Abstract
The MHC class I-like Fc receptor (FcRn) is an intracellular trafficking Fc receptor that is uniquely responsible for the extended serum half-life of antibodies of the IgG subclass and their ability to transport across cellular barriers. By performing these functions, FcRn affects numerous facets of antibody biology and pathobiology. Its critical role in controlling IgG pharmacokinetics has been leveraged for the design of therapeutic antibodies and related biologics. FcRn also traffics serum albumin and is responsible for the enhanced pharmacokinetic properties of albumin-conjugated therapeutics. The understanding of FcRn and its therapeutic applications has been limited by a paucity of reliable serological reagents against human FcRn. Here, we describe the properties of a new panel of highly specific monoclonal antibodies (mAbs) directed against human FcRn with diverse epitope specificities. We show that this antibody panel can be used to study the tissue expression pattern of human FcRn, to selectively block IgG and serum albumin binding to human FcRn in vitro and to inhibit FcRn function in vivo. This mAb panel provides a powerful resource for probing the biology of human FcRn and for the evaluation of therapeutic FcRn blockade strategies.
Key words:
Introduction
Although originally referred to as the neonatal Fc receptor, FcRn influences IgG serum levels and tissue distribution at all stages of life.Citation1–Citation5 This divergent member of the MHC class I family molecule is a heterodimer composed of an evolutionally distinct α-chain in complex with the β2-microglobulin (β2M) light chain that is common to most other class I molecules. FcRn is an intracellular trafficking, integral membrane Fc receptor for IgG. It resides primarily in the early acidic endosomes where it captures endocytosed IgG by binding to the Fc region only at a low pH.Citation6–Citation8 FcRn rescues bound IgG from degradation in the lysosomal compartment and transports its ligand to the cell surface for release at neutral extracellular pH. Through this mechanism, FcRn is responsible for the long serum half-life of IgG.Citation9 FcRn transports IgG across the tight epithelial barriers of the endothelium and mucosa, thus influencing its bioavailability.Citation7,Citation10 In antigen presenting cells, FcRn controls the presentation of antigens in IgG immune complexes to T cellsCitation11,Citation12 and IgG-Fc fusion protein vaccines.Citation13,Citation14 FcRn also controls serum albumin homeostasis by an analogous trafficking mechanism as for IgG.Citation5,Citation15,Citation16 These properties have proven to be critical for maximizing the pharmacokinetics immunofluorescence and bioavailability of IgG monoclonal antibodies (mAbs) and IgG-Fc fusion proteins and albumin conjugated protein therapeutics.Citation5,Citation17,Citation18 While the biological and therapeutic importance of FcRn's varied and expanding functional attributes are increasingly documented, major gaps remain in the understanding of the cell biology of FcRn, its tissue sites of action, and methods to exploit this biology for therapeutic purposes.
Of particular relevance is the need for reliable serological reagents for detection of human FcRn (hFcRn) and for probing its biology in normal and pathological states. This report describes the generation, characterization and applications of a part of mAbs with varied epitope specificities directed against human (h)FcRn. Our data demonstrate that the mAbs can readily detect hFcRn by flow cytometry and on tissue sections, which makes them valuable tools for defining the sites of hFcRn expression. Furthermore, the ability of certain mAbs to differentially block hIgG or human serum albumin binding supports a model of distinct binding sites for these ligands to hFcRn.Citation5,Citation16 Finally, treatment of hFcRn transgenic mice with a hIgG blocking mAb provides a proof-of-principle that anti-hFcRn mAbs can be applied therapeutically to specifically control the serum persistence of human IgG in vivo.
Results
Screening assay validation and antisera generation.
We first established a flow cytometry-based assay suitable for detecting antibody activity specific for human and mouse FcRn and for the assessment of the binding and functional blockade of hIgG binding. An enhanced green fluorescent protein (GFP) was spliced between the signal sequences and the N-terminus of FcRn constructs with truncated cytoplasmic, endosomal targeting domains (). This truncation resulted in robust expression of the hFcRn-GFP chimeric proteins on the plasma membrane as first validated by confocal immunofluorescence (IF) imaging of transiently transfected HeLa cells () and then by flow cytometric analysis of HEK293 cells stably transfected with the same construct (). Epitope tagging with GFP did not alter the normal pH-dependent binding of hFcRn to hIgG. The hFcRn-GFP fusion protein bound hIgG at acidic pH 6 but not neutral pH ( and B). Similar acidic pH-dependent binding of hIgG was observed for HeLa and 293 cells transfected with the mFcRn-GFP construct (data not shown).
To generate anti-hFcRn antisera, we immunized B6 wild-type (WT) and FcRn-/- mice with splenocytes from hFcRn transgenic mice. Plasma samples from immunized mice were first screened for their ability to bind hFcRn at a neutral pH to ensure that the antibodies bound hFcRn by their antigen binding domains rather than through the Fc region. Next, to determine whether the antisera could inhibit the binding of hIgG, we assessed their ability to compete with Alexa Fluor® 647-labeled hIgG (hIgGAF647) for binding to GFP-tagged human FcRn expressed on the surface of 293 cells (293hFcRn-GFP) cells at pH 6.0. The antisera showed different binding and blocking properties. For example, antiserum 26,162 bound hFcRn at pH 7.2 and blocked hIgGAF647 binding at pH 6.0 (, top). In contrast, antiserum 26898 bound, but did not block, hIgGAF647 binding to hFcRn (, middle). Lastly, some antisera, such as antiserum 26902, showed neither hFcRn binding nor blocking activity (, bottom).
Generation of anti-FcRn antibody-producing hybridomas.
Mice with antisera showing hFcRn binding or blocking activity were used for hybridoma generation. Supernatants from hybridomas were assayed for their binding to hFcRn using 293hFcRn-GFP cells in a cellular enzyme-linked immunosorbent assay (ELISA), and those showing activity against hFcRn in this assay were subcloned to ensure stable antibody production. mAbs from these hybridomas were purified, conjugated to Alexa Fluor 647, and tested in the previously described flow cytometry assay for their ability to bind at pH 7.2 to human or mouse FcRn. In total, nine unique hybridoma clones were established. Only one, DVN24, showed appreciable cross-reactivity to mFcRn (). The relative binding affinity of each mAb for hFcRn was estimated by its ability to bind 293hFcRn-GFP cells by quantifying the mean fluorescent intensity (MFI) of titered mAb concentrations. Most of the mAbs demonstrated comparably high affinity for hFcRn, with the exception of ADM12, which demonstrated a somewhat lower affinity ().
To evaluate the epitopes of hFcRn targeted by the mAbs, the ability of the mAbs to cross-compete for binding to hFcRn on 293hFcRn-GFP cells was determined (). The binding patterns demonstrated a spectrum of epitopes that were consistent with both unique and shared hFcRn binding sites. ADM11, DVN21 and DVN24 appear to have unique epitopes on hFcRn since they did not compete with the other mAbs in the competitive binding assay. DVN1, ADM31 and ADM32 were able to compete with each other, which suggests that they bind to overlapping epitopes. DVN22 and DVN23 were each capable of blocking the other's binding to hFcRn. Taken together, this mAb panel identified at least five distinct epitopes on hFcRn.
Confocal IF imaging of hFcRn using the mAbs.
We then assessed the ability of the mAbs to stain hFcRn in fresh frozen tissue sections from hFcRn transgenic mice (on a mFcRn-/- background) or from patient samples (). ADM31AF647 readily detected hFcRn in the hFcRn transgenic mouse and human kidney sections ( and D). Using ADM31AF647, the FcRn signal was strongest in the proximal tubules and the the glomerulus ( and E), consistent with the known expression pattern of FcRn in the kidney.Citation19,Citation20 As a negative control, ADM31 failed to stain kidney sections from B6 mice, confirming its binding specificity for hFcRn (). Other tissue types, including heart, brain, liver, muscle and spleen, similarly demonstrated patterns of hFcRn staining consistent with previous reports (data not shown). The other mAbs in the part displayed similar tissue staining profiles for hFcRn, with the exception of DNV24, which cross-reacted with mFcRn (data not shown). These results demonstrate the utility of the mAbs for defining the cellular and tissue distribution of human FcRn.
Inhibition of hIgG and HSA binding in vitro.
The potential of each mAb clone to inhibit the pH-dependent binding of hIgG or human serum albumin (HSA) to hFcRn was analyzed using an in vitro binding competition assay. In this assay, 293hFcRn-GFP cells were treated with increasing concentrations of each mAb clone before labeled hIgG or HSA was added. Under these competitive conditions, the bound hIgG or HSA was quantified by flow cytometry (). Both DVN24 and DVN21 potently blocked hIgG binding to hFcRn, with the maximum effect at 3 µg/mL and 10 µg/mL, respectively (; representative scatter plots results for DVN24, B). In contrast, DVN23, ADM31 and ADM32 most efficiently blocked HSA binding, leaving IgG binding intact (; representative scatter plots results for ADM31, D). These results showed that certain mAbs block the pH-dependent binding of hIgG and albumin selectively. Thus, the different epitopes on hFcRn recognized by our mAb part are consistent with published results that support spatially distinct IgG-Fc and HSA binding sites on FcRn.Citation5,Citation15,Citation16,Citation21
Treatment with anti-hFcRn mAbs in vivo.
We next investigated whether the newly generated mAbs that selectively blocked hIgG binding to hFcRn could accelerate the clearance of IgG antibodies in vivo. For these studies, we focused primarily on DVN24 because it was the most effective inhibitor of hIgG binding to hFcRn in vitro (). Tracer amounts of hIgG, mIgG1 and mIgA were injected into hFcRn transgenic mice to establish β-phase clearance baselines before the mice were treated with varying doses of DVN24 or an isotype-matched mAb ( and B). Mice treated with DVN24 showed 2.6- to 8.2-fold reduced concentrations of hIgG in serum compared with mice treated with a similar amount of isotype control mAb, with the maximal effect observed at 1 mg dosages. We also determined whether the therapeutic administration of DVN24 affected the serum persistence of HSA. DVN24 failed to promote the clearance of tracer HSA while promoting the clearance of hIgG in the same mice (). These results are consistent with the in vitro blockade studies described above (), and support the binding specificity of DVN24 to a site on hFcRn that specifically interacts with hIgG and results in the acceleration of hIgG clearance in vivo without affecting the half-life of HSA.
Since DVN24 cross-reacts with mFcRn (), we addressed whether administration of this mAb could influence the serum persistence of mIgG in mice. Treatment of B6 WT mice with DVN24 resulted in an approximate 2.8-fold reduction of tracer mIgG1 compared with treatment with the HSA-blocking mAb ADM31, which had a negligible effect (). While the treatment was not as efficient as that found in the hFcRn studies, the results indicate that the mAbs show both species and epitope specificity in this in vivo system.
Discussion
The overall goal of this study was to develop and characterize a diverse panel of mAbs with specificity for hFcRn and to explore their applications in vitro and in vivo. This effort was motivated primarily by the need for reliable serological reagents to probe the biology of hFcRn. Immunization of mice with cells from hFcRn-transgenic mice resulted in a panel of nine independently derived, stable hybridoma clones that secrete mAbs specific for native heavy chain determinants of hFcRn. The applications of the antibody part are summarized in . We show here that these mAbs have promise as incisive reagents capable of unlocking a deeper understanding of the tissue and cellular expression patterns of hFcRn, which may facilitate a better understanding of FcRn biology and lead to development of improved therapeutics. Whether the mAb panel may be applied to the study of FcRn in other mammalian species, including primates, remains to be determined.
The major function of FcRn is to bind IgG and serum albumin in endosomal compartments and rescue these ligands by returning them intact to the cell surface. This trafficking is highly dependent on pH because FcRn requires an acidic pH (∼6) for either ligand to bind with high affinity. While binding of both IgG and HSA are pH-dependent, their binding to FcRn appear to occur at geographically distinct sites.Citation5,Citation15,Citation16,Citation21 FcRn binding to the Fc region of IgG is mediated by protonation of histidine residues on the Fc fragment that then bind to acidic residues on the a1 and a2 domains of FcRn.Citation22,Citation23 Less is known about the FcRn-albumin interaction surface, but recent studies support a similar histidine-dependent mechanism in which protonated histidine residue (H166) on FcRn engages acidic Fc residues.Citation15,Citation21 As summarized in , the binding patterns of the mAb part observed in competition experiments in vitro identified both unique and overlapping epitopes on hFcRn. These studies, in combination with the ability of certain anti-hFcRn mAbs to inhibit the pH-dependent binding of hFcRn to hIgG (DVN21 and DVN24) with others inhibiting the binding of HSA (DVN23, ADM31 and ADM32), suggest that the mAb epitope footprints are discrete and spatially distinct. Direct biochemical measurements using methods including surface plasmon resonance could be used more to exactly define the mAb affinities and epitope specificities. The anti-FcRn mAb panel can therefore serve as a novel and useful tool for probing interactions of hFcRn with its ligands and their trafficking patterns. For example, two mAbs, DVN1 and DVN22, require a neutral pH to bind hFcRn. At this neutral pH, one (DVN1) cross-competes with ADM31 and ADM32. The fact that ADM31 and ADM32 are able to bind hFcRn and block HSA binding at an acidic pH while DVN1 binds this site only at a neutral pH may be of use for probing pH-sensitive HSA/FcRn interactions along the recycling pathway using ultramicroscopic/ultrastructural techniques.
Autoantibodies with IgG isotypes contribute to a wide range of autoimmune disorders, including Goodpasture's syndome, myasthenia gravis, idiopathic thrombocytopenic purpura (ITP), lupus and rheumatoid arthritis. Through its ability to rescue IgG from destruction in the lysosomal compartment, FcRn is responsible for the maintenance of high concentrations of IgG in circulation. FcRn-deficient mice are more resistant to autoimmune diseases caused by pathogenic IgG autoantibodies because they are unable to maintain the high concentrations of serum IgG.Citation24–Citation26 These studies lead to the concept that the therapeutic blockade of FcRn can be used to treat disease. The palliative effects of high dose administration of IgG in a number of autoimmune disorders can be explained at least partially by the ability of this treatment to saturate FcRn-mediated protection of IgG, resulting in rapid clearance of pathogenic IgG autoantibodies.Citation27–Citation29 Blockade strategies specifically directed at FcRn include the use of IgG mAbs genetically engineered in the Fc region for high affinity binding to FcRn (referred to as AbDegs),Citation30–Citation32 and synthetic peptides that bind the Fc engagement site of FcRn.Citation33
Blockade using mAbs specifically directed against FcRn is an obvious alternative therapeutic approach to promote the clearance of potentially pathogenic IgG. Proof of principle for mAb blockade of FcRn has been demonstrated previously in a rat model of experimentally-induced autoimmune myasthenia gravis in which treatment of the FcRn heavy chain-specific mAb 1G3 lead to a transient reduction of serum IgG concentrations as well as the severity of disease.Citation34 However, similar studies were not conducted for hFcRn. This approach is therapeutically relevant because hFcRn has more stringent binding requirements for IgG than rodent FcRn. Therefore, a ‘humanized’ FcRn system is needed to test antibodies and therapies that may be applied to patients. To this end, we utilized hFcRn-transgenic mice to test the feasibility of hFcRn blockade by epitope-specific antibodies. We first screened each anti-hFcRn mAb that we had generated for its ability to block IgG binding to hFcRn in vitro. IgG binding blockade was not a consistent feature in that of the seven anti-hFcRn mAbs tested that bound hFcRn at an acidic (endosomal) pH, only DVN21 and DVN24 effectively blocked hIgG binding in vitro (Table 2), and only DVN24 demonstrated therapeutic efficacy by accelerating the clearance of hIgG in vivo (; negative data for DVN21 are not shown). The lack of therapeutic effect with the other anti-hFcRn mAbs, which bind hFcRn avidly and, therefore, presumably remain bound to hFcRn but do not impede the endosomal trafficking of its normal molecular cargo, can be interpreted as support for the durability of the FcRn recycling pathway.
DVN24 is the only mAb identified to have a therapeutic affect on hIgG clearance in the hFcRn Tg model. Since the Fc fragment of mIgG minimally binds hFcRn,Citation35 this therapeutic effect is facilitated solely by its antigen specificity for FcRn. However, the effect proved to be transient and required multiple dosages of >50/mg/kg to achieve an appreciable therapeutic effect. This transience is very likely the result of the short serum persistence of native DVN24, which virtually disappears from the circulation within hours after administration (results not shown). Whether the therapeutic efficiency of DVN24 will be enhanced by the replacement of the mouse Fc with the human IgG-Fc, thus enabling it to capitalize on hFcRn's normal recycling function, remains to be determined. Finally, it is of interest that DVN24 was also the only mAb identified that cross-reacts with mFcRn. Administration of this mAb accelerated the clearance of mIgG1 in B6 WT mice, albeit less efficiently than its effect on hIgG clearance in hFcRn Tg mice. DVN24, therefore, has the dual potential for investigating the effects of the therapeutic blockade of FcRn in humanized and standard mouse models.
Materials and Methods
Mice.
C57BL6/J (B6) mice. FcRn knockout mice on the C57BL6/J background (designated B6.129X1-Fcgrttm1Dcr/DcrJ) carrying a null allele of the α-chain of FcRn have been previously described (FcRn-/-).Citation9,Citation36 Mice carrying hFcRn transgene (designated B6.Cg-Fcgrttm1Dcr Tg(CAG-FCGRT)276Dcr/DcrJ) carry the null allele of mFcRn and the hFcRn cDNA transgene under the control of the CAG promoter and are referred to as Tg276 or hFcRn transgenic mice.Citation31,Citation36 All mice were maintained under specific pathogen-free conditions and the procedures were approved by the Jackson Laboratory Animal Care and Use Committee.
Generation of hFcRn and mFcRn constructs for transfection.
To produce the hFcRn construct, the 5′ non-coding sequence (118 bp) including the sequence encoding the 23 AA hFcRn signal sequence was PCR-amplified from hFcRn cDNA (kindly provided by Clark Anderson, Ohio State University) using forward CCC CCC CCG CTA GCG AAG CCC CTC CTC GGC GTC CTG GT (NheI site underlined) and reverse CCC CCC CCA CCG GTC CGC CCA GGC TCC CAG GAA GGA GAA A (AgeI site underlined) primers. Extra cytosine nucleotides were included at the 5′ ends of primers to increase the efficiency of restriction endonuclease activity. This PCR product was digested with NheI and AgeI, and inserted downstream of the CMV-IE promoter, between the NheI and AgeI restriction sites upstream and in-frame with the EGFP coding sequence of the pEGFP-Cl vector (Clontech, 6084-1) to produce an N-terminal EGFP-tagged hFcRn construct lacking the C-terminal endosomal targeting domain. The mFcRn construct was generated from RNA isolated from ten day old C57BL/6J neonatal proximal small intestine and PCR amplified using forward (CCC CCC CCC TCG AGG GTC AGA GAC CCG CCC CCA, XhoI site underlined) and reverse (CCC CCC CCG AAT TCG CGC ATC CTG CCC CAC AA, EcoRI site underlined) primers. As described above, cytosine nucleotides were added to the 5′ ends of the primers to improve restriction endonuclease cleavage of the PCR product. A 944 bp product was digested with XhoI and EcoRI, and cloned into the corresponding sites of the pEGFP-C1 vector into which we had already inserted 118 bp of sequence including the sequence encoding the 23 amino acid hFcRn signal sequence downstream of the CMV-IE promoter, and upstream and in-frame with GFP to produce an N-terminal GFP-tagged tailless mFcRn construct. All PCR-amplified inserts were bi-directionally sequence-verified across the cloning sites.
Cell lines.
Purified hFcRn-GFP and mFcRn-GFP plasmids were transfected into HeLa or HEK293 cells using Lipofectamine Plus (Invitrogen, 15338-100) following the manufacturer's protocol. Stably transfected HEK293 cells were selected using 400 µg/mL G418 (Sigma-Aldrich, G5013) in 10% FBS-supplemented DMEM. Since the constructs lacked the endosomal targeting domain, the resulting 293hFcRn-GFP and 293mFcRn-GFP cells expressed FcRn at high levels on their plasma membrane, as verified by immunofluorescence imaging and FACS analysis after staining with anti-FcRn mAbs.
Antisera, hybridoma generation and screening.
To generate anti-hFcRn antibody responses, B6 WT or FcRn null mice were immunized multiple times via intraperitoneal (i.p.) injection with spleen cells from Tg276 mice in and complete or incomplete Freund's adjuvant. Plasma samples collected from boosted mice were assayed by flow cytometry for anti-FcRn binding and the functional blockade of IgG binding. To assess hFcRn binding, 293hFcRn-GFP cells were incubated with antisera, stained with goat anti-mouse IgG-R-phycoeythrin (Southern Biotech, 1031-09), and then analyzed using a FACSCalibur (BD Biosciences, San Jose, CA USA). The ability to block hFcRn function was assessed by adding antisera to 293hFcRn-EGFP cells in FACS buffer (PBS with 1% BSA and 0.05% NaN3), pH 6 or pH 7.2, followed by the addition of hIgGAF647, and data were similarly acquired using a FACSCalibur.
Spleen cells from immunized mice with high binding or blocking activity were fused with SP2/0-Ag14 myeloma cells according to standard protocols. Selection for hybridomas was provided by refeeding cells on days 2, 3, 4, 5, 7, 9 and 11 with DMEM-20 supplemented with HAT and 100 U/mL IL-6. A cellular ELISA was used to screen the hybridomas for hFcRn binding activity by incubating culture supernatant with 293hFcRn-GFP cells, staining with goat anti-mouse IgG-alkaline phosphatase (Southern Biotech, 1031-04), and measured by adding p-nitrophenyl phosphate (AMRESCO, 0617) at 1 mg/mL in substrate buffer (50 mM sodium bicarbonate, 5 mM MgCl2, pH 8.6) followed by the addition of 1% sodium dodecyl sulfate (Sigma-Aldrich, L4390) to solubilize cells prior to OD405 determination on a SpectraMAX 190 plate reader (Molecular Devices, Sunnyvale, CA). Productive hybridomas were expanded from anti-hFcRn positive wells, and cloned 3 times by limiting dilution to verify stable productive secretion of the resulting subclones. All mAb supernatants were protein G (GE Healthcare, 17-0404-01) purified and labeled with Alexa Fluor 674 (Invitrogen, A-20173). Specificity and relative affinity were measured by staining 293hFcRn-GFP, 293mFcRn-GFP and 293 cells with AF647-labeled anti-hFcRn mAbs at a fixed concentration of 200 ng/106 cells to determine specificity, or with a range of indicated concentrations to determine affinity. Data were acquired using a FACSCalibur and relative affinities for hFcRn were represented as ratios of the Alexa Fluor 647 MFI from each mAb concentration to the MFI yielded from its highest concentration.
Competitive binding assay.
Anti-hFcRn mAbs were screened for their ability to compete for binding to hFcRn on ice. In matrix fashion, unlabeled mAbs (blocking) were individually added to 106 293hFcRn-EGFP cells at a final concentration of 500 µg/mL for 30 min, and cells were washed with 4 mL FACS buffer, pH 7.2. The same set of mAbs labeled with AF647 were then added individually to antibody treated 293hFcRn-GFP cells at a final concentration of 500 ng/mL, incubated for 30 min, and cells were washed with 4 mL FACS buffer, pH 7.2. Events were acquired on a FACSCalibur and the percent inhibition of labeled antibody binding was calculated from the Alexa Fluor 647 MFI of each hFcRn-GFP+-gated cell population as follows: % Inhibition = (1 − ((MFIEXP − MFISELF)/MFINONE)) × 100, where MFIEXP is the MFI of a given labeled mAb in the presence of a competing unlabeled blocking mAb, MFISELF is the MFI of a given labeled mAb in the presence of the same unlabeled blocking mAb, MFINONE is the MFI of a given labeled mAb in the absence of an unlabeled blocking mAb.
Antibody blockade of hIgG and HSA binding in vitro.
Antisera and mAbs were screened for the ability to block hFcRn function on ice by adding either 2 µL of heteroantisera, or defined amounts of purified mAbs, to 106 293hFcRn-GFP cells in 25 µL FACS buffer, pH 6 or 7.2 for 30 min, then washed with corresponding pH FACS buffer. The cells were incubated for 60 min with hIgG-Alexa Fluor 647 (1 µg) or 10 µg human serum albumin-biotin (HSA-biotin), and washed with corresponding pH FACS buffer. For assessment of HSA-biotin binding, cells were incubated for 30 min with streptavidin-phycoerythrin (Southern Biotech, 7100-09) followed by washing with corresponding pH FACS buffer. HSA binding FACS buffer was albumin-free. MFI was determined by acquiring events on a FACSCalibur.
Immunofluorescence and confocal microscopy.
The expression pattern of hFcRn in various tissues was determined using immunofluorescence (IF) techniques. Organs from age and sex-matched mice were collected and immediately embedded with OCT and frozen using Precision Cryo-embedding System (IHC world, Woodstock, MD). Sectioning of these tissues was performed with a Leica CM 1950 Cryostat (Buffalo Grove, IL). Seven to 10 µm sections were applied to Fisherbrand Superfrost® Plus slides and stored at −20°C. Quality control of the preparations was evaluated by H&E staining of consecutive sections. Normal human kidney tissue was harvested from nephrectomy specimens and snap frozen in OCT medium and sectioned as described previously. Use of human tissue specimens was approved by the Washington University Institutional Review Board (IRB #201107288).
For the IF studies shown, hFcRn was detected using Alexa Fluor 647 labeled mAbs. Mouse kidney tissues were counterstained with rat anti-mouse podocalyxin (clone 192703) (R&D Systems Inc., MAB1556) coupled with a goat anti-rat FITC (Invitrogen, 629511) and biotin Lotus Tetragonolobus Lectin (LTL) (Vector Laboratories, B-1325) coupled with streptavidin DyLite 488 (Thermo Scientific, 21832) to highlight podocytes and proximal convoluted tubules. IF labeling was performed using standard protocols and imaged with a Leica SP5 confocal microscope within 24 h of mounting. Human tissues were stained in a similar fashion.
mAb blockade of FcRn in vivo.
hFcRn Tg276 mice carrying one copy of the hFcRn or B6 WT mice were first injected i.p. with 100 µg of tracer mIgG1 (1B7-11, anti-TNP, American Type Culture Collection, TIB-191), mIgA (2F11-15, anti-TNP, American Type Culture Collection, TIB-194), human IgG (Gammagard, Baxter, 1501375) or 1 mg HSA (Sigma-Aldrich, A-7284) as tracers. ELISA of serial blood samples was used to assess the plasma concentrations of each tracer. The tracer antibodies were captured with TNP-bovine IgG (EMD Chemicals, 324101), mouse anti-human IgG Fc (Southern Biotech, 9040-01), or rabbit anti-HSA (United States Biological, A1327-46), respectively. Goat anti-mouse IgG1-AP (Southern Biotech, 1070-04), rat anti-mouse IgA-AP (Southern Biotech, 1165-04), mouse anti-human kappa-AP (Southern Biotech, 9230-04) or goat anti-HSA-AP (Bethy Laboratories, A80-229AP), respectively, were used for detection. The ELISA plates were washed 3 times with 250 µL ELISA wash (PBS, 0.05% Tween 20, 0.05% NaN3) between each step, and both the block and all dilutions, were made in ELISA wash with 1% BSA. The ELISA was developed using p-nitrophenyl phosphate substrate added at 1 mg/mL in substrate buffer prior to OD405 measurement on a SpectraMAX 190 plate reader.
Disclosure of Potential Conflicts of Interest
D.C.R and S.A. have applied for US and European patents that include some of the mAbs described.
Authors' Contributions
G.J.C., V.Z.S., S.A., E.P. and S.P. designed and performed experiments and analyzed data. D.C.R. designed experiments and analyzed data. G.J.C., V.Z.S., S.A., G.P. and D.C.R. contributed to the writing of the manuscript.
Abbreviations
| MHC | = | major histocompatibility complex |
| IgG | = | immunoglobulin G (h for human, m for mouse) |
| FcRn | = | Fc receptor neonatal (h for human, m for mouse) |
| mAb | = | monoclonal antibody |
| β2M | = | beta-2 microglobulin |
| HSA | = | human serum albumin |
| GFP | = | enhanced green fluorescent protein |
| IF | = | immunofluorescence |
Figures and Tables
Figure 1 hFcRn binding and blocking activity of anti-hFcRn antisera. (A) Representation of the construct design lacking the cytoplasmic targeting domain (left). Confocal images of HeLa cells transiently transfected with the hFcRn-GFP after incubation with hIgG at the indicated pH, fixed and stained with goat anti-hIgGAF568 (right). (B) Flow cytometric data of 293hFcRn-GFP cells incubated with hIgGAF647 at pH 6.0 (left) or 7.2 (right). (C) Representative results from evaluation of antisera from mice primed with hFcRn. Binding of the antisera to hFcRn-GFP was detected with goat anti-mouse IgG-PE at pH 7.2 (left). Functional blockade of hIgG binding to hFcRn was determined by the ability to inhibit the binding of hIgGAF647 at pH 6.0 (right). Values within scatter plots represent the AF647 mean fluorescence intensity (MFI) of each hFcRn-GFP+-gated cell population. Data are representative of at least two experiments.
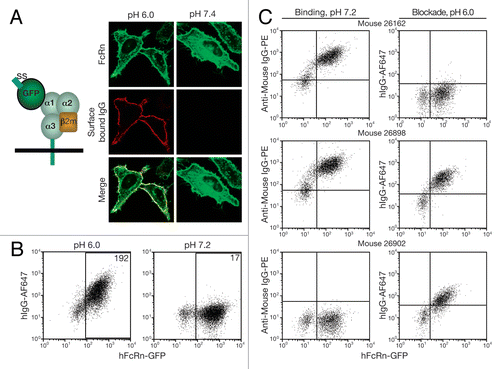
Figure 2 Anti-hFcRn mAb specificities and relative affinities. 293hFcRn-GFP, 293mFcRn-GFP, or control 293 cells were incubated with AF647-labeled anti-hFcRn mAbs (A) at a fixed concentration of 200 ng/106 cells to determine specificity or (B) with a range of concentrations to assess relative mAb affinities for hFcRn. Values within scatter plots represent the AF647 MFIs of each hFcRn-EGFP+-gated cell population. Data are representative of at least two experiments.
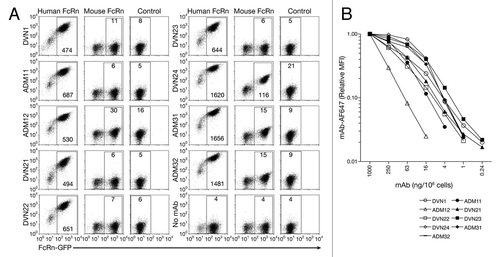
Figure 3 Immunofluorescent detection of hFcRn (red channel) in kidney tissue sections from (A) hFcRn transgenic mice, (B) hFcRn transgenic mice costained for proximal convoluted tubule (blue channel) and podocytes (green channel), (C) wild-type B6 mice costained for proximal convoluted tubule and podocytes, (D) human tissue, (E) human tissue costained for proximal convoluted tubule and podocytes, and (F) human tissue showing only proximal convoluted tubule and podocytes (GM, Glomerulus, PCT, proximal convoluted tubules).
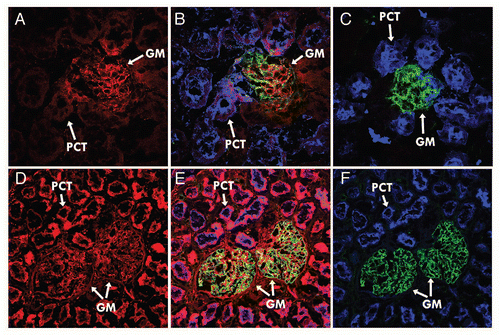
Figure 4 hFcRn blocking activity. 293hFcRn-GFP cells were incubated with a range of concentrations of DVN1 (○), ADM11 (●), DVN21 (▴), DVN22 (□), DVN23 (■), DVN24 (◊), ADM31 (◆) or ADM32 (−) at pH 6, then stained for functional binding with (A) 20 µg/mL hIgGAF647, or (B) 200 µg/mL HSAbiotin followed by streptavidin-PE. Nonfunctional binding was indicated by the AF647 or PE MFIs of cells stained with labeled ligands at pH 7.2 (X). (C) Scatter plots of 293hFcRn-GFP cells that were untreated at pH 6.0 (upper left) or at pH 7.2 (upper right), exposed to 200 ng/mL of DVN24 (lower left) or ADM11 (lower right) at pH 6.0, and then stained for functional binding of hIgGAF647 at the corresponding pH. (D) Scatter plots of 293hFcRn-GFP cells that were untreated at pH 6.0 (upper left) or at pH 7.5 (upper right), exposed to 200 ng/mL of ADM31 (lower left) or ADM11 (lower right), and then stained for functional binding with HSA-biotin and counterstained with streptavidin-PE at the corresponding pH. AF647 or PE MFI values of the hFcRn-GFP+-gated cell populations are indicated. Data are representative of three independent experiments.
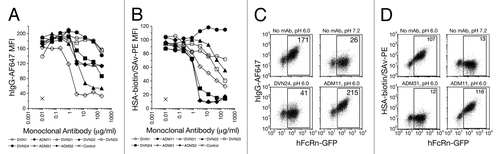
Figure 5 Blockade of IgG by anti-hFcRn mAbs in vivo. (A) B6 Tg276 mice were co-injected ip with 100 µg each of hIgG, mIgG1 (1B7.11) and mIgA (2F11–10) as tracers. The mice were then injected with 1,000 µg (◊), 300 µg (◆), 100 µg (△), 30 µg (▴) or 0 µg (□) of DVN24 or with 1,000 µg (X) of a control IgG2a mAb into groups of 3 mice on days 2, 3 and 4 d after tracer injection. (B) B6 Tg276 mice (n = 3) were co-injected ip with 100 µg hIgG and 1 mg of HSA tracers followed by injections with 1 mg doses of ADM32 (−), DVN24 (◊) or isotype matched mIgG2a (X). (C) B6 Tg276 mice (n = 4–5) were injected with 100 µg hIgG tracer and then treated on days 5–7 or 10–12 with 1 mg of DVN24 (◊) or mIgG2a (X). Significant differences (A–C) are indicated as *p < 0.05 and **p < 0.01 comparing 1,000 µg injections of DVN24 with the control isotype matched mIgG2a. (D) B6 WT mice (n = 8) were injected ip with 200 µg of mIgG1 (1B7.11) as a tracer, and then injected ip on days 4–6 with 1 mg doses of DVN24 (◊), ADM31 (◆) or vehicle only (X). Tracer plasma concentrations were determined by ELISA and plotted either as percent remaining as compared with the first time point plasma concentrations or as plasma concentrations ±SD. Significant differences (D) are indicated as **p < 0.01 comparing 1,000 µg injections of DVN24 with the vehicle only control.
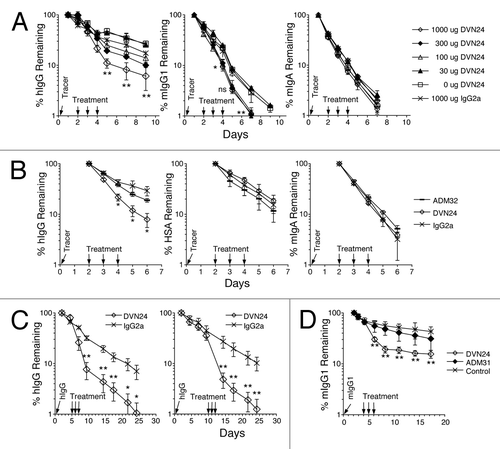
Table 1 Competitive binding of anti-hFcRn mAbsTable Footnotea
Table 2 Summary of anti-hFcRn mAb characteristics
Acknowledgments
This work was supported by the grants from the Alliance for Lupus Research and the National Institutes of Health (NIH RO1 DK56597).
Related Research Data
References
- Ghetie V, Ward ES. Multiple roles for the major histocompatibility complex class I-related receptor FcRn. Annu Rev Immunol 2000; 18:739 - 766; PMID: 10837074; http://dx.doi.org/10.1146/annurev.immunol.18.1.739
- Roopenian DC, Akilesh S. FcRn: the neonatal Fc receptor comes of age. Nat Rev Immunol 2007; 7:715 - 725; PMID: 17703228; http://dx.doi.org/10.1038/nri2155
- Ward ES, Ober RJ. Chapter 4: Multitasking by exploitation of intracellular transport functions the many faces of FcRn. Adv Immunol 2009; 103:77 - 115; PMID: 19755184; http://dx.doi.org/10.0016/S0065-2776(09)03004-1
- Kuo TT, Baker K, Yoshida M, Qiao SW, Aveson VG, Lencer WI, et al. Neonatal Fc receptor: from immunity to therapeutics. J Clin Immunol 2010; 30:777 - 789; PMID: 20886282; http://dx.doi.org/10.1007/s10875-010-9468-4
- Andersen JT, Sandlie I. The versatile MHC class I-related FcRn protects IgG and albumin from degradation: implications for development of new diagnostics and therapeutics. Drug Metab Pharmacokinet 2009; 24:318 - 332; PMID: 19745559; http://dx.doi.org/10.2133/dmpk.24.318
- Tesar DB, Björkman PJ. An intracellular traffic jam: Fc receptor-mediated transport of immunoglobulin G. Curr Opin Struct Biol 2010; 20:226 - 233; PMID: 20171874; http://dx.doi.org/10.1016/j.sbi.2010.01.010
- He W, Ladinsky MS, Huey-Tubman KE, Jensen GJ, McIntosh JR, Björkman PJ. FcRn-mediated antibody transport across epithelial cells revealed by electron tomography. Nature 2008; 455:542 - 546; PMID: 18818657; http://dx.doi.org/10.1038/nature07255
- Gan Z, Ram S, Vaccaro C, Ober RJ, Ward ES. Analyses of the recycling receptor, FcRn, in live cells reveal novel pathways for lysosomal delivery. Traffic 2009; 10:600 - 614; PMID: 19192244; http://dx.doi.org/10.1111/j.1600-0854.2009.00887.x
- Roopenian DC, Christianson GJ, Sproule TJ, Brown AC, Akilesh S, Jung N, et al. The MHC class I-like IgG receptor controls perinatal IgG transport, IgG homeostasis and fate of IgG-Fc-coupled drugs. J Immunol 2003; 170:3528 - 3533; PMID: 12646614
- Baker K, Qiao SW, Kuo T, Kobayashi K, Yoshida M, Lencer WI, et al. Immune and non-immune functions of the (not so) neonatal Fc receptor, FcRn. Semin Immunopathol 2009; 31:223 - 236; PMID: 19495758; http://dx.doi.org/10.1007/s00281-009-0160-9
- Baker K, Qiao SW, Kuo TT, Aveson VG, Platzer B, Andersen JT, et al. Neonatal Fc receptor for IgG (FcRn) regulates cross-presentation of IgG immune complexes by CD8-CD11b+ dendritic cells. Proc Natl Acad Sci USA 2011; 108:9927 - 9932; PMID: 21628593; http://dx.doi.org/10.1073/pnas.1019037108
- Liu X, Lu L, Yang Z, Palaniyandi S, Zeng R, Gao LY, et al. The neonatal FcR-mediated presentation of immune-complexed antigen is associated with endosomal and phagosomal pH and antigen stability in macrophages and dendritic cells. J Immunol 2011; 186:4674 - 4686; PMID: 21402891; http://dx.doi.org/10.4049/jimmunol.1003584
- Ye L, Zeng R, Bai Y, Roopenian DC, Zhu X. Efficient mucosal vaccination mediated by the neonatal Fc receptor. Nat Biotechnol 2011; 29:158 - 163; PMID: 21240266; http://dx.doi.org/10.1038/nbt.1742
- Lu L, Palaniyandi S, Zeng R, Bai Y, Liu X, Wang Y, et al. A neonatal Fc receptor-targeted mucosal vaccine strategy effectively induces HIV-1 antigen-specific immunity to genital infection. J Virol 2011; 85:10542 - 10553; PMID: 21849464; http://dx.doi.org/10.1128/JVI.05441-11
- Andersen JT, Dee Qian J, Sandlie I. The conserved histidine 166 residue of the human neonatal Fc receptor heavy chain is critical for the pH-dependent binding to albumin. Eur J Immunol 2006; 36:3044 - 3051; PMID: 17048273; http://dx.doi.org/10.1002/eji.200636556
- Anderson CL, Chaudhury C, Kim J, Bronson CL, Wani MA, Mohanty S. Perspective—FcRn transports albumin: relevance to immunology and medicine. Trends Immunol 2006; 27:343 - 348; PMID: 16731041; http://dx.doi.org/10.1016/j.it.2006.05.004
- Neumann E, Frei E, Funk D, Becker MD, Schrenk HH, Müller-Ladner U, et al. Native albumin for targeted drug delivery. Expert Opin Drug Deliv 2010; 7:915 - 925; PMID: 20586704; http://dx.doi.org/10.1517/17425247.2010.498474
- Andersen JT, Pehrson R, Tolmachev V, Daba MB, Abrahmsén L, Ekblad C. Extending half-life by indirect targeting of the neonatal Fc receptor (FcRn) using a minimal albumin binding domain. J Biol Chem 2011; 286:5234 - 5241; PMID: 21138843; http://dx.doi.org/10.1074/jbc.M110.164848
- Akilesh S, Huber TB, Wu H, Wang G, Hartleben B, Kopp JB, et al. Podocytes use FcRn to clear IgG from the glomerular basement membrane. Proc Natl Acad Sci USA 2008; 105:967 - 972; PMID: 18198272; http://dx.doi.org/10.1073/pnas.0711515105
- Haymann JP, Levraud JP, Bouet S, Kappes V, Hagíge J, Nguyen G, et al. Characterization and localization of the neonatal Fc receptor in adult human kidney. J Am Soc Nephrol 2000; 11:632 - 639; PMID: 10752522
- Andersen JT, Dalhus B, Cameron J, Daba MB, Plumridge A, Evans L, et al. Structure-based mutagenesis reveals the albumin-binding site of the neonatal Fc receptor. Nat Commun 2012; 3:610; PMID: 22215085; http://dx.doi.org/10.1038/ncomms1607
- Burmeister WP, Huber AH, Bjorkman PJ. Crystal structure of the complex of rat neonatal Fc receptor with Fc. Nature 1994; 372:379 - 383; PMID: 7969498; http://dx.doi.org/10.1038/372379a0
- Martin WL, West AP Jr, Gan L, Bjorkman PJ. Crystal structure at 2.8 A of an FcRn/heterodimeric Fc complex: mechanism of pH-dependent binding. Mol Cell 2001; 7:867 - 877; PMID: 11336709; http://dx.doi.org/10.1016/S1097-2765(01)00230-1
- Christianson GJ, Blankenburg RL, Duffy TM, Panka D, Roths JB, Marshak-Rothstein A, et al. beta2-microglobulin dependence of the lupus-like autoimmune syndrome of MRL-lpr mice. J Immunol 1996; 156:4932 - 4939; PMID: 8648144
- Ghetie V, Hubbard JG, Kim JK, Tsen MF, Lee Y, Ward ES. Abnormally short serum half-lives of IgG in beta 2-microglobulin-deficient mice. Eur J Immunol 1996; 26:690 - 696; PMID: 8605939; http://dx.doi.org/10.1002/eji.1830260327
- Israel EJ, Wilsker DF, Hayes KC, Schoenfeld D, Simister NE. Increased clearance of IgG in mice that lack beta 2-microglobulin: possible protective role of FcRn. Immunology 1996; 89:573 - 578; PMID: 9014824; http://dx.doi.org/10.1046/j.1365-2567.1996.d01-775.x
- Jin F, Balthasar JP. Mechanisms of intravenous immunoglobulin action in immune thrombocytopenic purpura. Hum Immunol 2005; 66:403 - 410; PMID: 15866704; http://dx.doi.org/10.1016/j.humimm.2005.01.029
- Akilesh S, Petkova S, Sproule TJ, Shaffer DJ, Christianson GJ, Roopenian D. The MHC class I-like Fc receptor promotes humorally mediated autoimmune disease. J Clin Invest 2004; 113:1328 - 1333; PMID: 15124024
- Li N, Zhao M, Hilario-Vargas J, Prisayanh P, Warren S, Diaz LA, et al. Complete FcRn dependence for intravenous Ig therapy in autoimmune skin blistering diseases. J Clin Invest 2005; 115:3440 - 3450; PMID: 16284651; http://dx.doi.org/10.1172/JCI24394
- Vaccaro C, Zhou J, Ober RJ, Ward ES. Engineering the Fc region of immunoglobulin G to modulate in vivo antibody levels. Nat Biotechnol 2005; 23:1283 - 1288; PMID: 16186811; http://dx.doi.org/10.1038/nbt1143
- Petkova SB, Akilesh S, Sproule TJ, Christianson GJ, Al Khabbaz H, Brown AC, et al. Enhanced half-life of genetically engineered human IgG1 antibodies in a humanized FcRn mouse model: potential application in humorally mediated autoimmune disease. Int Immunol 2006; 18:1759 - 1769; PMID: 17077181; http://dx.doi.org/10.1093/intimm/dxl110
- Patel DA, Puig-Canto A, Challa DK, Perez Montoyo H, Ober RJ, Ward ES. Neonatal Fc receptor blockade by Fc engineering ameliorates arthritis in a murine model. J Immunol 2011; 187:1015 - 1022; PMID: 21690327; http://dx.doi.org/10.4049/jimmunol.1003780
- Mezo AR, Sridhar V, Badger J, Sakorafas P, Nienaber V. X-ray crystal structures of monomeric and dimeric peptide inhibitors in complex with the human neonatal Fc receptor, FcRn. J Biol Chem 2010; 285:27694 - 27701; PMID: 20592032; http://dx.doi.org/10.1074/jbc.M110.120667
- Liu L, Garcia AM, Santoro H, Zhang Y, McDonnell K, Dumont J, et al. Amelioration of experimental autoimmune myasthenia gravis in rats by neonatal FcR blockade. J Immunol 2007; 178:5390 - 5398; PMID: 17404325
- Ober RJ, Radu CG, Ghetie V, Ward ES. Differences in promiscuity for antibody-FcRn interactions across species: implications for therapeutic antibodies. Int Immunol 2001; 13:1551 - 1559; PMID: 11717196; http://dx.doi.org/10.1093/intimm/13.12.1551
- Roopenian DC, Christianson GJ, Sproule TJ. Human FcRn transgenic mice for pharmacokinetic evaluation of therapeutic antibodies. Methods Mol Biol 2010; 602:93 - 104; PMID: 20012394; http://dx.doi.org/10.1007/978-1-60761-058-8_6