
Abstract
Conventional chemo- and radiotherapies for the treatment of cancer target rapidly dividing cells in both tumor and non-tumor tissues and can exhibit severe cytotoxicity in normal tissue and impair the patient's immune system. Novel targeted strategies aim for higher efficacy and tumor specificity. The role of ATM protein in the DNA damage response is well known and ATM deficiency frequently plays a role in tumorigenesis and development of malignancy. In addition to contributing to disease development, ATM deficiency also renders malignant cells heavily dependent on other pathways that cooperate with the ATM-mediated DNA damage response to ensure tumor cell survival. Disturbing those cooperative pathways by inhibiting critical protein components allows specific targeting of tumors while sparing healthy cells with normal ATM status. We review druggable candidate targets for the treatment of ATM-deficient malignancies and the mechanisms underlying such targeted therapies.
Introduction
In response to the increasing number of patients with life-threatening malignant tumors, a major focus of the life sciences has become understanding how cancers develop in order to develop better treatments.
Genome instability and mutations, together with tumor-promoting inflammation, are the enabling characteristics of cancers.Citation1 The human body possesses well-organized systems to detect and respond to impaired genome integrity. These survey systems constantly signal and kill cells with an abnormal genome to avoid damage to the whole organism. Deficiencies in critical proteins that are involved in the response to DNA damage can disable these survey mechanisms and serve as potential biomarkers for cancers. Because of the crucial role of the ataxia telangiectasia mutated (ATM) protein in many pathways of the DNA damage response, defects in the ATM gene are considered driver deficiencies in malignancies. The functions of ATM in maintaining genome stability are well documented, and a vast number of studies using ATM-deficient cell lines and animal models have been published (for a review see ref. Citation2–Citation4). Only recently, however, have attempts been made to translate results of these studies into personalized therapies for patients with defective ATM. The present review aims to give an update on how the Achilles' heel of ATM-deficient malignancies can be targeted and explain the mechanisms underlying individual approaches.
Role of ATM in the DNA damage response
Genomic integrity is constantly being challenged by endogenous and exogenous factors. DNA lesions can be either caused by ubiquitous agents such as UV radiation, gamma radiation, or reactive oxygen species, or intentionally induced by treatment with chemotherapeutic drugs and radiation therapy. Cells recruit a host of proteins to the lesion site to sense and relay the damage signal. This cellular response, termed the DNA damage response (DDR), is crucial for the fate of the cells: the outcome of the DDR decides whether cells survive and re-enter the cell cycle or undergo programmed cell death (apoptosis).Citation5
Cell cycle arrest, DNA repair, apoptosis, and chromatin remodeling are the four critical events of DDR that ensure and maintain genomic stability.Citation6 These four events are not independent of each other, but share many common factors. Mutations in many of the genes associated with the DDR have been found in the germ line of patients suffering from cancer-prone syndromes.
As an immediate response to DNA damage, checkpoint pathways are activated to prevent cell cycle progression. Cells can be arrested at the G1/S, intra-S, or G2/M phases, depending on the type of DNA lesion.Citation7 Checkpoint pathways allow cells to slow their growth in order to repair the lesions and further ensure that the DNA has been fully repaired prior to replication and distribution to the daughter cells.Citation6 Depending on the type of insult and the resulting lesion, different repair mechanisms are activated. At least four highly conserved, partially overlapping damage repair pathways operate in mammals – nucleotide excision repair (NER), base excision repair (BER), homologous recombination (HR), and non- homologous end joining (NHEJ).Citation7 In addition, the chromatin is remodeled for better access of signaling and repair factors to sites of damage. If genotoxic stress is excessive and the damage is beyond repair, pathways leading to apoptosis are activated. In contrast to unicellular organisms, apoptosis in multicellular organisms is beneficial because the organism survives at the cost of a few somatic cells.
The DDR consists of two main branches: the ATM pathway and the ataxia telangiectasia and Rad3-related (ATR) pathway. The ATR pathway is activated by single-strand breaks (SSBs) and bulky DNA lesions induced by UV-light and stalled replication forks during S-phase, whereas the ATM pathway is activated in response to DNA double-strand breaks (DSBs), either primary DSBs such as those induced by ionizing radiation (IR) or topoisomerase II inhibitors, or secondary DSBs resulting from replication of SSBs or collapsed replication forks. Rapid and effective repair of DSBs is of utmost importance to cell survival because a single unrepaired DSB can be lethal. The ATM and ATR pathways have distinct functions but partially overlap. For example, during processing of a DSB, ATM activity is required to generate a single-strand intermediate, which may result in activation of the ATR pathway.Citation8 Furthermore, in response to UV treatment or replication fork stalling, ATM is activated in an ATR-dependent manner and cooperates with ATR to ensure an effective G2/M checkpoint.Citation9 Interplay between the two pathways occurs particularly when one pathway is partially or completely deficient and the other pathway is able to execute the damage response through phosphorylation of their common DDR effectors (for a review see ref. Citation2).
ATM is a master regulator of the DDR. The ATM gene was first discovered as an allele that was mutated and inactivated in patients with ataxia telangiectasia (A-T).Citation10 A-T patients are radiosensitive and have a predisposition to cancers, especially lymphoid malignancies (for a review see refs. Citation3,Citation10). ATM is a member of the PI3K-like kinase (PIKK) family and has a highly conserved C-terminal kinase domain;Citation3 other family members include ATR, DNA-PK, and mTOR. PIKK family members phosphorylate substrates at serine or threonine residues in the context of SQ/TQ consensus sites. The substrates of ATM, including ATM itself, histone H2AX, checkpoint kinase 1 and 2 (Chk1, Chk2), p53, and BRCA1,Citation11 can also be phosphorylated by ATR. This sharing of common substrates forms the basis for the interplay between ATM and ATR.Citation2
DSBs are immediately sensed by the Mre11-Rad50-Nbs1 (MRN) complex and ATM is recruited by the C-terminus of Nbs1, thereby activating ATM kinase.Citation12 Upon autophosphorylation and acetylation, the conformation of ATM changes from an inactive dimer to an activated monomer that can then function in the DDR.Citation5,Citation13
ATM is engaged in all four pathways of the DDR through phosphorylation of different effectors. Beginning with auto-phosphorylation and H2AX phosphorylation, a cascade of phosphorylation events and protein recruitment is initiated to form ionizing radiation-induced foci (IRIF) in the vicinity of the break. IRIFs maintain and amplify DSB signals for the recruitment of checkpoint and repair factors in situ.Citation14 Cell cycle arrest is the first cellular response to DSBs. ATM first effects a G1/S phase arrest that blocks replication of damaged DNA and thus prevents amplification of damaged DNA. Together with its substrate Chk2, ATM phosphorylates p53, which then acts as a transcription factor to induce p21 expression. p21 in turn inhibits the cyclin-dependent kinases (Cdk)2, Cdk4, or Cdk6, which are required for entry into S phase.Citation6 G1/S phase arrest forms a barrier prior to replication to ensure that damaged DNA is not amplified. ATM can also effect a later G2/M phase arrest as a final barrier to ensure that DNA lesions are repaired prior to mitosis, when unrepaired DNA damage would cause mitotic catastrophe and cell death.Citation15 Entry into mitosis is controlled by Cdk1, which is active in its dephosphorylated cyclin B-bound state. The phosphatase cell division cycle 25 homolog (Cdc25) dephosphorylates and thus activates Cdk1, whereas the kinase Wee1 counteracts Cdc25 by phosphorylating and inactivating Cdk1. The ATM/Chk2 complex phosphorylates both Wee1 and Cdc25, leading to inactivation of Cdk1 and preventing mitotic entry.Citation16,Citation17 At later stages of the DSB response or in ATM-deficient cells, ATR/Chk1 kinases can be activated to phosphorylate Cdc25 and Wee1, which then synergistically effect a G2/M arrest.Citation18,Citation19
Once the cell cycle is stalled, cells activate two major pathways for repairing DSBs: non-homologous end joining and homologous recombination. NHEJ is a rapid repair pathway in which the Ku70 and Ku80 subunits of the DNA-dependent protein kinase (DNA-PK) complex first bind and protect the broken termini. DNA-bound Ku70/80 recruits additional NHEJ proteins to the site of the lesion, including the catalytic subunit of DNA-PK (DNA-PKcs), X-ray repair cross complementing protein 4 (XRCC4), and DNA Ligase IV. Ligase IV is a flexible ligase that joins incompatible DNA ends, thus making NHEJ error-prone (for a review see ref. Citation20).
Although NHEJ is the preferred pathway for repairing DSBs in G1 phase, in S and G2 phases sister chromatids are available and DSBs are repaired by slower but error free ATM-dependent HR using the intact sister chromatid as a template. Upon recruitment to the DSBs, ATM phosphorylates CtBP interacting protein (CtIP) and increases its 5′-3′ exonuclase activity. Together with the exonucleases Exo1 and Mre11, CtIP resects DSB ends to generate 3′ single-strand overhangs. Replication protein A (RPA) initially binds these ssDNA ends but is later replaced by Rad51 to form nuclear filaments. Rad51 filaments initiate homology search and strand invasion on the intact sister chromatid, core steps in HR repair. The HR process is completed by formation and subsequent resolution of Holliday junctions.Citation7 The breast cancer susceptibility gene 2 (BRCA2) protein binds and facilitates Rad51 function in HR,Citation21 whereas ATM-phosphorylated BRCA1 participates in various steps of HR from DSB signaling to Rad51 filament formation.Citation22 Activation of either the HR or the NHEJ repair pathway represses the other pathway: DNA-PKcs blocks the necessary recruitment of Exo1 for HR repair, whereas the MRN complex involved in HR repair inhibits DNA ligase/XRCC4-mediated end joining. Moreover, ATM phosphorylates DNA-PKcs to promote the dissociation of DNA-PKcs from the DSB termini, thus improving Exo1 recruitment.Citation23 In addition, ATM phosphorylates factors required for chromatin remodeling such as KAP1, RNF-20/RNF40, NuRD, and CHD4 to open up highly compacted chromatin around lesions, thus improving accessibility for signaling and repair proteins (for a review see ref. Citation4).
When DNA damage in a cell is beyond repair, ATM phosphorylates p53 in a process called DSB-induced apoptosis. Upon its activation by phosphorylation, p53 induces the expression of pro-apoptotic target genes such as PUMA, Bax, BAK, and Noxa.Citation24 These factors increase mitochondrial membrane permeability, leading to cytochrome c release and activation of caspases, the direct executors of apoptosis.
ATM defects
ATM is a malignancy susceptibility gene, and patients with ATM deficiency are predisposed to malignant tumors. Early in tumorigenesis (i.e., prior to genomic instability and malignant conversion), activated oncogenes enhance Cdk activity and enable rapid and uncontrolled cell proliferation as a result of deregulated entry to the cell cycle. Such hyperproliferation of cells frequently results in replication stress and continuous formation of DNA DSBs, as observed in precancerous lesions and cancers.Citation25-Citation27 Cells possess two anticancer barriers to cope with constitutive DNA damage in precancerous lesions: the ATR/ATM-regulated DNA damage response network and oncogene-induced senescence, in which ATM also plays an important role.Citation25,Citation28 In the absence of functional ATM, critical barriers of tumorigenesis are lost. ATM deficiency contributes to the progression of precancerous cells to malignancy and, once established, ATM-deficient cancers are resistant to chemo- and radiotherapies.Citation15 Defective ATM is an indicator of reduced disease-free survival, reduced cancer-specific survival and lower overall survival.Citation29-Citation31
ATM is altered in approximately 10% of human tumors.Citation15 These alterations include deletions and mutations that often vary between alleles, leading to ATM allelic heterozygotes.Citation32 Detection of deletion of chromosome 11q (where ATM is located) by fluorescence in situ hybridization (FISH) was traditionally the standard method of diagnosing ATM loss; nowadays, FISH is often used in conjunction with next-generation sequencing to verify the status of the remaining allele. Single allelic ATM loss does not necessarily lead to cells with deficient ATM: in the case of a single allelic 11q deletion (wt/del), normal ATM activity is maintained by the remaining wild type allele and cells show deficient responses toward DNA damage only when the second ATM allele is also mutated or deleted (del/del or del/mut).Citation33,Citation34 Nonsense mutations in ATM with often result in a truncated ATM (ATMtrunc), whereas missense mutations (ATMmis) result in full-length, albeit mutated, forms of ATM. ATMtrunc and ATMmis are proposed to have different effects on ATM function, with ATMtrunc (and ATMdel) having a recessive effect and ATMmis having a dominant negative effect on the other allele.Citation32
ATM deficiency refers to its functional loss, which is attributed to inactivated ATM protein or low protein levels. Biallelic ATM mutations (del/del, del/mut, mut/mut), as well as single allelic missense mutations (ATMmis/wt), have been shown to inactivate ATM.Citation33,Citation35,Citation36 ATM protein levels can be reduced by overexpression of tumor protein D52, an ATM interaction partner and negative regulator, via a currently unknown mechanism.Citation37 Moreover, micro-RNAs (miRNAs) miR203, miR181a/b, and miR18 are negative post-transcriptional regulators of ATM expression by either mediating degradation or inhibiting translation of ATM mRNA. The level of these miRNAs inversely correlates with cellular ATM protein levels. High levels of miR203, miR181a/b, and miR18 have been detected in aggressive and drug-resistant cancer samples with reduced ATM level and reduced ATM function with regard to the DDR.Citation38-Citation40
ATM kinase activity is used as the indicator of ATM function in DDR and can be assayed by detecting the phosphorylation level of ATM substrates (including ATM itself, p53, H2AX, Nbs1, SMC1, and Kap1) in response to DSB-inducing agents such as IR or topoisomerase II inhibitors. As ATM phosphorylates and upregulates p53 expression and consequently induces p21 expression, increased levels of p53 and p21 have also been used as readouts (for a review see ref. Citation41).
Personalized therapy
Conventional therapies based on chemotherapeutics and radiation aim to introduce DNA damage into rapidly dividing cells. Both target tissues (cancer cells) as well as normal tissues with rapid cell division (e.g., hematopoietic cells, hair cells, and mucosa) are affected, and the resulting severe cytotoxicity in normal tissues limits the dose and duration of such therapies. Targeted therapies using antibodies and their conjugates against tumor specific antigens are potentially more selective and have fewer side effects; the drawbacks, however, are high cost and in many cases insufficient efficacy.Citation42
As defects in ATM and other DNA damage response proteins (e.g., p53, ATM, BRCA1/2, Nbs1, ATR) are involved in malignant transformation of cells,Citation43 these defects can also be exploited for the treatment of such malignancies. Although normal cells can arrest their cell cycle and repair DNA damage induced by radio- and chemotherapeutic treatment, cancer cells that lack certain functional checkpoints or repair pathways depend heavily on alternative pathways to ensure their survival. Targeting these remaining functional checkpoints and repair pathways offers a chance of developing personalized therapies that can complement conventional first-line therapies by increasing the susceptibility of tumor cells. When the remaining checkpoint pathways are inhibited by drugs, cancer cells progress into mitosis despite the presence of DSBs and then die as a result of mitotic catastrophe, a form of cell death caused by aberrant mitosis. If alternative repair pathways are inhibited, cells are unable to repair lesions in time and the persistent DNA lesions can activate apoptosis pathways. In both cases, cancer cells are killed selectively.Citation44,Citation45
This scenario of selective cancer cell death is based on the concept of synthetic lethality (SL), in which cell death occurs when two genes are mutated whereas cells with a single mutation in one of these genes remain viable. Genes involved in synthetic lethal interactions usually govern critical steps in parallel pathways required for cell survival and are called synthetic lethal interactors (SLIs).Citation44 Exploiting synthetic lethality for the treatment of malignancies is a recent approach to personalized therapy as many cancer cells with mutations in genes participating in key events of the DDR are vulnerable to DNA damage-inducing therapies when their corresponding SLIs are inhibited. Inhibition of poly(adenosine diphosphate [ADP]–ribose) polymerase 1 (PARP1) in BRCA1/2-mutated cancers is a prime example of clinical application of the SL concept. PARP1 is involved in the repair of SSBs by addition of a polymer of ADP-ribose (PAR) onto acceptor proteins, thus enabling recruitment of the SSB repair scaffold protein XRCC1/Ligase III to the site of the nick. PAR-modified histones H1 and H2B mediate relaxation of the chromatin superstructure to facilitate repair.Citation46 PARP1 inhibition blocks SSB repair, and unrepaired SSBs are converted to DSBs upon DNA replication. The DSBs converted from replicating SSBs have a one-ended structure, as opposed to two-ended DSBs arising from treatment with IR or topoisomerase II inhibitors in which a duplex DNA is fractured into two parts. Repair of one-ended DSBs using NHEJ to join ends from independent loci will inevitably result in large-scale sequence rearrangements and is thus deleterious.Citation20 One-ended DSBs are therefore predominantly repaired by HR, in which BRCA1/2 plays a critical role.Citation47 PARP1 and BRCA1/2 are thus SLIs with regard to the repair of SSBs. In addition to its role in SSB repair, PARP1 is involved in multiple other repair pathways (BER, NER, HR, NHEJ) as well as other critical cellular functions including cell survival (for a review see ref. Citation48). In addition to the synthetic lethality of PARP1/BRCA observed in SSBR/DSBR, PARP1 and BRCA2 synergistically protect stalled replication forks and promote replication restart.Citation49,Citation50 PARP1 inhibition was first shown to be selectively cytotoxic to BRCA-deficient breast and ovarian cancer cells.Citation51,Citation52 More recently, PARP1 inhibitors have been shown to selectively kill BRCA1/2-deficient cells from patients with other solid tumors.Citation53
Therapeutic targets in ATM-deficient malignancies
ATM-deficient cancer cells fail to enter DNA damage-induced apoptosis due to an impaired ATM/Chk2/p53 pathway. This dysfunctional apoptotic response is responsible for their resistance to chemo- and radiotherapeutic intervention as well as the relapses observed in patients.Citation15 Surviving cancer cells rely on SLIs of ATM for effective checkpoint controls and DNA damage repair. Genes that cooperate with ATM either in checkpoint control or in DNA damage repair can be used as targets for personalized therapy of ATM-deficient malignancies.
More synthetic lethal interactors of ATM have been described than can be reviewed here. Some of them are not druggable and therefore cannot be used for therapies. 53BP1 is one such non-druggable proteins that is critical for NHEJ in addition to DNA-PKcs. Loss of 53BP1 markedly decreases the survival of lymphomas in Atm−/− mice.Citation54 Druggable targets can be specifically modulated by small molecules that are bioavailable and safe for human application.Citation55 Protein kinases are one family of druggable proteins. Kinases bind ATP through their catalytic core structure and catalyze the transfer of the terminal phosphate of ATP to substrate residues. Most small-molecule kinase inhibitors are either ATP mimetic compounds that compete with the binding of ATP to the kinases or compounds that bind outside the ATP binding site and inhibit kinase activity in an allosteric manner. Kinase inhibitors are approved for cancer treatment (for a review see ref. Citation56).
In ATM-deficient cells, repair by HR is impaired as a result of insufficient signaling of DSBs and disturbed Rad51 nuclear filament formation.Citation45,Citation57 These cells rely on DNA-PKcs-dependent NHEJ for their survival. Activity of the NHEJ pathway is increased in these cells and can compensate for the HR defects in DSB repair.Citation15 Upon inhibition of DNA-PKcs, DSB termini in ATM-deficient cells are resected to form ssDNA ends but remain unrepaired. RPA binds to the accumulated ssDNAs and recruits ATR through ATR-interacting protein (ATRIP).Citation19,Citation45 ATR then activates its downstream kinase Chk1 and further phosphorylates p53. The proapoptotic protein PUMA is induced by phosphorylated p53 to execute apoptosis. Although binding of RPA to ssDNA overhangs is also required for HR repair, the RPA foci in ATM-deficient cells are significantly larger than those formed during HR, suggesting the presence of longer stretches of RPA-ssDNA.Citation45 ATR is not normally activated as part of the DSB response, and it is unclear whether these longer stretches of RPA-ssDNA are a specific signal to recruit ATR for induction of apoptosis. The activated ATR/Chk1/p53 substitutes for the defective ATM/Chk2/p53 apoptotic pathway and exerts cell death. The effectiveness of inhibiting DNA-PKcs in ATM-deficient cancers has been demonstrated in a mouse model of ATM-deficient lymphoma xenografts. Grafted lymphomas were resistant to the DSB-inducing topoisomerase II inhibitor etoposide. However, the lymphomas were highly sensitized to apoptotic death after treatment with the DNA-PKcs inhibitor KU-0060648 alone or combined with etoposide, demonstrating that DNA-PKcs is a possible therapeutic target for ATM-deficient malignancies.Citation45
The clinical success of PARP1 inhibitors in the treatment of BRCA1/2-deficient tumors has encouraged searches for additional SLIs based on the synergistic function of SSB repair and DSB repair pathways. Single-strand breaks can either be caused directly by agents such as free radicals or indirectly as intermediates of BER. As lesions that are repaired by BRE affect only one DNA strand, BER is considered a sub-pathway of SSBR.Citation58 BER is the main guardian against alteration of bases by endogenous products of cellular metabolism including reactive oxygen species (ROS), methylation, deamination, and hydroxylation. In one of the first steps of BER, DNA glycosylase removes damaged bases and generates an apurinic/apyrimidinic (AP) site. The sugar residue of an AP site is cleaved by AP endonuclease 1 (APE1), resulting in a nicked strand. PARP1 is responsible for signaling and subsequent repairing of the nick. If APE1 is inhibited, AP sites are not processed and the subsequent repair steps are blocked, leading to an accumulation of AP sites that stall replication forks. When replication forks are not re-initiated or bypassed for a long time, they eventually collapse and one-ended DSBs are generated. The result of inhibiting APE1 is similar to that of inhibiting PARP1: in both cases, one-ended DSBs are generated from unrepaired single strand lesions, and a functional HR pathway is required for complete DNA repair. ATM and BRCA1/2 are two of the non-abundant proteins critically required for HR, and it is tempting to assume that impairing SSBR by inhibitors of either PARP1 or APE1 would effectively kill cancer cells that are deficient for ATM or BCRA1/2. Indeed, data from cell lines show that an APE1 inhibitor specifically inhibits the growth of ATM- and BRCA1/2-deficient cells (). These cells show elevated levels of unrepaired DSBs and decreased viability,Citation59 but it is unclear whether cell death is due to apoptosis or mitotic catastrophe. Furthermore, PARP1 has been suggested as an SLI for many HR proteins, including ATM.Citation60 Cell lines that have either de novo mutated ATM or ATM deficiency due to siRNA expression are sensitive to PARP1 inhibitors.Citation60,Citation61 A PARP1 inhibitor selectively killed cells from ATM-deficient lymphoma patients or xenografts through mitotic catastrophe,Citation62 further supporting the concept of SL of PARP1 and ATM.
Figure 1. Model for synthetic lethal interactions between ATM and APE1 and between ATM and FANC. (A) Replication forks (RF) stall when they encounter base lesions. Inhibition of APE1 blocks the PARP1-mediated SSBR pathway (grayed out), leading to collapsed replication forks. The FA pathway is required for stabilization of sporadic stalled replication forks. Blocking the FA pathway and inhibiting the monoubiquitination of FANCD2 therefore destabilizes stalled replication forks (grayed out) and leads to fork collapse. The resulting DSB lesions are repaired predominantly by ATM-dependent HR. (B) When APE1 or FANC are inhibited in ATM-deficient cancer cells, the cells fail to repair DSBs caused by collapsed RFs. Accumulation of unrepaired DNA lesions causes cell death. APE1, AP endonuclease 1; ATM, ataxia telangiectasia mutated; DSB, double-strand break; FA, Fanconi anemia; FANC, FA complementation group; HR, homologous recombination; PARP1, Poly(adenosine diphosphate [ADP]–ribose) polymerase 1; RF, replication fork; SSBR, SSB repair.
![Figure 1. Model for synthetic lethal interactions between ATM and APE1 and between ATM and FANC. (A) Replication forks (RF) stall when they encounter base lesions. Inhibition of APE1 blocks the PARP1-mediated SSBR pathway (grayed out), leading to collapsed replication forks. The FA pathway is required for stabilization of sporadic stalled replication forks. Blocking the FA pathway and inhibiting the monoubiquitination of FANCD2 therefore destabilizes stalled replication forks (grayed out) and leads to fork collapse. The resulting DSB lesions are repaired predominantly by ATM-dependent HR. (B) When APE1 or FANC are inhibited in ATM-deficient cancer cells, the cells fail to repair DSBs caused by collapsed RFs. Accumulation of unrepaired DNA lesions causes cell death. APE1, AP endonuclease 1; ATM, ataxia telangiectasia mutated; DSB, double-strand break; FA, Fanconi anemia; FANC, FA complementation group; HR, homologous recombination; PARP1, Poly(adenosine diphosphate [ADP]–ribose) polymerase 1; RF, replication fork; SSBR, SSB repair.](/cms/asset/3cbef6a2-2537-47f8-bd6e-6f7d57e0e770/kmco_a_10929905_f0001.gif)
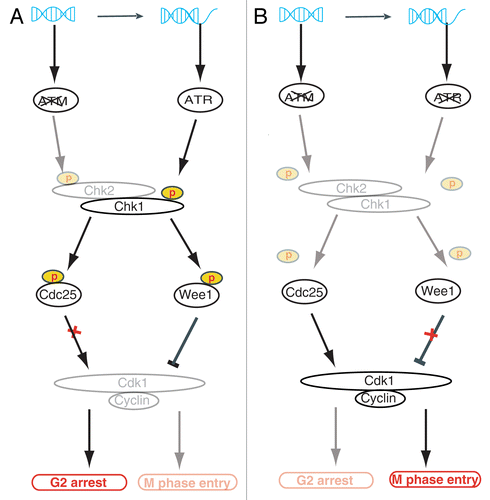
ATM null cells show reduced checkpoints in S/G2/M phases and rely heavily on ATR for a functional cell cycle arrest in response to DSBs.Citation18 Cells from A-T patients and ATM-deficient oral squamous cell carcinoma (OSCC) lines show decreased sensitivity to IR and upregulated ATR-Chk1 activity.Citation63,Citation64 The decreased sensitivity to IR is possibly due to a defective ATM/Chk2/p53 apoptosis pathway.Citation15 Both ATR/Chk1 and ATM/Chk2 can phosphorylate Cdc25 and Wee1 to allow G2 progression to M phase. Increased activity of the ATR/Chk1 pathway allows the cells to regain the capacity for G2 arrest, allowing time for DNA damage repair.Citation63,Citation64 Activated ATR could potentially relay apoptotic signaling through p53 to compensate for the defective ATM/p53 apoptosis pathway;Citation45 however, apoptotic cell death was not observed in these experiments. When ATR or Chk1 is depleted by siRNA, ATM-defective OSCC cells lose G2 arrest and die from mitotic catastrophe.Citation63,Citation64 ATM and ATR thus act cooperatively for effective G2/M arrest after DNA damage ().Citation18 The ATR inhibitor VE821 increases the sensitivity of ATM-defective cells toward cisplatin, the first line treatment for OSCC.Citation18 Previous concerns regarding inhibition of ATR related to the fact that ATR is involved in normal DNA metabolism (e.g., for preventing replication fork collapse) and ATR knockout mice are embryonic lethal.Citation65,Citation66 This concern is now considered less relevant because VE821-mediated growth arrest in normal cells is reversible.Citation18
Figure 2. Model of ATM and ATR as synthetic lethal interactors for controlling G2 cell cycle arrest. (A) In ATM-defective cancer cells, DSBs are processed to ssDNA ends and activate ATR. ATR phosphorylates Chk1, which can substitute for Chk2 in phosphorylation of Cdc25 and Wee1 to induce G2 arrest. Activated Chk1 can phosphorylate Cdc25, thus suppressing its phosphatase function and preventing Cdk1 activation. Chk1 can also phosphorylate Wee1 to exert an inhibitory effect on Cdk1. Inactive Cdk1 (grayed out) prevents progression from G2 to M phase. (B) When ATR inhibitor is applied to ATM-deficient cancer cells, neither Chk1 nor Chk2 are activated (grayed out), and Cdc25 and Wee1 are not phosphorylated. Non-phosphorylated Cdc25 has phosphatase activity and activates Cdk1. Moreover, non-phosphorylated Wee1 fails to inhibit Cdk1. Cells with active Cdk1 enter M phase despite the presence of DNA damage, and cell death occurs by mitotic catastrophe. ATM, ataxia telangiectasia mutated; ATR, ataxia telangiectasia and Rad3-related; Chk, checkpoint kinase; Cdc25, cell division cycle 25 homolog; DSB, double-strand break.
The Fanconi anemia (FA)-pathway might also be a target for the treatment of ATM-deficient malignancies. Cells from FA patients have a defective response to DNA interstrand crosslinking (ICL) agents and are therefore very sensitive to mitomycin, cisplatin, and other platinum-based chemotherapeutics. Fourteen FA complementation (FANC) groups whose mutations are responsible for Fanconi anemia have been described—FANCA, B, C, D1, D2, E, F, G, I, J, L, M, N, P—together with accessory proteins, including FAAP20, FAAP24, and FAAP100. The key event in the FA pathway is monoubiquitination of FANC-I/FANC-D2 dimers. Monoubiquitinated FANC-I/D2 interacts with FANCD1(BRCA2)/FANCJ/FANCN for signaling and repair of ICL lesions. The function of BRCA2 in the FA pathway might be different from the canonical function of BRCA2 in HR. ICL lesions are mainly repaired via coordinated NER, translesion synthesis, and HR pathways after the crosslinks are incised and unhooked (for a review see ref. Citation67). In addition to its role in the ICL damage response, monoubiquitinated FANCD2(FANCD2-Ub) is also recruited to stalled replication forks in an ATR/Chk1-dependent manner.Citation68 Replication forks stall in S phase when they encounter DNA lesions or when dNTP levels do not match the replication speed, which can occur during normal cellular metabolism.Citation69 This type of sporadic S-phase DNA damage is common in dividing cells, but more frequent in cancerous cells due to their hyperproliferation. Although it is known that cells with defects in the FA pathway are sensitive to stalled replication forks,Citation70 the function of FANCD2-Ub at stalled replication forks is unclear. It has been proposed that FANCD2-Ub contributes to the role of BRCA2 in stabilizing and protecting stalled replication forks from degradation through interaction with BRCA2.Citation68,Citation71 Stabilized replication forks are resumed after bypass of the lesions or after the fork is reinitiated.Citation69 After prolonged stalls, however, the replication forks collapse and replication proteins dissociate from the DNA. The resulting structure makes chromosomes vulnerable to breakage and generation of deleterious DSBs. Cells therefore rely on a functional HR pathway for the repair of secondary DSBs resulting from collapsed replication forks. Indeed, FANC-deficient cells have elevated DSB levels and require constitutive activation of ATM to prevent accumulation of spontaneous DNA breakage. These cells die after treatment with ATM inhibitors as a result of dramatically increased frequencies of chromosomal breaks, an indicator of unrepaired DSB termini,Citation72 suggesting parallel and compensatory roles of ATM and FANC in coping with sporadic DNA damage (). The concept of ATM and FANCs as SLIs is supported by observations that Fancg−/−Atm−/− mice exhibit embryonic lethality, whereas mice with individual gene knockouts are viable.Citation72 ATM-deficient cells are selectively killed by EF24, a derivative of curcumin that inhibits FANCD2-Ub via a poorly-defined mechanism,Citation73 further supporting a role of FANC proteins as targets for ATM-deficient cancers.
Perspective
Malignancies with genetic defects in the DDR frequently rely on remaining DNA damage response pathways to repair the DNA damage induced by radio- and chemotherapies. Synthetic lethality exploits the reliance of cancer cells on these remaining pathways to develop personalized therapies that block these pathways and thus sensitize cancer cells to DNA damage. Identifying these critical survival pathways is the first step toward developing personalized therapies. A combination of functional genomics and high-throughput screening with RNAi could produce a candidate list of SLIs for malignancies with a particular mutation. To date, no such systematic screen of SLIs has been reported for human ATM; the SLIs of ATM known so far (PARP1, ATR, p53, 53BP1, DNA-PKcs, FANC, and APE1) were identified either in individual experiments or by screening SLIs of other mutations. Once identified in a screen, SLIs of ATM need to be validated in cell lines and animal models to determine their usefulness as targets for the treatment of ATM-deficient cancers. When inhibitors of DNA repair proteins are used in combination with genotoxic drugs, DNA damage can also accumulate in normal tissues with rapid cell division and eventually lead to harmful DNA lesions, as observed after long-term application of PARP1 inhibitor.Citation53 To avoid these problems, treatment regimens with appropriate intervals to allow regeneration of normal tissues or use of inhibitors as single agents can be considered. As an example, PARP1 inhibitor alone can effectively kill BRCA1/2-deficient cell lines.Citation51,Citation52 Similarly, DNA-PKcs inhibitor alone prevents ATM-deficient lymphoma growth in a mouse model.Citation45 This effect might be because cancer cells produce higher levels of ROS, which attack DNA molecules, and have higher replication stress due to uncontrolled hyperproliferation, both of which result in endogenous DNA damage that could substitute for DNA damage induced by chemotherapeutic drugs or radiotherapy.
Several selective inhibitors of ATM kinase activity have been developed (e.g., KU60019, KU59403, KU55933, CP466722). ATM kinase inhibitors sensitize tumor cells and xenografts toward IR and DNA damage-inducing chemicals; however, no clinical trials have been reported (for a review see ref. Citation74). It is tempting to speculate that ATM kinase inhibitors would be effective for treating cancers that are deficient in known synthetic lethal interactors of ATM. For example, the ATM inhibitor KU59403 selectively inhibited the growth of p53-defective tumor xenografts in a mouse model.Citation75 The combination of low doses of ATM kinase inhibitors and Chk1 or PARP1 inhibitors could offer a novel perspective to the treatment of cancers with other deficiencies in the DNA damage response. As an example, combined treatment with PARP1 and ATM kinase inhibitors preferentially sensitizes cancer cell lines deficient in p53,Citation76,Citation77 whereas combined treatment with an ATM kinase inhibitor and a Chk1 inhibitor increases the specific cytotoxicity to FA-deficient tumor cells compared with treatment with individual inhibitors.Citation78
In the case of complex malignancies such as chronic lymphocytic leukemia with clonal evolution and the resulting heterogeneity, targeted therapies relying on ATM deficiency would exert a selective pressure. Inhibiting SLIs of ATM can decrease the number of ATM-defective cells, however subcolonies with other mutations will eventually proliferate and dominate during later disease stages.Citation79,Citation80 Therefore the fight against malignancy is a long-term battle that requires ever-changing strategies.
| Abbreviations: | ||
| AP | = | apurinic/apyrimidinic |
| APE1 | = | AP endonuclease 1 |
| ATM | = | ataxia telangiectasia mutated |
| ATR | = | ataxia telangiectasia and Rad3-related |
| BER | = | base excision repair |
| BRCA | = | breast cancer susceptibility gene |
| Chk | = | checkpoint kinase |
| Cdc25 | = | cell division cycle 25 homolog |
| Cdk | = | cyclin-dependent kinase |
| CtIP | = | C-terminal-binding protein interacting protein |
| DDR | = | DNA damage response |
| DSB | = | double-strand break |
| DSBR | = | DSB repair |
| DNA-PK | = | DNA-dependent protein kinase |
| DNA-PKcs | = | catalytic subunit of DNA-PK |
| FA | = | Fanconi anemia |
| FANC | = | FA complementation group |
| FANCD2-Ub | = | monoubiquitinated FANC-D2 |
| FISH | = | fluorescence in situ hybridization |
| HR | = | homologous recombination |
| ICL | = | interstrand crosslinking |
| IR | = | ionizing radiation |
| miRNA | = | micro-RNA |
| MRN | = | Mre11-Rad50-Nbs1 |
| NER | = | nucleotide excision repair |
| NHEJ | = | non-homologous end joining |
| OSCC | = | oral squamous cell carcinoma |
| PAR | = | polymer of ADP-ribose |
| PARP1 | = | Poly(adenosine diphosphate [ADP]–ribose) polymerase 1 |
| PIKK | = | PI3K-like kinase |
| ROS | = | reactive oxygen species |
| RPA | = | replication Protein A |
| SL | = | synthetic lethality |
| SLI | = | synthetic lethal interactor |
| SSB | = | single-strand break |
| SSBR | = | SSB repair |
| XRCC | = | X-ray repair cross complementing |
Disclosure of Potential Conflicts of Interest
The authors declare no potential conflicts of interest.
Financial disclosure statement
This work was supported by the DFG Collaborative Research Center SFB670
Acknowledgment
This work was supported by the Collaborative research center 670 (SFB670)
References
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144:646 - 74; http://dx.doi.org/10.1016/j.cell.2011.02.013; PMID: 21376230
- Shiloh Y. ATM and related protein kinases: safeguarding genome integrity. Nat Rev Cancer 2003; 3:155 - 68; http://dx.doi.org/10.1038/nrc1011; PMID: 12612651
- Lavin MF, Scott S, Gueven N, Kozlov S, Peng C, Chen P. Functional consequences of sequence alterations in the ATM gene. DNA Repair (Amst) 2004; 3:1197 - 205; http://dx.doi.org/10.1016/j.dnarep.2004.03.011; PMID: 15279808
- Shiloh Y, Ziv Y. The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat Rev Mol Cell Biol 2013; 14:197 - 210; http://dx.doi.org/10.1038/nrm3546
- Bakkenist CJ, Kastan MB. Initiating cellular stress responses. Cell 2004; 118:9 - 17; http://dx.doi.org/10.1016/j.cell.2004.06.023; PMID: 15242640
- Kastan MB, Bartek J. Cell-cycle checkpoints and cancer. Nature 2004; 432:316 - 23; http://dx.doi.org/10.1038/nature03097; PMID: 15549093
- Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature 2001; 411:366 - 74; http://dx.doi.org/10.1038/35077232; PMID: 11357144
- Jazayeri A, Falck J, Lukas C, Bartek J, Smith GCM, Lukas J, Jackson SP. ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat Cell Biol 2006; 8:37 - 45; http://dx.doi.org/10.1038/ncb1337; PMID: 16327781
- Stiff T, Walker SA, Cerosaletti K, Goodarzi AA, Petermann E, Concannon P, O’Driscoll M, Jeggo PA. ATR-dependent phosphorylation and activation of ATM in response to UV treatment or replication fork stalling. EMBO J 2006; 25:5775 - 82; http://dx.doi.org/10.1038/sj.emboj.7601446; PMID: 17124492
- Savitsky K, Bar-Shira A, Gilad S, Rotman G, Ziv Y, Vanagaite L, Tagle DA, Smith S, Uziel T, Sfez S, et al. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science 1995; 268:1749 - 53; http://dx.doi.org/10.1126/science.7792600; PMID: 7792600
- Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER 3rd, Hurov KE, Luo J, Bakalarski CE, Zhao Z, Solimini N, Lerenthal Y, et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007; 316:1160 - 6; http://dx.doi.org/10.1126/science.1140321; PMID: 17525332
- D’Amours D, Jackson SP. The Mre11 complex: at the crossroads of dna repair and checkpoint signalling. Nat Rev Mol Cell Biol 2002; 3:317 - 27; http://dx.doi.org/10.1038/nrm805; PMID: 11988766
- Sun Y, Jiang X, Xu Y, Ayrapetov MK, Moreau LA, Whetstine JR, Price BD. Histone H3 methylation links DNA damage detection to activation of the tumour suppressor Tip60. Nat Cell Biol 2009; 11:1376 - 82; http://dx.doi.org/10.1038/ncb1982; PMID: 19783983
- Bekker-Jensen S, Mailand N. Assembly and function of DNA double-strand break repair foci in mammalian cells. DNA Repair (Amst) 2010; 9:1219 - 28; http://dx.doi.org/10.1016/j.dnarep.2010.09.010; PMID: 21035408
- Jiang H, Reinhardt HC, Bartkova J, Tommiska J, Blomqvist C, Nevanlinna H, Bartek J, Yaffe MB, Hemann MT. The combined status of ATM and p53 link tumor development with therapeutic response. Genes Dev 2009; 23:1895 - 909; http://dx.doi.org/10.1101/gad.1815309; PMID: 19608766
- Boutros R, Dozier C, Ducommun B. The when and wheres of CDC25 phosphatases. Curr Opin Cell Biol 2006; 18:185 - 91; http://dx.doi.org/10.1016/j.ceb.2006.02.003; PMID: 16488126
- Falck J, Mailand N, Syljuåsen RG, Bartek J, Lukas J. The ATM-Chk2-Cdc25A checkpoint pathway guards against radioresistant DNA synthesis. Nature 2001; 410:842 - 7; http://dx.doi.org/10.1038/35071124; PMID: 11298456
- Reaper PM, Griffiths MR, Long JM, Charrier J-D, Maccormick S, Charlton PA, Golec JMC, Pollard JR. Selective killing of ATM- or p53-deficient cancer cells through inhibition of ATR. Nat Chem Biol 2011; 7:428 - 30; http://dx.doi.org/10.1038/nchembio.573; PMID: 21490603
- Shiotani B, Zou L. Single-stranded DNA orchestrates an ATM-to-ATR switch at DNA breaks. Mol Cell 2009; 33:547 - 58; http://dx.doi.org/10.1016/j.molcel.2009.01.024; PMID: 19285939
- Helleday T, Lo J, van Gent DC, Engelward BP. DNA double-strand break repair: from mechanistic understanding to cancer treatment. DNA Repair (Amst) 2007; 6:923 - 35; http://dx.doi.org/10.1016/j.dnarep.2007.02.006; PMID: 17363343
- Yuan SS, Lee SY, Chen G, Song M, Tomlinson GE, Lee EY. BRCA2 is required for ionizing radiation-induced assembly of Rad51 complex in vivo. Cancer Res 1999; 59:3547 - 51; PMID: 10446958
- Caestecker KW, Van de Walle GR. The role of BRCA1 in DNA double-strand repair: past and present. Exp Cell Res 2013; 319:575 - 87; http://dx.doi.org/10.1016/j.yexcr.2012.11.013; PMID: 23200932
- Zhou Y, Paull TT. DNA-dependent protein kinase regulates DNA end resection in concert with Mre11-Rad50-Nbs1 (MRN) and ataxia telangiectasia-mutated (ATM). J Biol Chem 2013; 288:37112 - 25; http://dx.doi.org/10.1074/jbc.M113.514398; PMID: 24220101
- Benchimol S. p53-dependent pathways of apoptosis. Cell Death Differ 2001; 8:1049 - 51; http://dx.doi.org/10.1038/sj.cdd.4400918; PMID: 11687883
- Bartkova J, Horejsí Z, Koed K, Krämer A, Tort F, Zieger K, Guldberg P, Sehested M, Nesland JM, Lukas C, et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 2005; 434:864 - 70; http://dx.doi.org/10.1038/nature03482; PMID: 15829956
- Gorgoulis VG, Vassiliou L-VF, Karakaidos P, Zacharatos P, Kotsinas A, Liloglou T, Venere M, Ditullio RA Jr., Kastrinakis NG, Levy B, et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 2005; 434:907 - 13; http://dx.doi.org/10.1038/nature03485; PMID: 15829965
- Vafa O, Wade M, Kern S, Beeche M, Pandita TK, Hampton GM, Wahl GM. c-Myc can induce DNA damage, increase reactive oxygen species, and mitigate p53 function: a mechanism for oncogene-induced genetic instability. Mol Cell 2002; 9:1031 - 44; http://dx.doi.org/10.1016/S1097-2765(02)00520-8; PMID: 12049739
- Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, Vassiliou L-VF, Kolettas E, Niforou K, Zoumpourlis VC, et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 2006; 444:633 - 7; http://dx.doi.org/10.1038/nature05268; PMID: 17136093
- Bueno RC, Canevari RA, Villacis RAR, Domingues MAC, Caldeira JRF, Rocha RM, Drigo SA, Rogatto SR. ATM down-regulation is associated with poor prognosis in sporadic breast carcinomas. Ann Oncol 2014; 25:69 - 75; http://dx.doi.org/10.1093/annonc/mdt421; PMID: 24285016
- Döhner H, Stilgenbauer S, Benner A, Leupolt E, Kröber A, Bullinger L, Döhner K, Bentz M, Lichter P. Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med 2000; 343:1910 - 6; http://dx.doi.org/10.1056/NEJM200012283432602; PMID: 11136261
- Kim JW, Im S-A, Kim MA, Cho HJ, Lee DW, Lee K-H, Kim T-Y, Han S-W, Oh D-Y, Lee H-J, et al. Ataxia-telangiectasia-mutated protein expression with microsatellite instability in gastric cancer as prognostic marker. Int J Cancer 2014; 134:72 - 80; http://dx.doi.org/10.1002/ijc.28245; PMID: 23649938
- Gatti RA, Tward A, Concannon P. Cancer risk in ATM heterozygotes: a model of phenotypic and mechanistic differences between missense and truncating mutations. Mol Genet Metab 1999; 68:419 - 23; http://dx.doi.org/10.1006/mgme.1999.2942; PMID: 10607471
- Austen B, Skowronska A, Baker C, Powell JE, Gardiner A, Oscier D, Majid A, Dyer M, Siebert R, Taylor AM, et al. Mutation status of the residual ATM allele is an important determinant of the cellular response to chemotherapy and survival in patients with chronic lymphocytic leukemia containing an 11q deletion. J Clin Oncol 2007; 25:5448 - 57; http://dx.doi.org/10.1200/JCO.2007.11.2649; PMID: 17968022
- Navrkalova V, Sebejova L, Zemanova J, Kminkova J, Kubesova B, Malcikova J, Mraz M, Smardova J, Pavlova S, Doubek M, et al. ATM mutations uniformly lead to ATM dysfunction in chronic lymphocytic leukemia: application of functional test using doxorubicin. Haematologica 2013; 98:1124 - 31; http://dx.doi.org/10.3324/haematol.2012.081620; PMID: 23585524
- Chenevix-Trench G, Spurdle AB, Gatei M, Kelly H, Marsh A, Chen X, Donn K, Cummings M, Nyholt D, Jenkins MA, et al. Dominant negative ATM mutations in breast cancer families. J Natl Cancer Inst 2002; 94:205 - 15; http://dx.doi.org/10.1093/jnci/94.3.205; PMID: 11830610
- Oguchi K, Takagi M, Tsuchida R, Taya Y, Ito E, Isoyama K, Ishii E, Zannini L, Delia D, Mizutani S. Missense mutation and defective function of ATM in a childhood acute leukemia patient with MLL gene rearrangement. Blood 2003; 101:3622 - 7; http://dx.doi.org/10.1182/blood-2002-02-0570; PMID: 12511424
- Chen Y, Kamili A, Hardy JR, Groblewski GE, Khanna KK, Byrne JA. Tumor protein D52 represents a negative regulator of ATM protein levels. Cell Cycle 2013; 12:3083 - 97; http://dx.doi.org/10.4161/cc.26146; PMID: 23974097
- Bisso A, Faleschini M, Zampa F, Capaci V, De Santa J, Santarpia L, Piazza S, Cappelletti V, Daidone M, Agami R, et al. Oncogenic miR-181a/b affect the DNA damage response in aggressive breast cancer. Cell Cycle 2013; 12:1679 - 87; http://dx.doi.org/10.4161/cc.24757; PMID: 23656790
- Song L, Lin C, Wu Z, Gong H, Zeng Y, Wu J, Li M, Li J. miR-18a impairs DNA damage response through downregulation of ataxia telangiectasia mutated (ATM) kinase. PLoS One 2011; 6:e25454; http://dx.doi.org/10.1371/journal.pone.0025454; PMID: 21980462
- Zhou Y, Wan G, Spizzo R, Ivan C, Mathur R, Hu X, Ye X, Lu J, Fan F, Xia L, et al. miR-203 induces oxaliplatin resistance in colorectal cancer cells by negatively regulating ATM kinase. Mol Oncol 2014; 8:83 - 92; http://dx.doi.org/10.1016/j.molonc.2013.09.004; PMID: 24145123
- Stankovic T, Skowronska A. The role of ATM mutations and 11q deletions in disease progression in chronic lymphocytic leukemia. Leuk Lymphoma 2014; 55:1227 - 39; http://dx.doi.org/10.3109/10428194.2013.829919; PMID: 23906020
- Panowksi S, Bhakta S, Raab H, Polakis P, Junutula JR. Site-specific antibody drug conjugates for cancer therapy. MAbs 2014; 6:34 - 45; http://dx.doi.org/10.4161/mabs.27022; PMID: 24423619
- Lavin MF. ATM and the Mre11 complex combine to recognize and signal DNA double-strand breaks. Oncogene 2007; 26:7749 - 58; http://dx.doi.org/10.1038/sj.onc.1210880; PMID: 18066087
- Nijman SMB. Synthetic lethality: general principles, utility and detection using genetic screens in human cells. FEBS Lett 2011; 585:1 - 6; http://dx.doi.org/10.1016/j.febslet.2010.11.024; PMID: 21094158
- Riabinska A, Daheim M, Herter-Sprie GS, Winkler J, Fritz C, Hallek M, Thomas RK, Kreuzer K-A, Frenzel LP, Monfared P, et al. Therapeutic targeting of a robust non-oncogene addiction to PRKDC in ATM-defective tumors. Sci Transl Med 2013; 5:89ra78; http://dx.doi.org/10.1126/scitranslmed.3005814; PMID: 23761041
- Schreiber V, Dantzer F, Ame J-C, de Murcia G. Poly(ADP-ribose): novel functions for an old molecule. Nat Rev Mol Cell Biol 2006; 7:517 - 28; http://dx.doi.org/10.1038/nrm1963; PMID: 16829982
- Arnaudeau C, Lundin C, Helleday T. DNA double-strand breaks associated with replication forks are predominantly repaired by homologous recombination involving an exchange mechanism in mammalian cells. J Mol Biol 2001; 307:1235 - 45; http://dx.doi.org/10.1006/jmbi.2001.4564; PMID: 11292338
- Michels J, Vitale I, Saparbaev M, Castedo M, Kroemer G. Predictive biomarkers for cancer therapy with PARP inhibitors. Oncogene 2014;; 33::; 3894 - -907; PMID: 24037533
- Bryant HE, Petermann E, Schultz N, Jemth A-S, Loseva O, Issaeva N, Johansson F, Fernandez S, McGlynn P, Helleday T. PARP is activated at stalled forks to mediate Mre11-dependent replication restart and recombination. EMBO J 2009; 28:2601 - 15; http://dx.doi.org/10.1038/emboj.2009.206; PMID: 19629035
- Ying S, Hamdy FC, Helleday T. Mre11-dependent degradation of stalled DNA replication forks is prevented by BRCA2 and PARP1. Cancer Res 2012; 72:2814 - 21; http://dx.doi.org/10.1158/0008-5472.CAN-11-3417; PMID: 22447567
- Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005; 434:913 - 7; http://dx.doi.org/10.1038/nature03443; PMID: 15829966
- Farmer H, McCabe N, Lord CJ, Tutt ANJ, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005; 434:917 - 21; http://dx.doi.org/10.1038/nature03445; PMID: 15829967
- Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, Mortimer P, Swaisland H, Lau A, O’Connor MJ, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med 2009; 361:123 - 34; http://dx.doi.org/10.1056/NEJMoa0900212; PMID: 19553641
- Rybanska-Spaeder I, Reynolds TL, Chou J, Prakash M, Jefferson T, Huso DL, Desiderio S, Franco S. 53BP1 is limiting for NHEJ repair in ATM-deficient model systems that are subjected to oncogenic stress or radiation. Mol Cancer Res 2013; 11:1223 - 34; http://dx.doi.org/10.1158/1541-7786.MCR-13-0252-T; PMID: 23858098
- Hopkins AL, Groom CR. The druggable genome. Nat Rev Drug Discov 2002; 1:727 - 30; http://dx.doi.org/10.1038/nrd892; PMID: 12209152
- Zhang J, Yang PL, Gray NS. Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer 2009; 9:28 - 39; http://dx.doi.org/10.1038/nrc2559; PMID: 19104514
- Yuan S-SF, Chang H-L, Lee EY-HP. Ionizing radiation-induced Rad51 nuclear focus formation is cell cycle-regulated and defective in both ATM(-/-) and c-Abl(-/-) cells. Mutat Res 2003; 525:85 - 92; http://dx.doi.org/10.1016/S0027-5107(03)00009-5; PMID: 12650908
- Caldecott KW. Single-strand break repair and genetic disease. Nat Rev Genet 2008; 9:619 - 31; PMID: 18626472
- Sultana R, McNeill DR, Abbotts R, Mohammed MZ, Zdzienicka MZ, Qutob H, Seedhouse C, Laughton CA, Fischer PM, Patel PM, et al. Synthetic lethal targeting of DNA double-strand break repair deficient cells by human apurinic/apyrimidinic endonuclease inhibitors. Int J Cancer 2012; 131:2433 - 44; http://dx.doi.org/10.1002/ijc.27512; PMID: 22377908
- McCabe N, Turner NC, Lord CJ, Kluzek K, Bialkowska A, Swift S, Giavara S, O’Connor MJ, Tutt AN, Zdzienicka MZ, et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res 2006; 66:8109 - 15; http://dx.doi.org/10.1158/0008-5472.CAN-06-0140; PMID: 16912188
- Gilardini Montani MS, Prodosmo A, Stagni V, Merli D, Monteonofrio L, Gatti V, Gentileschi MP, Barilà D, Soddu S. ATM-depletion in breast cancer cells confers sensitivity to PARP inhibition. J Exp Clin Cancer Res 2013; 32:95; http://dx.doi.org/10.1186/1756-9966-32-95; PMID: 24252502
- Weston VJ, Oldreive CE, Skowronska A, Oscier DG, Pratt G, Dyer MJS, Smith G, Powell JE, Rudzki Z, Kearns P, et al. The PARP inhibitor olaparib induces significant killing of ATM-deficient lymphoid tumor cells in vitro and in vivo. Blood 2010; 116:4578 - 87; http://dx.doi.org/10.1182/blood-2010-01-265769; PMID: 20739657
- Sankunny M, Parikh RA, Lewis DW, Gooding WE, Saunders WS, Gollin SM. Targeted inhibition of ATR or CHEK1 reverses radioresistance in oral squamous cell carcinoma cells with distal chromosome arm 11q loss. Genes Chromosomes Cancer 2014; 53:129 - 43; http://dx.doi.org/10.1002/gcc.22125; PMID: 24327542
- Wang X, Khadpe J, Hu B, Iliakis G, Wang Y. An overactivated ATR/CHK1 pathway is responsible for the prolonged G2 accumulation in irradiated AT cells. J Biol Chem 2003; 278:30869 - 74; http://dx.doi.org/10.1074/jbc.M301876200; PMID: 12791699
- Brown EJ, Baltimore D. ATR disruption leads to chromosomal fragmentation and early embryonic lethality. Genes Dev 2000; 14:397 - 402; PMID: 10691732
- de Klein A, Muijtjens M, van Os R, Verhoeven Y, Smit B, Carr AM, Lehmann AR, Hoeijmakers JH. Targeted disruption of the cell-cycle checkpoint gene ATR leads to early embryonic lethality in mice. Curr Biol 2000; 10:479 - 82; http://dx.doi.org/10.1016/S0960-9822(00)00447-4; PMID: 10801416
- Kottemann MC, Smogorzewska A. Fanconi anaemia and the repair of Watson and Crick DNA crosslinks. Nature 2013; 493:356 - 63; http://dx.doi.org/10.1038/nature11863; PMID: 23325218
- Andreassen PR, D’Andrea AD, Taniguchi T. ATR couples FANCD2 monoubiquitination to the DNA-damage response. Genes Dev 2004; 18:1958 - 63; http://dx.doi.org/10.1101/gad.1196104; PMID: 15314022
- Osborn AJ, Elledge SJ, Zou L. Checking on the fork: the DNA-replication stress-response pathway. Trends Cell Biol 2002; 12:509 - 16; http://dx.doi.org/10.1016/S0962-8924(02)02380-2; PMID: 12446112
- Pichierri P, Franchitto A, Rosselli F. BLM and the FANC proteins collaborate in a common pathway in response to stalled replication forks. EMBO J 2004; 23:3154 - 63; http://dx.doi.org/10.1038/sj.emboj.7600277; PMID: 15257300
- Schlacher K, Wu H, Jasin M. A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell 2012; 22:106 - 16; http://dx.doi.org/10.1016/j.ccr.2012.05.015; PMID: 22789542
- Kennedy RD, Chen CC, Stuckert P, Archila EM, De la Vega MA, Moreau LA, Shimamura A, D’Andrea AD. Fanconi anemia pathway-deficient tumor cells are hypersensitive to inhibition of ataxia telangiectasia mutated. J Clin Invest 2007; 117:1440 - 9; http://dx.doi.org/10.1172/JCI31245; PMID: 17431503
- Landais I, Hiddingh S, McCarroll M, Yang C, Sun A, Turker MS, Snyder JP, Hoatlin ME. Monoketone analogs of curcumin, a new class of Fanconi anemia pathway inhibitors. Mol Cancer 2009; 8:133; http://dx.doi.org/10.1186/1476-4598-8-133; PMID: 20043851
- Cremona CA, Behrens A. ATM signalling and cancer. Oncogene 2003; PMID: 23851492
- Batey MA, Zhao Y, Kyle S, Richardson C, Slade A, Martin NMB, Lau A, Newell DR, Curtin NJ. Preclinical evaluation of a novel ATM inhibitor, KU59403, in vitro and in vivo in p53 functional and dysfunctional models of human cancer. Mol Cancer Ther 2013; 12:959 - 67; http://dx.doi.org/10.1158/1535-7163.MCT-12-0707; PMID: 23512991
- Williamson CT, Kubota E, Hamill JD, Klimowicz A, Ye R, Muzik H, Dean M, Tu L, Gilley D, Magliocco AM, et al. Enhanced cytotoxicity of PARP inhibition in mantle cell lymphoma harbouring mutations in both ATM and p53. EMBO Mol Med 2012; 4:515 - 27; http://dx.doi.org/10.1002/emmm.201200229; PMID: 22416035
- Kubota E, Williamson CT, Ye R, Elegbede A, Peterson L, Lees-Miller SP, Bebb DG. Low ATM protein expression and depletion of p53 correlates with olaparib sensitivity in gastric cancer cell lines. Cell Cycle 2014; 13:2129 - 37; http://dx.doi.org/10.4161/cc.29212; PMID: 24841718
- Chen CC, Kennedy RD, Sidi S, Look AT, D’Andrea A. CHK1 inhibition as a strategy for targeting Fanconi Anemia (FA) DNA repair pathway deficient tumors. Mol Cancer 2009; 8:24; http://dx.doi.org/10.1186/1476-4598-8-24; PMID: 19371427
- Forbes SA, Bindal N, Bamford S, Cole C, Kok CY, Beare D, Jia M, Shepherd R, Leung K, Menzies A, et al. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res 2011; 39:D945 - 50; http://dx.doi.org/10.1093/nar/gkq929; PMID: 20952405
- Schuh A, Becq J, Humphray S, Alexa A, Burns A, Clifford R, Feller SM, Grocock R, Henderson S, Khrebtukova I, et al. Monitoring chronic lymphocytic leukemia progression by whole genome sequencing reveals heterogeneous clonal evolution patterns. Blood 2012; 120:4191 - 6; http://dx.doi.org/10.1182/blood-2012-05-433540; PMID: 22915640