Abstract
We describe protein synthesis, folding and assembly of antibody fragments and full-length aglycosylated antibodies using an Escherichia coli-based open cell-free synthesis (OCFS) system. We use DNA template design and high throughput screening at microliter scale to rapidly optimize production of single-chain Fv (scFv) and Fab antibody fragments that bind to human IL-23 and IL-13α1R, respectively. In addition we demonstrate production of aglycosylated immunoglobulin G (IgG1) trastuzumab. These antibodies are produced rapidly over several hours in batch mode in standard bioreactors with linear scalable yields of hundreds of milligrams/L over a 1 million-fold change in scales up to pilot scale production. We demonstrate protein expression optimization of translation initiation region (TIR) libraries from gene synthesized linear DNA templates, optimization of the temporal assembly of a Fab from independent heavy chain and light chain plasmids and optimized expression of fully assembled trastuzumab that is equivalent to mammalian expressed material in biophysical and affinity based assays. These results illustrate how the open nature of the cell-free system can be used as a seamless antibody engineering platform from discovery to preclinical development of aglycosylated monoclonal antibodies and antibody fragments as potential therapeutics.
Keywords: :
Introduction
Engineered single chain antibody variable fragments (scFv; e.g., blinatumomab), antigen-binding fragments (Fab; ranibizumab) and aglycosylated full-length antibodies (IgG; BMS-945429 and monovalent IgG; MetMab) are in clinical trials, or have been approved, and represent unique opportunities for the development of engineered non-glycosylated therapeutics.Citation1,Citation2 The choices for large scale expression of engineered antibodies are often dictated by economic and competitive pressures that require rapid, high yield production with short development times.Citation3,Citation4 For example, the biopharmaceutical industry has seen a rapid increase in the volumetric yields of recombinant IgGs, with fed-batch titers of several g/L in multi-week mammalian cell culture fermentations following extensive process optimization. Regardless of the antibody format, the ability to produce the protein in high yield, in an active purified form is essential.
Although scFvs, Fabs and aglycosylated IgGs have been produced in E. coli,Citation5–Citation8 bacterial expression is not considered ideal as many recombinant antibody fragments tend to accumulate as insoluble proteins due to aggregation of folding intermediates.Citation3,Citation9,Citation10 Secreted expression to the periplasm results in lower yields of soluble scFvs and IgGs,Citation8 although the expression of g/L levels of correctly folded IgGs in high-density E. coli cell culture has been reported in reference Citation5.
For soluble expression of Fabs and IgGs, the requirement for coordinated expression of independent heavy (HC) and light chains (LC) with correct folding and assembly can be challenging. Typically this is addressed using bicistronic vectors that require an ad hoc combinatorial screen of intergenic sequences varying the codon usage of the translation initiation region (TIR) or by altering the promoter strength to optimize translation.Citation8,Citation11–Citation13 Alternatively, methods have been developed to co-express proteins from independent plasmids.Citation14 This often requires recloning various open reading frames (ORFs) into each different replicating plasmid or combinatorial recombineeringCitation15 to determine which combination gives good expression. Unfortunately, the rules for which construct will work for a particular protein are not well understood, and thus each new protein requires further combinatorial plasmid screening.
Transient transfection of mammalian cells results in mAb-producing cells.Citation16 However, since the product DNA is maintained/replicated as an extrachromosomal unit, the expression ability is rapidly lost, only allowing the production of small quantities of mAbs.Citation17 Furthermore, yields of some mAbs expressed in small amounts often do not scale in high-density cell culture fermentations; in these cases, extensive and time-consuming reoptimization of stable cell lines and process parameters for large scale production of clinical material is required.
Here, we extend a scalable in vitro biochemical protein synthesis system, termed open cell-free synthesis (OCFS),Citation18,Citation19 to the production of correctly folded antibody fragments and an aglycosylated version of anti-HER2 IgG1 trastuzumab. In vitro protein synthesis systems whereby a cell extract from E. coli cells harvested in exponential phase serves as a source of ribosomes and other cellular factors for translationCitation20 offer an alternative expression system that avoids many of the problems of conventional cell-based expression technologies. The cell extract is mixed with template DNA (plasmid or linear PCR fragments), amino acids, nucleotides, T7 RNA polymerase (RNAP), and an energy source (). Appropriate disulfide isomerase chaperones can be added to aid correct formation of disulfide bonds. All of the metabolic resources can then be directed to the exclusive synthesis of only protein encoded by the template DNA, thereby generating antibodies at high titer in hours without the need for cloning and transformation of different plasmid constructs. Because the system is “open” and scalable from microliter to large-scale bioreactors, it is easily adapted for high throughput, automation or miniaturization procedures, followed by rapid process development of clinical material using a single GMP-banked cell lineCitation19 for facility flexibility and faster turnaround for multiple drug product production.
Results
High throughput screening of TIR libraries using linear DNA templates.
Translation initiation, not elongation, is rate-limiting in cell-free synthesis,Citation19 so we optimized translational efficiency by varying sequences in the translational initiation region (TIR),Citation13 upstream of a gene coding an anti-human IL-23 scFv using in vitro scanning mutagenesis of linear DNA templates.Citation21 We screened mutations in the 5′ untranslated region (UTR) that binds the S1 protein in the translational initiation complex of the 30S ribosomal subunit.Citation22 This rapid, high-throughput (HT) gene synthesis approach takes advantage of the rapidly declining costs of gene synthesis and eliminates the need to propagate, sequence and purify individual plasmid DNA subclones.
A schematic of our TIR library synthesis technique performed in 96-well format is shown in . Synthetic DNA sequences of about 200 bp containing single point mutations in the TIR were amplified by overlapping PCR with the corresponding 3′ scFv ORF and T7 terminator 1 kb fragment to yield a ca. 1,250 bp product that was used as a template for OCFS. Because linear DNA templates are susceptible to degradation by the ExoV nuclease activity of RecBCD, we added an inhibitor of this activity, the λ bacteriophage GamS protein,Citation23 for efficient high-yield protein expression (Figs. S1 and S2).
shows that for linear DNA templates, anti-hIL-23 scFv yields varied between 500 and 950 mg/L, suggesting that single point mutations in the TIR can change the translational efficiency, presumably due to differences in the binding free energy between the ribosomal S1 protein and 5′ UTR RNA. As the gene sequence design rules for protein expression are not well understood,Citation24 this predictable and scalable expression library approach offers the opportunity to rapidly screen DNA synthesized mutants for optimal expression.
Proteins expressed in microtiter plates from independent linear DNA templates show excellent reproducibility, have comparable yields to corresponding plasmid-based yields (Fig. S1), and have yields of scFvs and Fabs that are ∼10- to 1,000-fold higher than in other E. coli-based high-throughput screening (HTS) expression systems.Citation14,Citation21,Citation25,Citation26 Antibodies produced in this format are easily purified and tested using functional in vitro and cell-based assays. For example, the cell-free produced anti-hIL-23 scFv bound specifically to the p19 subunit of human IL-23,Citation27 with a KD of 10 nM, as measured by surface plasmon resonance (SPR; ). Finally, we note that the addition of λ-GamS required for linear template stability does not require reengineering the host strain.Citation28,Citation29 Thus, production of antibody variants identified from transcription-translation of PCR templates in HTS can be accurately scaled () using the same cell-free extract to rapidly produce gram-scale quantities of proteins for more detailed functional characterization, including in vivo animal models.
Rapid optimization of disulfide bond formation and Fab assembly.
Conditions of the OCFS reaction can be directly manipulated to provide an optimized environment for both protein expression and folding. In contrast, the ability of E. coli to correctly fold recombinant proteins containing multiple disulfide bonds is limited, due the reducing environment of the cytosol and the limited capacity for co-translational folding.Citation10,Citation30–Citation34 We used combinatorial optimization methods to rapidly and predictably optimize expression of a Fab that binds to the hIL-13 α1 receptor.Citation25 We optimized redox conditions with added glutathione buffers and disulfide isomerase chaperone () and tuned multiple gene expression and assembly of the heavy and light chains based on the known effect of optimizing HC:LC genes for antibody assembly in vivo.Citation5,Citation13 We determined the dependence of Fab assembly on both the ratio and absolute amounts of HC to LC genes added in the cell-free synthesis reaction (). Independent expression of HC and LC shows that both are produced at similar rates, but HC synthesis alone is subject to aggregation and proteolytic degradation, in the absence of LC expression (Fig. S3) and this may account for the relatively smaller amount of LC plasmid required for co-expression.
We took advantage of this observation by transcription-translation of the LC for 1 h, followed by addition of the HC plasmid for an additional 7 h (). A significant increase in functional Fab titer was observed under these conditions, resulting in ca. 300 mg/L Fab expression and covalent assembly. This is consistent with a requirement for coordinated folding of the LC first that serves as a scaffold template for correct folding of the HC and subsequent assembly of the Fab heterodimer. The fully purified Fab fragment shows high affinity binding to IL-13α1R by BIACORE (), and again using the optimized parameters determined at small scale, shows excellent linear scalability to gram scale production ().
In contrast to these results, in vivo optimization requires multiple genetic constructs to identify conditions for optimized assembly,Citation5 and there are the limitations of negative-selection pressure for plasmid retention and screening results that often do not scale to high-density fermentation. The ability to rapidly screen antibody heavy and light chain assembly from separate plasmids should allow VH − VL chain shufflingCitation14,Citation35 without recloning steps and without in vivo expression bias.
Production and characterization of full-length IgG.
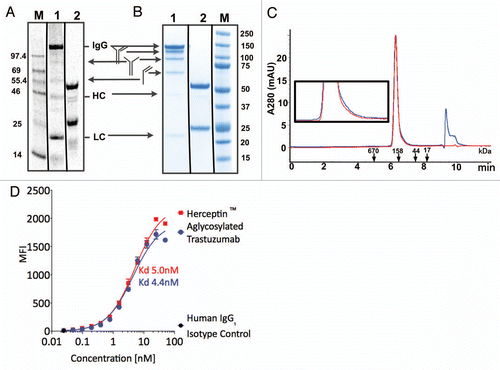
Using similar optimization procedures to those described above for production of scFv and Fab, we co-expressed the HC and LC of trastuzmab, in the presence of added disulfide isomerase chaperones DsbC or PDI. shows that a significant fraction of protein produced from the cell-free extract is fully assembled, with trace levels of non-fully assembled proteins corresponding to the sizes of the LC (ca. 25 kDa), the HC or a LC homodimer (ca. 50 kDa), a heterodimer of the LC and HC (ca 75 kDa), heterotrimer of one LC and two HC (125 kD), as well as the completely assembled heterotetrameric antibody (ca. 150 kDa). After three step purification via Protein A affinity chromatography, hydrophobic interaction chromatography and size exclusion chromatography, SDS-PAGE gel analysis () and analytical size exclusion chromatography () showed homogenous product, with a mixture of covalent and non-covalent heterotetrameric species. Endotoxin levels (2.9 EU/mg) and host cell proteins were within specifications after this purification process.
Differential scanning calorimetry analysis of thermal unfolding at pH 7.6 (Fig. S6) showed transitions corresponding to CH2 domain unfolding with melting temperatures Tm = 62°C for aglycosylated trastuzumab and Tm = 68°C for Herceptin®, as well as a second transition corresponding to the unfolding of the Fab fragment and the CH3 domains (Tm = 81°C for aglycosylated IgG1 and Tm = 83°C for Herceptin®). The 6 degree decrease in Tm for CH2 domain unfolding of the aglycosylated antibody relative to glycosylated antibody shows that glycosylation of N297 stabilizes the CH2 domain toward thermal unfolding.
The binding of IgGs to HER2 antigen that is overexpressed on the surface of SK-BR-3 cells was confirmed using Alexa-647-labeled secondary antibody () with similar equilibrium binding affinities for aglycosylated and glycosylated trastuzumab IgGs to cell-surface antigen. Similarly, both IgGs bound to immobilized Fc-Erb2 antigen with equivalent affinities as measured by biolayer interferometry (data not shown).
Discussion
We describe here a scalable approach to expression of antibody fragments and aglycosylated IgGs for discovery and preclinical development. The OCFS system produces antibodies with predictable yields of several hundreds of mg/L in hours over a million-fold range in scales in simple batch processes. Antibodies are produced at linearly scalable rates of ∼1 peptide bond per second per ribosome,Citation19 maintained over many hours of production. The antibodies are produced with high fidelity (<10−5 missense errors/codon; unpublished results), are fully soluble, non-aggregated, correctly folded, functional in biochemical and cell-based assays, and are free of inherent biological contamination such as viruses.Citation36 The open nature of the system allows us to profile the cell-free metabolome and proteome (unpublished results) and to model and rapidly test the effects of addition and subtraction of various components for system optimization, without the requirements for tuning more complex cellular networks required to maintain cell viability. Finally, the linear scalability allows antibodies identified in ribosome display-based selectionsCitation31 and HTS to be immediately scaled for production of multiple gram quantities of antibodies, thus avoiding the delays and challenges of conventional mammalian cell line development.
Prokaryotic vs. eukaryotic combined transcription-translation of antibodies.
It has been suggested that cell-free synthesis and correct assembly of multi-domain eukaryotic proteins such as Fabs and IgGs are inherently limited in prokaryotic systems due to mRNA degradation and less efficient co-translational folding.Citation30,Citation32,Citation33 Indeed, previously reported yields of Fabs are less than 50 µg/mL,Citation37,Citation38 or for an IgG ca. 1 µg/mL,Citation6 in E. coli based cell-free systems. We found however, that careful optimization of heterodimer assembly coupled with the rapid ribosomal translation rate of the OCFS are keys to enhance the yields of Fabs and IgGs. We observe high level expression of fully soluble, correctly folded, multi-domain proteins with up to 16 disulfide bonds. The absence of any significant truncated or aggregated products ( and S5) from codon-optimized templates suggests that transcription, ribosomal synthesis and protein folding are well-coordinated in the optimized OCFS reaction. We conclude that with appropriate optimization, the significantly higher translational efficiency of E. coli-based cell-free synthesis, relative to eukaryotic cell-free systems, does not limit the expression of fully functional multi-domain eukaryotic proteins.Citation34
A rapid “make-test” cycle for in vitro produced antibody libraries.
The results described here indicate the possibility of rapidly producing designed libraries of engineered antibodies in high density formats.Citation14,Citation21,Citation39 Using scanning mutagenesis libraries produced by de novo DNA synthesis with automated parallel cell-free synthesis and micropurification in plates with off-the shelf robotics equipment and disposables, it is now possible to rapidly screen designed libraries of several thousand variants in a two week “make-test cycle” from DNA design/synthesis to cell-biology assays. Such in vitro produced antibody libraries should prove particularly valuable for antibody engineering studies exploring in vitro antibody affinity evolution,Citation21,Citation39 as a rapid way of identifying variants with interesting properties that can then be rapidly scaled for more detailed structural and functional characterization. Subsequent “make-test” rounds of combinatorial variants with multiple additive mutations could be rapidly identified. This same sequential mutation approach should be useful for improving other antibody attributes such as non-natural amino acid sites of incorporation, expression level and IgG stability.
Materials and Methods
T7 plasmid design.
The previously described high copy T7-based plasmid pYD317,Citation19 used for all plasmids in this work contains kanamycin resistance gene, an engineered high-copy ColE1 origin of replication, the T7 gene 10 translational enhancer,Citation42 the Shine-Dalgarno (SD) sequence as a binding site for prokaryotic ribosomes (RBS) with an optimum distance to the start AUG codon and a strong T7 promoter sequence driving transcription. Genes were cloned in frame with the ATG start codon using Nde1 and Sal1 cloning sites. A strong T7 transcription terminator hairpin prevents elongated transcripts and 3′-terminal exonucleolytic degradation of the mRNA.Citation43 Genes coding for an scFv that binds the p19 subunit of human IL-23,Citation27 a Fab that binds to human IL13R-1αCitation25 with a C-terminal hinge containing c-myc-tag and His-tags, and trastuzumabCitation44 IgG1 HC and LC were codon optimized and synthesized (DNA 2.0, Menlo Park, CA).
Linear DNA template design.
For high-throughput cell-free expression optimization of the anti hIL-23-scFv, a library of 28 linear templates with single point mutation in each A/U rich enhancer region were individually amplified from ca. 200 bp ds DNA synthesized fragments (Biosearch Technologies, Inc., Novato, CA) containing the TIR A/U rich sequence, Shine-Dalgarno (SD) sequence, and a portion of N-terminal anti-hIL-23 scFv sequence. The C-terminal portion of the anti-hIL-23 scFv gene and the T7 terminator sequence, was amplified by 5antiI: (CAT ATG CAA GTT CAA TTA GTT CAA AGT GGT GCG GAA G) and 3chiT2.317 (GCG TAC TAG CGT ACC ACG TGG CTG GTG GCC TCG GTG AGT TTT CTC CTT C) primers using pYD317-anti-hIL-23 scFv as template. We used a GC-rich sequence ChiT2 (GCG TAC TAG CGT ACC ACG TGG CTG GTG G) for subsequent overlapping PCR amplification in 96-well format using a single primer.Citation28 The resulting single band PCR fragments were purified using PureLink™ 96 PCR Purification Kit (Invitrogen) and quantitated using PicoGreen (Invitrogen, Carlsbad, CA). Based on the titration of linear DNA, an average of >30 nM linear template DNA with 1 µM GamS,Citation23,Citation45 was used for each cell-free reaction in the 96-well plate.
Accessory protein expression and purification.
The gamS gene was amplified from pKD46,Citation46 and GamS6XHis was expressed in a 5-ml OCFS reaction. The soluble fraction after centrifugation at 14,000 g for 15 min contained 500 µg/mL protein was purified using Histrap HP columns (GE Healthcare Life Sciences, Piscataway, NJ) connected in series according to the manufacturer's instructions. The >95% pure GamS6XHis protein was dialyzed against 10 mM TRIS-acetate (pH 8.2), 60 mM potassium acetate and stored in 20% sucrose at −80°C. Yeast protein disulfide isomerase (PDI; residues 28–522 with a C-terminal 6XHis tag),Citation47 was expressed at levels of ca. 500 µg/mL in a 25-ml OCFS reaction and purified on a 1 mL HisTrap FF column according to the manufacturer's instructions. After elution with 500 mM imidazole, the PDI was buffer exchanged into 10 mM TRIS-acetate, 60 mM potassium acetate, pH 8.2 via dialysis. His6X T7 RNAP and DsbC as a molecular chaperone were produced as previously described in reference Citation19.
Cell-free reactions.
The cell-free reactions were run at 30°C in Petri dishes or microtiter plates at small scale, or in stirred tank bioreactors up to 5 L scale, as previously described in reference Citation19. To facilitate disulfide bond formation, the extract was treated with 50 µM iodoacetamide prior to use. In addition, 1–10 µM E. coli DsbC or PDI and variable ratios of GSSG and GSH were added to the cell-free reaction to promote proper folding and antibody assembly.
Determination of antibody titers.
Antibodies produced at 250 µL scale or less were monitored by incorporation of l-[U-14C]-leucine as previously described in reference Citation19. Folding and assembly were analyzed by reducing or non-reducing 12% SDS-PAGE (Invitrogen, CA) according to the manufacturer's instructions. Non-reduced samples were not boiled prior to gel loading. The covalent assembled Fab and IgG concentrations was determined from the fraction of fully assembled antibody pixels under non-reducing conditions, relative to fully reduced HC, LC and clipped product bands that correspond to the soluble protein produced, as analyzed by autoradiography using a Storm 840 PhosphoImager with ImageQuantTL software (GE Life Sciences, NJ).
At larger scales, protein concentrations were determined directly from the cell-free extract by measuring the initial rate of antigen binding with cell-free produced samples diluted >1,000- fold, using biolayer interferometry with a ForteBio Octet 384 (ForteBio, Menlo Park, CA) and comparing to pure reference standards (Fig. S4).
Purification of antibody fragments.
The anti-hIL-23 scFv was captured by cation-exchange (Fractogel SE Hicap, EMD Biosciences), eluted fractions were subjected to Toyopearl Phenyl-650M HIC (Tosoh Bioscience) and eluted with a NaCl gradient. Fractions were pooled and concentrated via tangential flow filtration and subjected to final polishing steps using DEAE Sepharose Fast Flow, (GE Lifesciences) and size exclusion chromatography on Sephacryl S200 HR, (GE Lifesciences) (Fig. S5).
Anti-IL13α1R Fab was purified from the cell free reaction by 2-fold dilution in 50 mM sodium citrate, pH 7.0 (Buffer A), clarified via centrifugation, followed by 0.2 µm filtration. The resultant clarified material was loaded onto FAbsorbent™ F1P HF column (ProMetic BioSciences Ltd., United Kingdom) previously equilibrated in Buffer A. After washing the column extensively with Buffer A, the anti-IL13α1R Fab was eluted with a linear gradient to 50 mM sodium citrate, pH 2.7. Elution fractions were immediately pH neutralized with 0.15 mL of 1 M TRIS-HCl, pH 8.5 per 1 mL of eluate. Elution fractions containing anti-IL13R Fab were pooled and concentrated via 3 kD cut-off ultrafiltration. The concentrated material purified by SEC on a Superdex 75 10/300 GL (GE Healthcare Life Sciences, Piscataway, NJ) gel filtration column equilibrated in 50 mM TRIS-HCl, 500 mM NaCl, 0.1% Polysorbate 20, pH 8.0 (Fig. S4). The correct intact mass of Fab was confirmed by mass spec analysis (data not shown) and endotoxin levels were within specifications (data not shown).
Purification of trastuzumab.
Aglycosylated trastuzumab was purified from the cell-free reaction via a three column procedure. After dilution with 0.5 volumes of PBS, clarification via centrifugation and filtration on a 1.2 µm PES membrane, the material was loaded onto a Protein A HP (GE Healthcare Life Sciences, Piscataway, NJ) column with 5 min residence time. Elution was performed using a step to 0.1 M glycine-HCl, pH 3.0, followed by immediate neutralization with 0.15 mL of 1 M TRIS-HCl, pH 8.5 per 1 mL of eluate. Removal of soluble aggregates from the Protein A HP pool was accomplished by HIC chromatography on Phenyl (high sub) Sepharose FF (GE Healthcare Life Sciences, Piscataway, NJ), after adjustment of the pooled material to a concentration of 0.75 M (NH4)2SO4. Precipitates formed upon (NH4)2SO4 addition were removed by centrifugation. HIC chromatography was performed in 50 mM NaH2PO4-NaOH, pH 7.3 with a linear gradient from 0.75 M to 0 M (NH4)2SO4. Trastuzumab eluted from the column was pooled and subsequently concentrated 36-fold on a 10 kD ultrafilter for injection on a Superdex 200 10/300 GL (GE Healthcare Life Sciences, Piscataway, NJ) size exclusion column. As the final purification step SEC was performed in 50 mM L-histidine, 300 mM NaCl, 4% (w/v) trehalose dehydrate, pH 6.0.
Real-time biomolecular interaction analysis.
Surface plasmon resonance (SPR) spectroscopy was performed on a BIAcore T100 Instrument (BIAcore, Uppsala, Sweden). The target (5 µg/ml in 10 mM Na-acetate pH 4.5) was immobilized on CM5 sensor chip (GE Healthcare, Piscataway, NJ) resulting in a surface density of ca. 2,100 response units (RU). The scFv or Fab fragment was injected at a series of appropriate concentrations whereby HBS/T (Hepes buffered saline containing 0.005% (v/v) Tween 20) was used as running buffer. Complex formation was observed at a continuous flow rate of 40 µl/min and the kinetic parameters were determined by globally fitting the data to a 1:1 Langmuir binding model with the BIAevaluation software version 4.1 (BIAcore, Piscataway, NH). The sensorgrams were corrected by double subtraction of the corresponding signals measured for the in-line control blank channel and an averaged baseline determined from several buffer blank injections.Citation48
SEC and differential scanning calorimetry analysis.
Analytical SEC was performed on a Sepax Zenix SEC-300, 7.8 × 200 mm column, pore size 300 ¥, particle size 3 µm and flow rate 1 mL/min. Differential scanning calorimetry analysis was performed on VP-Capillary DSC system (GE Healthcare Bio-Sciences Corp., Piscataway, NJ USA) at 1 mg/ml protein concentration, pH 7.6, with a scan rate of 2°C/min. Data analysis (Fig. S6) was performed using Origin 7.0 DSC Analysis software.
HER2 binding analysis for purified trastuzumab.
The binding of the purified aglycosylated trastuzumab antibody to HER2 on SK-BR-3 cells, which overexpress the HER2/c-erb-2 gene product, with over 1.5 million receptor copies per cell (ATCC #HTB-30, Manassas, VA) was compared with clinical grade Herceptin® or human serum IgG1 as a negative control (Sigma-Aldrich). SK-BR-3 cells were cultured in DMEM:Ham's F-12 (50:50), high glucose (Cellgro-Mediatech) supplemented with 10% heat-inactivated fetal bovine serum (Hyclone), 2 mM glutamax (Invitrogen, Carlsbad, CA) and 1× Pencillin/streptomycin (Cellgro Mediatech). Adherent cells were washed twice with calcium and magnesium-free Hanks Balanced Salt Solution (HBSS), harvested with HyQTase (Hyclone) and 200,000 cells per sample were incubated with serial dilutions of either aglycosylated trastuzumab, clinical grade Herceptin®, or human serum IgG1 in FACS buffer (DPBS buffer supplemented with 1% bovine serum albumin) for 60 min on ice. Cells were washed twice with ice-cold FACS buffer and incubated with either 5 µg/ml Alexa 647 labeled goat anti-human IgG secondary antibody or 1:200 Alexa 647 labeled goat anti human (Fab')2 (Jackson Immunoresearch) on ice for 1 h. All samples were washed using FACS buffer and analyzed using a BD FACSCalibur system (BD Biosciences). Mean fluorescence intensities were fitted using nonlinear regression analysis with one site specific binding equation using GraphPad Prism (v. 5.00, GraphPad Software, San Diego, CA, www.graphpad.com).
Conclusion
Open cell-free protein synthesis efficiently produces antibody fragments and full-length aglycosylated antibodies, at scales sufficient for discovery research and preclinical development purposes. The therapeutic utility of aglycosylated antibodies, including indications where effector functionCitation40,Citation41 or site-specific small-molecule drug conjugation may be required, will be determined by pharmacological parameters, including stability, biodistribution and immunogenicity in human clinical trials in concert with ongoing process development efforts to further increase titers for rapid clinical production of potential therapeutic antibodies.
Abbreviations
| Fab | = | antigen-binding fragment |
| HC | = | immunoglobulin heavy chain |
| IgG | = | immunoglobulin G |
| KD | = | dissociation equilibrium constant |
| LC | = | immunoglobulin light chain |
| OCFS | = | open cell-free synthesis |
| PDI | = | yeast protein disulfide isomerase |
| RNAP | = | RNA polymerase |
| scFv | = | single chain antibody fragment |
| SPR | = | surface plasmon resonance |
| TIR | = | translation initiation region |
| UTR | = | 5′ untranslated region |
Figures and Tables
Figure 1 Schematic diagram of the open cell-free synthesis (OCFS) system. Energy required for transcription-translation is driven by glutamate catabolism via the TCA cycle to produce reducing equivalents, primarily in the form of NADH, which fuels oxidative phosphorylation providing a stable supply of ATP over the course of the 10 h cell-free protein synthesis reaction. Addition of T7-RNA polymerase (RNAP), individual HC and LC T7-based plasmids or linear template for scFv, with amino acids drives transcription and subsequent ribosomal translation followed by protein folding, assisted by added glutathione and disulfide isomerase chaperone.
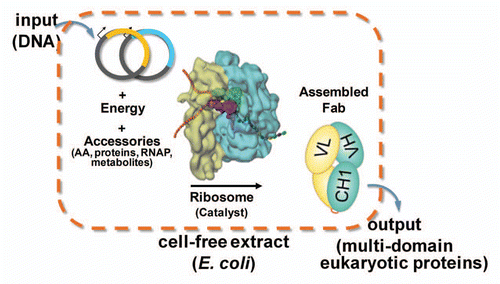
Figure 2 Schematic diagram illustrating high throughput optimization of translation initiation and scalable scFv expression. (A) T7-based TIR linear template libraries from gene synthesized 200-mer ds DNA fragments were (B) individually assembled by overlap extension PCR, purified and quantitated using PicoGreen. (C) Cell-free expression yields were significantly improved for several TIR library clones, relative to the starting, reference Citation5′ UTR sequence. Expression of duplicate clones from assembled linear DNA templates was measured by [14C]-leucine incorporation at 5 h in a 96-well format. Reactions contained ≥30 nM linear DNA template stabilized by 1 µM λ-GamS protein. Error bars correspond to 1 sec.d. of replicate protein expression yields from independently synthesized and assembled linear DNA templates. (D) BIACORE analysis confirms the functional activity of the anti-IL-23 scFv produced by the OCFS system with KD = 2 nM. (E) The kinetics of soluble production of anti-hIL-23 scFv up to 5 L is linearly scalable in the OCFS system. The concentrations of anti-hIL-23 scFv were determined by [14C]-leucine incorporation at 250 µL or less, and by BIACORE analysis at larger scales, as described in the Materials and Methods. CVs were ≤10% and the proteins were ≥95% soluble for all time points. The solid lines are for illustration purposes only.
![Figure 2 Schematic diagram illustrating high throughput optimization of translation initiation and scalable scFv expression. (A) T7-based TIR linear template libraries from gene synthesized 200-mer ds DNA fragments were (B) individually assembled by overlap extension PCR, purified and quantitated using PicoGreen. (C) Cell-free expression yields were significantly improved for several TIR library clones, relative to the starting, reference Citation5′ UTR sequence. Expression of duplicate clones from assembled linear DNA templates was measured by [14C]-leucine incorporation at 5 h in a 96-well format. Reactions contained ≥30 nM linear DNA template stabilized by 1 µM λ-GamS protein. Error bars correspond to 1 sec.d. of replicate protein expression yields from independently synthesized and assembled linear DNA templates. (D) BIACORE analysis confirms the functional activity of the anti-IL-23 scFv produced by the OCFS system with KD = 2 nM. (E) The kinetics of soluble production of anti-hIL-23 scFv up to 5 L is linearly scalable in the OCFS system. The concentrations of anti-hIL-23 scFv were determined by [14C]-leucine incorporation at 250 µL or less, and by BIACORE analysis at larger scales, as described in the Materials and Methods. CVs were ≤10% and the proteins were ≥95% soluble for all time points. The solid lines are for illustration purposes only.](/cms/asset/65d088f8-0fc6-45fd-95bc-bc44af4c2289/kmab_a_10919202_f0002.gif)
Figure 3 Rapid combinatorial optimization of Fab assembly in the OCFS system. (A) The expression and covalent assembly of soluble anti-IL13aR Fab was measured by autoradiography of non-reducing SDS PAGE of [14C]-leucine incorporated product as a function of added GSSG and GSH with 10 µM DsbC chaperone. (B) Response surface of the fraction of heavy chain (HC) plasmid, fHC and sum of added heavy and light chain (LC) plasmids on covalent assembly of anti-IL-13α1R Fab. Fab assembly was monitored by [14C]-autoradiography of non-reducing SDS-PAGE gels to quantitate the soluble Fab assembled protein yield. (C) Staggered expression of LC for 1 h, followed by addition of HC plasmid for an additional 7 h. Under optimized conditions, the yield of Fab was 300 mg/L and was >95% soluble at all time points. (D) BIAcore analysis of purified anti-hIL-13Rα1 Fab binding to immobilized hIL-13α1 R. Fab concentrations were 3.9 pM, 11.7 pM, 35 pM, 105 pM and 315 pM. The data in red were globally fit to a 1:1 isotherm as indicated by the black lines, with kon = 2 × 108 M−1s−1, koff = 4 × 10−4 s−1, KD = 2 pM. (E) The kinetics of soluble production of anti-IL-13α1R Fab up to 4 L is linearly scalable in the OCFS system. The concentrations of Fab were determined by [14C]-leucine incorporation at 250 µL or less, and by ForteBio analysis at larger scales (Fig. S4). CVs were ≤10% and the proteins were ≥95% soluble for all time points. The solid line is for illustration purposes only.
![Figure 3 Rapid combinatorial optimization of Fab assembly in the OCFS system. (A) The expression and covalent assembly of soluble anti-IL13aR Fab was measured by autoradiography of non-reducing SDS PAGE of [14C]-leucine incorporated product as a function of added GSSG and GSH with 10 µM DsbC chaperone. (B) Response surface of the fraction of heavy chain (HC) plasmid, fHC and sum of added heavy and light chain (LC) plasmids on covalent assembly of anti-IL-13α1R Fab. Fab assembly was monitored by [14C]-autoradiography of non-reducing SDS-PAGE gels to quantitate the soluble Fab assembled protein yield. (C) Staggered expression of LC for 1 h, followed by addition of HC plasmid for an additional 7 h. Under optimized conditions, the yield of Fab was 300 mg/L and was >95% soluble at all time points. (D) BIAcore analysis of purified anti-hIL-13Rα1 Fab binding to immobilized hIL-13α1 R. Fab concentrations were 3.9 pM, 11.7 pM, 35 pM, 105 pM and 315 pM. The data in red were globally fit to a 1:1 isotherm as indicated by the black lines, with kon = 2 × 108 M−1s−1, koff = 4 × 10−4 s−1, KD = 2 pM. (E) The kinetics of soluble production of anti-IL-13α1R Fab up to 4 L is linearly scalable in the OCFS system. The concentrations of Fab were determined by [14C]-leucine incorporation at 250 µL or less, and by ForteBio analysis at larger scales (Fig. S4). CVs were ≤10% and the proteins were ≥95% soluble for all time points. The solid line is for illustration purposes only.](/cms/asset/de79285d-caa2-489a-b691-0b856a365807/kmab_a_10919202_f0003.gif)
Figure 4 Analytical characterization of aglycosylated trastuzumab produced in the OCFS system. (A) Cell-free reaction product pool was visualized, using SDS-PAGE, by 14C autoradiography of leucine incorporation at 10 h. that measures only protein produced. Lane M: MW Marker, Lane 1, non-reducing, Lane 2, reducing. Autoradiography of the cell-free reaction product indicates 14C-leucine incorporation corresponding to ca. 400 mg/L production of full-length trastuzumab IgG1, as well as HC and LC, and confirms that only antibody was produced because the T7 RNA polymerase limits transcription to the T7 promoter on the plasmid added to the reaction, with no evidence for aberrant protein products produced from native RNA polymerase transcription. (B) SDS-PAGE analysis of purified protein. Following purification, non-reduced and reduced samples were analyzed with Coomassie blue staining following electrophoresis. Lane 1: 2 µg aglycosylated trastuzumab-non-reducing, Lane 2: 2 µg aglycosylated trastuzumab-reducing, Lane M: Precision Plus (BioRad) Marker Lane. (C) Analytical size exclusion chromatography of Herceptin® (red) and aglycosylated trastuzumab (blue). The peak at 9.5–10 min corresponds to buffer. (Inset) Enlarged view of traces. (D) FACS analysis of aglycosylated trastuzumab produced by OCFS and Herceptin® binding to HER2 overexpressing SK-BR-3 cells. The equilibrium KD corresponds to 5.0 nM and 4.4 nM, respectively. Control experiments showed that binding of aglycosylated trastuzumab is specifically competed with Herceptin® (data not shown).
Additional material
Download Zip (1.2 MB)Acknowledgments
We would like to thank Albert Wong from Biosearch Technologies for DNA library design, Carolyn Koo for BIACORE analysis, and Tyler Heibeck, Dean Ng, Evan Green and Albert Boot for excellent technical assistance. Special thanks to Leslie McEvoy, Christopher Thanos, Ramesh Baliga and Henry Heinsohn for scientific discussions and guidance.
Related Research Data
References
- Nelson AL, Reichert JM. Development trends for therapeutic antibody fragments. Nat Biotechnol 2009; 27:331 - 337; PMID: 19352366; http://dx.doi.org/10.1038/nbt0409-331
- Nelson AL. Antibody fragments: hope and hype. MAbs 2010; 2:77 - 83; PMID: 20093855; http://dx.doi.org/10.4161/mabs.2.1.10786
- Swartz JR. Advances in Escherichia coli production of therapeutic proteins. Curr Opin Biotechnol 2001; 12:195 - 201; PMID: 11287237; http://dx.doi.org/10.1016/S0958-1669(00)00199-3
- Andersen DC, Reilly DE. Production technologies for monoclonal antibodies and their fragments. Curr Opin Biotechnol 2004; 15:456 - 462; PMID: 15464378; http://dx.doi.org/10.1016/j.copbio.2004.08.002
- Simmons LC, Reilly D, Klimowski L, Raju TS, Meng G, Sims P, et al. Expression of full-length immunoglobulins in Escherichia coli: rapid and efficient production of aglycosylated antibodies. J Immunol Methods 2002; 263:133 - 147; PMID: 12009210; http://dx.doi.org/10.1016/S0022-1759(02)00036-4
- Frey S, Haslbeck M, Hainzl O, Buchner J. Synthesis and characterization of a functional intact IgG in a prokaryotic cell-free expression system. Biol Chem 2008; 389:37 - 45; PMID: 18095868; http://dx.doi.org/10.1515/BC.2008.007
- Hakim R, Benhar I. “Inclonals”: IgGs and IgG-enzyme fusion proteins produced in an E. coli expression-refolding system. MAbs 2009; 1:281 - 287; PMID: 20065645; http://dx.doi.org/10.4161/mabs.1.3.8492
- Makino T, Skretas G, Kang TH, Georgiou G. Comprehensive engineering of Escherichia coli for enhanced expression of IgG antibodies. Metab Eng 2011; 13:241 - 251; PMID: 21130896; http://dx.doi.org/10.1016/j.ymben.2010.11.002
- Glockshuber R, Schmidt T, Plückthun A. The disulfide bonds in antibody variable domains: effects on stability, folding in vitro and functional expression in Escherichia coli. Biochemistry 1992; 31:1270 - 1279; PMID: 1736986; http://dx.doi.org/10.1021/bi00120a002
- Baneyx F, Mujacic M. Recombinant protein folding and misfolding in Escherichia coli. Nat Biotechnol 2004; 22:1399 - 1408; PMID: 15529165; http://dx.doi.org/10.1038/nbt1029
- Pfleger BF, Pitera DJ, Smolke CD, Keasling JD. Combinatorial engineering of intergenic regions in operons tunes expression of multiple genes. Nat Biotechnol 2006; 24:1027 - 1032; PMID: 16845378; http://dx.doi.org/10.1038/nbt1226
- Levy R, Weiss R, Chen G, Iverson BL, Georgiou G. Production of correctly folded Fab antibody fragment in the cytoplasm of Escherichia coli trxB gor mutants via the coexpression of molecular chaperones. Protein Expr Purif 2001; 23:338 - 347; PMID: 11676610; http://dx.doi.org/10.1006/prep.2001.1520
- Simmons LC, Yansura DG. Translational level is a critical factor for the secretion of heterologous proteins in Escherichia coli. Nat Biotechnol 1996; 14:629 - 634; PMID: 9630956; http://dx.doi.org/10.1038/nbt0596-629
- Mao H, Graziano JJ, Chase TM, Bentley CA, Bazirgan OA, Reddy NP, et al. Spatially addressed combinatorial protein libraries for recombinant antibody discovery and optimization. Nat Biotechnol 2010; 28:1195 - 1202; PMID: 20972421; http://dx.doi.org/10.1038/nbt.1694
- Bieniossek C, Nie Y, Frey D, Olieric N, Schaffitzel C, Collinson I, et al. Automated unrestricted multigene recombineering for multiprotein complex production. Nat Methods 2009; 6:447 - 450; PMID: 19412171; http://dx.doi.org/10.1038/nmeth.1326
- Rita Costa A, Elisa Rodrigues M, Henriques M, Azeredo J, Oliveira R. Guidelines to cell engineering for monoclonal antibody production. Eur J Pharm Biopharm 2010; 74:127 - 138; PMID: 19853660; http://dx.doi.org/10.1016/j.ejpb.2009.10.002
- Kunaparaju R, Liao M, Sunstrom NA. Epi-CHO, an episomal expression system for recombinant protein production in CHO cells. Biotechnol Bioeng 2005; 91:670 - 677; PMID: 15948170; http://dx.doi.org/10.1002/bit.20534
- Jewett MC, Calhoun KA, Voloshin A, Wuu JJ, Swartz JR. An integrated cell-free metabolic platform for protein production and synthetic biology. Mol Syst Biol 2008; 4:220; PMID: 18854819; http://dx.doi.org/10.1038/msb.2008.57
- Zawada JF, Yin G, Steiner AR, Yang J, Naresh A, Roy SM, et al. Microscale to manufacturing scale-up of cell-free cytokine production—a new approach for shortening protein production development timelines. Biotechnol Bioeng 2011; 108:1570 - 1578; PMID: 21337337; http://dx.doi.org/10.1002/bit.23103
- Zubay G. In vitro synthesis of protein in microbial systems. Annu Rev Genet 1973; 7:267 - 287; PMID: 4593305; http://dx.doi.org/10.1146/annurev.ge.07.120173.001411
- Burks EA, Chen G, Georgiou G, Iverson BL. In vitro scanning saturation mutagenesis of an antibody binding pocket. Proc Natl Acad Sci USA 1997; 94:412 - 417; PMID: 9012796; http://dx.doi.org/10.1073/pnas.94.2.412
- Boni IV, Isaeva DM, Musychenko ML, Tzareva NV. Ribosome-messenger recognition: mRNA target sites for ribosomal protein S1. Nucleic Acids Res 1991; 19:155 - 162; PMID: 2011495; http://dx.doi.org/10.1093/nar/19.1.155
- Sitaraman K, Esposito D, Klarmann G, Le Grice SF, Hartley JL, Chatterjee DK. A novel cell-free protein synthesis system. J Biotechnol 2004; 110:257 - 263; PMID: 15163516; http://dx.doi.org/10.1016/j.jbiotec.2004.02.014
- Allert M, Cox JC, Hellinga HW. Multifactorial determinants of protein expression in prokaryotic open reading frames. J Mol Biol 2010; 402:905 - 918; PMID: 20727358; http://dx.doi.org/10.1016/j.jmb.2010.08.010
- Su B, Hrin R, Harvey BR, Wang YJ, Ernst RE, Hampton RA, et al. Automated high-throughput purification of antibody fragments to facilitate evaluation in functional and kinetic based assays. J Immunol Methods 2007; 322:94 - 103; PMID: 17362979; http://dx.doi.org/10.1016/j.jim.2007.02.006
- Ryabova LA, Desplancq D, Spirin AS, Plückthun A. Functional antibody production using cell-free translation: effects of protein disulfide isomerase and chaperones. Nat Biotechnol 1997; 15:79 - 84; PMID: 9035111; http://dx.doi.org/10.1038/nbt0197-79
- Beyer BM, Ingram R, Ramanathan L, Reichert P, Le HV, Madison V, et al. Crystal structures of the pro-inflammatory cytokine interleukin-23 and its complex with a high-affinity neutralizing antibody. J Mol Biol 2008; 382:942 - 955; PMID: 18708069; http://dx.doi.org/10.1016/j.jmb.2008.08.001
- Woodrow KA, Swartz JR. A sequential expression system for high-throughput functional genomic analysis. Proteomics 2007; 7:3870 - 3879; PMID: 17960738; http://dx.doi.org/10.1002/pmic.200700471
- Michel-Reydellet N, Woodrow K, Swartz J. Increasing PCR fragment stability and protein yields in a cell-free system with genetically modified Escherichia coli extracts. J Mol Microbiol Biotechnol 2005; 9:26 - 34; PMID: 16254443; http://dx.doi.org/10.1159/000088143
- Endo Y, Sawasaki T. Cell-free expression systems for eukaryotic protein production. Curr Opin Biotechnol 2006; 17:373 - 380; PMID: 16828277; http://dx.doi.org/10.1016/j.copbio.2006.06.009
- Jermutus L, Ryabova LA, Plückthun A. Recent advances in producing and selecting functional proteins by using cell-free translation. Curr Opin Biotechnol 1998; 9:534 - 548; PMID: 9821285; http://dx.doi.org/10.1016/S0958-1669(98)80042-6
- Maier T, Ferbitz L, Deuerling E, Ban N. A cradle for new proteins: trigger factor at the ribosome. Curr Opin Struct Biol 2005; 15:204 - 212; PMID: 15837180; http://dx.doi.org/10.1016/j.sbi.2005.03.005
- Kawasaki T, Gouda MD, Sawasaki T, Takai K, Endo Y. Efficient synthesis of a disulfide-containing protein through a batch cell-free system from wheat germ. Eur J Biochem 2003; 270:4780 - 4786; PMID: 14622267; http://dx.doi.org/10.1046/j.1432-033.2003.03880.x
- Kolb VA, Makeyev EV, Spirin AS. Co-translational folding of an eukaryotic multidomain protein in a prokaryotic translation system. J Biol Chem 2000; 275:16597 - 16601; PMID: 10748063; http://dx.doi.org/10.1074/jbc.M002030200
- Collet TA, Roben P, O'Kennedy R, Barbas CF 3rd, Burton DR, Lerner RA. A binary plasmid system for shuffling combinatorial antibody libraries. Proc Natl Acad Sci USA 1992; 89:10026 - 10030; PMID: 1438192; http://dx.doi.org/10.1073/pnas.89.21.10026
- Bethencourt V. Virus stalls Genzyme plant. Nat Biotechnol 2009; 27:681; PMID: 19668157; http://dx.doi.org/10.1038/nbt0809-681a
- Oh IS, Lee JC, Lee MS, Chung JH, Kim DM. Cell-free production of functional antibody fragments. Bioprocess Biosyst Eng 2010; 33:127 - 132; PMID: 19701777; http://dx.doi.org/10.1007/s00449-009-0372-3
- Jiang X, Ookubo Y, Fujii I, Nakano H, Yamane T. Expression of Fab fragment of catalytic antibody 6D9 in an Escherichia coli in vitro coupled transcription/translation system. FEBS Lett 2002; 514:290 - 294; PMID: 11943168; http://dx.doi.org/10.1016/S0014-5793(02)02383-9
- Rajpal A, Beyaz N, Haber L, Cappuccilli G, Yee H, Bhatt RR, et al. A general method for greatly improving the affinity of antibodies by using combinatorial libraries. Proc Natl Acad Sci USA 2005; 102:8466 - 8471; PMID: 15939870; http://dx.doi.org/10.1073/pnas.0503543102
- Sazinsky SL, Ott RG, Silver NW, Tidor B, Ravetch JV, Wittrup KD. Aglycosylated immunoglobulin G1 variants productively engage activating Fc receptors. Proc Natl Acad Sci USA 2008; 105:20167 - 20172; PMID: 19074274; http://dx.doi.org/10.1073/pnas.0809257105
- Jung ST, Reddy ST, Kang TH, Borrok MJ, Sandlie I, Tucker PW, et al. Aglycosylated IgG variants expressed in bacteria that selectively bind FcgammaRI potentiate tumor cell killing by monocyte-dendritic cells. Proc Natl Acad Sci USA 2010; 107:604 - 609; PMID: 20080725; http://dx.doi.org/10.1073/pnas.0908590107
- Olins PO, Devine CS, Rangwala SH, Kavka KS. The T7 phage gene 10 leader RNA, a ribosome-binding site that dramatically enhances the expression of foreign genes in Escherichia coli. Gene 1988; 73:227 - 235; PMID: 3072257; http://dx.doi.org/10.1016/0378-1119(88)90329-0
- Ahn JH, Kang TJ, Kim DM. Tuning the expression level of recombinant proteins by modulating mRNA stability in a cell-free protein synthesis system. Biotechnol Bioeng 2008; 101:422 - 427; PMID: 18404770; http://dx.doi.org/10.1002/bit.21884
- Cho HS, Mason K, Ramyar KX, Stanley AM, Gabelli SB, Denney DW Jr, et al. Structure of the extracellular region of HER2 alone and in complex with the Herceptin Fab. Nature 2003; 421:756 - 760; PMID: 12610629; http://dx.doi.org/10.1038/nature01392
- Sitaraman K, Chatterjee DK. High-throughput protein expression using cell-free system. Methods Mol Biol 2009; 498:229 - 244; PMID: 18988029; http://dx.doi.org/10.1007/978-1-59745-196-3_15
- Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA 2000; 97:6640 - 6645; PMID: 10829079; http://dx.doi.org/10.1073/pnas.120163297
- Tian G, Xiang S, Noiva R, Lennarz WJ, Schindelin H. The crystal structure of yeast protein disulfide isomerase suggests cooperativity between its active sites. Cell 2006; 124:61 - 73; PMID: 16413482; http://dx.doi.org/10.1016/j.cell.2005.10.044
- Myszka DG. Improving biosensor analysis. J Mol Recognit 1999; 12:279 - 284; PMID: 10556875; http://dx.doi.org/10.1002/(SICI)1099-1352(199909/10)12:5<279::AID-JMR473>3.0.CO;2-3