.1038/nature11111
- Jaakola VP, Griffith MT, Hanson MA, Cherezov V, Chien EY, Lane JR, Ijzerman AP, Stevens RC. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science 2008; 322:1211-7; PMID:18832607; http://dx.doi.org/10.1126/science.1164772
- Wu B, Chien EY, Mol CD, Fenalti G, Liu W, Katritch V, Abagyan R, Brooun A, Wells P, Bi FC, et al. Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science 2010; 330:1066-71; PMID:20929726; http://dx.doi.org/10.1126/science.1194396
- Jacoby E, Bouhelal R, Gerspacher M, Seuwen K. The 7 TM G-protein-coupled receptor target family. ChemMedChem 2006; 1:761-82; PMID:16902930; http://dx.doi.org/10.1002/cmdc.200600134
- Ajuebor MN, Zagorski J, Kunkel SL, Strieter RM, Hogaboam CM. Contrasting roles for CXCR2 during experimental colitis. Exp Mol Pathol 2004; 76:1-8; PMID:14738862; http://dx.doi.org/10.1016/j.yexmp.2003.08.004
- Matsuo Y, Raimondo M, Woodward TA, Wallace MB, Gill KR, Tong Z, Burdick MD, Yang Z, Strieter RM, Hoffman RM, et al. CXC-chemokine/CXCR2 biological axis promotes angiogenesis in vitro and in vivo in pancreatic cancer. Int J Cancer 2009; 125:1027-37; PMID:19431209; http://dx.doi.org/10.1002/ijc.24383
- Wente MN, Keane MP, Burdick MD, Friess H, Buchler MW, Ceyhan GO, Reber HA, Strieter RM, Hines OJ. Blockade of the chemokine receptor CXCR2 inhibits pancreatic cancer cell-induced angiogenesis. Cancer Lett 2006; 241:221-7; PMID:16458421; http://dx.doi.org/10.1016/j.canlet.2005.10.041
- Bleicher KH, Green LG, Martin RE, Rogers-Evans M. Ligand identification for G-protein-coupled receptors: a lead generation perspective. Curr Opin Chem Biol 2004; 8:287-96; PMID:15183327; http://dx.doi.org/10.1016/j.cbpa.2004.04.008
- Jimonet P, Jager R. Strategies for designing GPCR-focused libraries and screening sets. Curr Opin Drug Discov Devel 2004; 7:325-33; PMID:15216936
- Howl J, Langel U, Hawtin SR, Valkna A, Yarwood NJ, Saar K, Wheatley M. Chimeric strategies for the rational design of bioactive analogs of small peptide hormones. Faseb J 1997; 11:582-90; PMID:9212082
- Ja WW, Roberts RW. G-protein-directed ligand discovery with peptide combinatorial libraries. Trends Biochem Sci 2005; 30:318-24; PMID:15950876; http://dx.doi.org/10.1016/j.tibs.2005.04.001
- Lee B, Sharron M, Montaner LJ, Weissman D, Doms RW. Quantification of CD4, CCR5, and CXCR4 levels on lymphocyte subsets, dendritic cells, and differentially conditioned monocyte-derived macrophages. Proc Natl Acad Sci U S A 1999; 96:5215-20; PMID:10220446; http://dx.doi.org/10.1073/pnas.96.9.5215
- Ohno Y, Suda K, Masuko K, Yagi H, Hashimoto Y, Masuko T. Production and characterization of highly tumor-specific rat monoclonal antibodies recognizing the extracellular domain of human L-type amino-acid transporter 1. Cancer Sci 2008; 99:1000-7; PMID:18294274; http://dx.doi.org/10.1111/j.1349-7006.2008.00770.x
- Gupta A, Decaillot FM, Gomes I, Tkalych O, Heimann AS, Ferro ES, Devi LA. Conformation state-sensitive antibodies to G-protein-coupled receptors. J Biol Chem 2007; 282:5116-24; PMID:17148456; http://dx.doi.org/10.1074/jbc.M609254200
- Lecat S, Bucher B, Mely Y, Galzi JL. Mutations in the extracellular amino-terminal domain of the NK2 neurokinin receptor abolish cAMP signaling but preserve intracellular calcium responses. J Biol Chem 2002; 277:42034-48; PMID:12185075; http://dx.doi.org/10.1074/jbc.M203606200
- Levin MC, Marullo S, Muntaner O, Andersson B, Magnusson Y. The myocardium-protective Gly-49 variant of the beta 1-adrenergic receptor exhibits constitutive activity and increased desensitization and down-regulation. J Biol Chem 2002; 277:30429-35; PMID:12034720; http://dx.doi.org/10.1074/jbc.M200681200
- Houimel M, Mazzucchelli L. Identification of biologically active peptides that inhibit binding of human CXCL8 to its receptors from a random phage-epitope library. J Leukoc Biol 2009; 85:728-38; PMID:19118103; http://dx.doi.org/10.1189/jlb.0608380
- Bannert N, Craig S, Farzan M, Sogah D, Santo NV, Choe H, Sodroski J. Sialylated O-glycans and sulfated tyrosines in the NH2-terminal domain of CC chemokine receptor 5 contribute to high affinity binding of chemokines. J Exp Med 2001; 194:1661-73; PMID:11733580; http://dx.doi.org/10.1084/jem.194.11.1661
- Comerford I, Nibbs RJ. Post-translational control of chemokines: a role for decoy receptors? Immunol Lett 2005; 96:163-74; PMID:15585320; http://dx.doi.org/10.1016/j.imlet.2004.08.018
- Mortier A, Gouwy M, Van Damme J, Proost P. Effect of posttranslational processing on the in vitro and in vivo activity of chemokines. Exp Cell Res 2011; 317:642-54; PMID:21146523; http://dx.doi.org/10.1016/j.yexcr.2010.11.016
- Slootstra JW, Puijk WC, Ligtvoet GJ, Langeveld JP, Meloen RH. Structural aspects of antibody-antigen interaction revealed through small random peptide libraries. Mol Divers 1996; 1:87-96; PMID:9237197; http://dx.doi.org/10.1007/BF01721323
- Timmerman P, Puijk WC, Meloen RH. Functional reconstruction and synthetic mimicry of a conformational epitope using CLIPS technology. J Mol Recognit 2007; 20:283-99; PMID:18074397; http://dx.doi.org/10.1002/jmr.846
- Fields CG, Lloyd DH, Macdonald RL, Otteson KM, Noble RL. HBTU activation for automated Fmoc solid-phase peptide synthesis. Pept Res 1991; 4:95-101; PMID:1815783
- Timmerman P, Beld J, Puijk WC, Meloen RH. Rapid and quantitative cyclization of multiple peptide loops onto synthetic scaffolds for structural mimicry of protein surfaces. Chembiochem 2005; 6:821-4; PMID:15812852; http://dx.doi.org/10.1002/cbic.200400374
- Vaughan TJ, Williams AJ, Pritchard K, Osbourn JK, Pope AR, Earnshaw JC, McCafferty J, Hodits RA, Wilton J, Johnson KS. Human antibodies with sub-nanomolar affinities isolated from a large non-immunized phage display library. Nat Biotechnol 1996; 14:309-14; PMID:9630891; http://dx.doi.org/10.1038/nbt0396-309
- Lloyd C, Lowe D, Edwards B, Welsh F, Dilks T, Hardman C, Vaughan T. Modelling the human immune response: performance of a 1011 human antibody repertoire against a broad panel of therapeutically relevant antigens. Protein Eng Des Sel 2009; 22:159-68; PMID:18974080; http://dx.doi.org/10.1093/protein/gzn058
- Groves M, Lane S, Douthwaite J, Lowne D, Rees DG, Edwards B, Jackson RH. Affinity maturation of phage display antibody populations using ribosome display. J Immunol Methods 2006; 313:129-39; PMID:16730741; http://dx.doi.org/10.1016/j.jim.2006.04.002
- Kristensen P, Winter G. Proteolytic selection for protein folding using filamentous bacteriophages. Fold Des 1998; 3:321-8; PMID:9806934; http://dx.doi.org/10.1016/S1359-0278(98)00044-3