Abstract
MicroRNAs (miRs) are small non-coding RNAs that generally function as negative regulators of target messenger RNAs (mRNAs) at the posttranscriptional level. MiRs bind to the 3′UTR of target mRNAs through complementary base pairing, resulting in target mRNA cleavage or translation repression. To date, over 15,000 distinct miRs have been identified in organisms ranging from viruses to man, and interest in miR research continues to intensify. Of note, the most enlightening aspect of miR function – the mRNAs they target – continues to be elusive. Descriptions of the molecular origins of independent miR molecules currently support the hypothesis that miR hairpin generation is based on the adjacent insertion of two related transposable elements (TEs) at one genomic locus. Thus transcription across such TE interfaces establishes many, if not the majority of functional miRs. The implications of these findings are substantial for understanding how TEs confer increased genomic fitness, describing miR transcriptional regulations, and making accurate miR target predictions. In this work, we have performed a comprehensive analysis of the genomic events responsible for the formation of all currently annotated miR loci. We find that the connection between miRs and transposable elements is more significant than previously appreciated, and more broadly, supports an important role for repetitive elements in miR origin, expression and regulatory network formation. Further, we demonstrate the utility of these findings in miR target prediction. Our results greatly expand the existing repertoire of defined miR origins, detailing the formation of 2,392 of 15,176 currently recognized miR genomic loci and supporting a mobile genetic element model for the genomic establishment of functional miRs.
Introduction
MicroRNAs (miRs) are small (∼20 nt) non-coding RNAs that regulate networks of genes.Citation1 While initially thought to be Caenorhabditis elegans specific, their small size merely masked their discovery and prevalence in higher organisms until nearly a decade later.Citation2,Citation3 MiRs are widespread in higher eukaryotes and similar in function to small interfering RNA (siRNAs).Citation4 Typically initially expressed as a portion of a several thousand nucleotide miR transcript, pri-miRs are substantially processed by Drosha to generate a ∼70 nt stem loop (pre-miR) in the nucleus.Citation5 Pre-miRs are exported to the cytoplasm where DICER cleaves and denatures these dsRNAs to produce the final mature single stranded miRCitation6 (). Partial sequence complementarity between miRs and target mRNAs mediates translational repression through multiple mechanisms with dramatic cellular consequences, clearly supported by the multiple pathologies now associated with miR misregulations (recently reviewed by ref. Citation7).
Progress in deciphering miR coordination has proven exceptionally challenging primarily due to the ability of miRs to target mRNAs that bear only partial sequence complementarity.Citation8 While numerous studies have attempted to characterize the specific determinants of miR targeting, no model for target recognition has proven entirely accurate. Unraveling this phenomenon is particularly important not only for identifying the specific mRNAs a miR regulates, but also for consideration when designing therapeutic inhibitory RNAs. Several studies suggest that the primary criterion for determining if a given siRNA or miR imparts message degradation or translational repression rests on the degree of complementarity between the small RNA and a mRNA.Citation9,Citation10
Gene regulation by small non-coding RNAs is governed by sequence complementarity shared with the target mRNA. While siRNAs require nearly perfect complementarity to bring about message degradation, miR target recognition and subsequent translational repression is commonly mediated through only 6 or 7 basepairs (bps).Citation9 Typically located in the 5′ miR sequence, the participating nts have become known as a miR “seed” and their reverse complement in a target mRNA as a “seed match”Citation10 (). The recurrent observation of perfect complementarity between a seed and seed match in the few characterized miR:target interactions is the basis for most target recognition algorithms. Following this, the principle algorithms differ predominantly through the significance assigned to seed match conservation across species, multiple seed matches within a given mRNA or the degree of complementarity between the remainder of a miR and proposed target.Citation11–Citation18
The molecular origins of miRs and corresponding mRNA targets are not established, but the abundance of mobile genetic elements in genomes of higher eukaryotes suggests a mechanism of functional miR establishment. Almost half of the human genome is comprised of transposonsCitation19 whereas transposable elements can constitute as much as eighty percent of plant genomes.Citation20 While insertion into coding regions are generally detrimental, intron and untranslated region (UTR) insertions are quite common, with ∼50% of metazoan loci harboring at least one co-transcribed transposable element.Citation21,Citation22 Smalheiser and TorvikCitation21 were the first to describe a potential molecular origin of miRs, suggesting a bias for miR loci to straddle the termini of two oppositely-oriented, related transposable elements (TEs) (). Now corroborated and expanded by several independent analyses,Citation23–Citation27 this research suggests that transcription across such mirrored-TE interfaces and subsequent RNAi processing gave rise to many if not the majority of functional miRs.
While TE colonization has given rise to a number of beneficial cellular regulatory mechanisms (e.g., RAG1/RAG2-mediated immunoglobulin transpositionCitation28 and murine B2 SINE repression of mRNA transcription in response to heat shockCitation29,Citation30), the principle effects of TE genome colonization have classically been believed to be sequence alterations at the site of integration. A TE-based miR origin, however, suggests another interesting (and perhaps particularly advantageous) role for TE domestication. Through processing a single TE sequence, the RNAi machinery can become loaded with a small RNA guide capable of targeting all RNAs containing that TE on the opposite strand.Citation4 This implies that prior to miR establishment, a network of targets has already been formed. MiRs therefore “arise”, when an advantageous regulatory niche has developed out of a series of random TE insertions. While additional implications of a TE-based miR origin may yet be realized, the most apparent utility is simply limiting miR target searches to transcripts containing a miR's progenitor TE. Here, we describe our analysis of the genomic events responsible for the formation of all currently annotated miR loci. We define the TE origins of over 2,000 distinct miRs and then demonstrate how to use these findings to predict targets for miRs with characterized TE origins. We propose that limiting miR target searches to transcripts containing a miR's progenitor mobile genetic elements can facilitate miR target identification.
Results
Fifteen percent of miR loci identified as being formed from TE sequences.
In order to test the model that miR establishment can arise from mobile DNA elements, we characterized the sequence relationships between all known miR loci and known repetitive elements. We screened the 15,176 currently recognized miR genomic loci against the principle datasets for TECitation31 and noncoding RNAsCitation32 using either Censor ServerCitation33 or an in-house, stand-alone BLAST server. In all, we identify 2,392 TE-based miR origins ( and Sup. Table 1). Averaging 82.9% identity over 85.7 bps, 1,741 of these relationships were identified directly through sequence based alignment while the remaining 651 annotations were based on familial inclusion ( and Sup. Table 2). Our alignment analysis clearly demonstrates the majority of TE-based miR origins occurred via the mechanism depicted in , and we conclude this represents the most common scenario for de novo miR locus formation. Therefore, our results are in agreement with several previous but more limited analyses,Citation23–Citation27 suggesting that transcription across such mirrored-TE interfaces and subsequent RNAi processing gave rise to many miRs.
Transposable element origins.
Transposable elements are generally classified as either transposons or retrotransposons, based on their mechanism of propagation. Transposons synthesize a DNA copy of themselves, while retrotransposons generate a RNA intermediate that is then reverse-transcribed into DNA and integrated into the genome. Transposable elements fall into three principle categories: DNA transposons, long terminal repeat (LTR) retrotransposons and non-LTR retrotransposons (recently reviewed in refs. Citation34–Citation36). DNA transposons are flanked by inverted repeats and typically contain two or more open reading frames (ORFs) corresponding to the proteins required for making copies of their sequences and distributing them through the genome (reviewed in ref. Citation36). We find DNA transposons constitute the transposable element most frequently responsible for miR loci formation identifying 891 origins from characterized DNA transposons and an additional 137 loci being formed from related DNA satellite repeat elements. While distinct in composition and mechanism from DNA transposons, LTR retrotransposons more closely resemble retroviral genomes. They are flanked by 250 to 600 bp direct repeats called long terminal repeats (LTRs) and contain ORFs for proteins related to viral Gag and Pol.Citation35 Based on sequence based alignment and familial inclusion, we identify 414 LTR retrotransposon:miR relationships. Finally, similar to LTR retrotransposons containing portions of Gag and Pol-like sequences,Citation34 we find non-LTR retrotransposons are collectively responsible for the formation of 814 miR loci. While several distinct categories of non-LTR retrotransposons have been identified, we find two of these groups most frequently responsible for miR locus formation, long interspersed repeated elements (LINEs), forming 312 distinct miR loci, and a second type of non-autonomous element that utilizes proteins encoded by LINEs for their own propagation: short interspersed repeated elements (SINEs) which we find responsible for the formation of 353 additional miR loci ().
Familial inclusions.
Approximately 25% of our miR locus annotations were determined through familial inclusion. Following sequence based annotation, all miRs were separated into familial clusters based on standard miRBase nomenclature.Citation37 All members of a family were said to have arisen from a common TE in the event that: (1) multiple members of a miR family were identified as being related to the same TE and (2) no other member sequences were identified as being more closely related to a different TE. In all, we describe shared TE-based genomic origins for 1,345 miRs, 48.4% (651) of which were not initially identified through sequence based alignment, defining 45 distinct miR families ranging from 3 to 97 member sequences ().
details the familial inclusions of 8 unique miR-284 hairpins. Thus far, the genomes of 12 distinct Drosophila species have been found to each contain a single, conserved miR-284 hairpin. Due to the particularly rigid stringency of our sequence based alignment criterion, initial computational analyses identified only 4 of the miR-284 hairpins as arising from a specific mariner DNA transposon (Mariner-35) (each bearing >75% sequence identity to over 50 nts). However, the high degree of hairpin and flanking sequence conservation () suggests that all 12 miR hairpins share a common origin—when miR-284 was initially formed in a common ancestral species. Therefore, all members of the miR-284 family were annotated as arising from the Mariner-35 DNA transposon.
Taxon-specific miR expansions.
Highly indicative of a transposable element origin, we identified several instances of robust taxon, even species-specific, expansions of individual miR families. Genomic analysis indicates 72 distinct miR-430 genomic loci [57 in the zebrafish (Danio rerio) and 15 in the Japanese killifish (Oryzias latipes)] were each formed from satellites. We readily identified the genomic elements responsible for the initial formation of this miR family as a fish-specific satellite repeat with individual hairpins aligning to the consensus sequence with as much as 100% identity over 71 bp. Similarly, our sequenced based alignments suggest that miR-1302 in horse and primate species were formed from MER53. Eleven, eight, five and six miR-1302 loci are characterized in the human, chimp (Pan troglodytes), orangutan (Pongo pygmaeus) and horse (Equus caballus) genomes respectively, accounting for 30 distinct miR-1302 hairpins. While the mature miR-1302 hairpins are nearly identical, the specific identification of miR-1302 hairpins in both equine and primate genomes argues against a common ancestral origin. Indeed, we find that while both sets of miR-1302 hairpins were formed from MER53 repeats, their genomic context argues against a common ancestry and instead suggests convergent evolution (data not shown).
Sequence analysis of the miR-466, miR-467 and miR-669 families further reveals taxon specific expansions. To date, 33 miR-466 genomic loci have been identified: one in human, one in chicken (Gallus gallus), four in rat (Rattus norvegicus) and 27 in mouse (Mus musculus). In addition, 17 highly-related miR-467 loci have also been described exclusively in the mouse genome. We find these 50 miR loci each arose from CR1 non-LTR retrotransposons sequences. Similarly expanded specifically in the mouse genome, 31 miR-669 loci have thus far been described, 30 in the mouse and one in cow (Bos taurus) all being formed from EnSpm DNA transposons.
Plant genomes also show evidence of a TE mediated proliferation of miR families. Stowaway elements in Oryza sativa and Sorghum bicolor correlate with multiple miR families. In all, 25 miR-437 hairpins formed from Stowaway elements have been identified with 22 encoded in the Sorghum bicolor genome and three others, each found in distinct plant species. Additionally, our results show Oryza sativa (rice) specific miR-441, -809, -812, -814, -818, -819 and -1,862 families, comprising 45 genomic loci, were each likely formed from Stowaway transposons. In legumes, 81 Medicago truncatula-specific miR hairpins corresponding to 13 miR families align with MuDR elements, connecting their origin with MuDR transposition events.
Our analyses describe several additional taxon-specific miR families of note: (1) 64 miR-548 loci identified in three primate genomes: 40 in humans, 18 in chimp, and six in the Rhesus monkey (Macca mulatta) each arose from MADE1 elements. (2) Alu and SVA elements formed the 162 primate specific miR hairpins comprising the miR-515 family. (3) 24 miR-2284 genomic loci unique to the cow genome were formed from an OOREP1-like element. And finally, (4) similarly species-specific, we find the 19 Populus trichocarpa miR-478 hairpins were formed from Gypsy element genomic insertions. Taken together, our alignments using genomic sequences from phylogenetically distinct eukaryotes strongly support the model that expansions of individual miR families within a genome correlate with unique genome TE compositions.
Non-transposable element origins.
While ∼95% of our sequence based annotations identified known transposable element progenitors, we also identified 136 miRs bearing significant sequence identity to known noncoding RNA sequences (e.g., snoRNAs, scaRNAs, tRNAs) (Sup. Table 1) indicated as “other” in . As each of the aligning noncoding RNAs are transcribed from RNA polymerase III (Pol III) promoters,Citation23,Citation31 the majority of these relationships most likely indicate origins from uncharacterized SINE elements as all SINEs are derived from Pol III transcribed noncoding RNA genes (e.g., tRNAs).Citation38–Citation40
MiR target prediction.
Having identified the repetitive elements responsible for the initial formation of over 2,000 miRs, we tested the utility of our dataset in facilitating target prediction. For this, we selected human miR-28 which we identified as being initially formed by LINE1 sequences to predict targets based on a common miR-and-target origin. As we strictly required miR target sites to contain both perfect seed matches and at least 50% identity between sequences flanking a mature miR and sequences flanking a predicted target site, the resulting target predictions are far from saturated. However, although these results must still be experimentally verified in subsequent studies, as miR locus and proposed miR target site sequences have each apparently been specifically maintained (), we conclude these likely represent functional interactions.
Discussion
Our objective was to test the model that functional miRs arose in part from mobile element insertion events using genomic database analysis. Our report comprehensively examines miR genomic origins in the context of TE sequence comparisons. Strikingly, we identify TE-based origins for nearly 1/6 of the 15,176 currently recognized miR genomic loci. Undoubtedly, additional bioinformatic analyses will expand the current repertoire of miR-repetitive element relationships for several reasons. (1) Principally, not all miRs have been identified, and miR discoveries are biased towards evolutionarily older, conserved and non-repetitive miRs.Citation8 For example, several large scale sequencing efforts aimed at identifying the full repertoire of human miRs missed sequences with alignment to repetitive elements.Citation41,Citation42 Additionally, a background pool of ∼20 nt tRNA “degradation” products complicates identification by cloning. Even so, the tRNA source of SINEsCitation39 and connections with miR origins described here, warrants a re-evaluation of these small RNAs as active miRs.Citation41,Citation42 (2) Not all consensus repetitive elements are described and RepBase is continually updated, so Censor Server annotations are limited to the set of reported TEs.Citation31,Citation33 Greater than 1,000 miR loci familial inclusion-based annotations were removed from this study due to an inability to conclusively determine their common TE progenitor, likely indicating their absence in the current RepBase dataset. (3) Finally, genomes are dynamic and sequences with no associated benefit eventually degenerate. Therefore, if a stable miR locus and regulation of host genes containing targeted TE components were to arise, then the associated benefit of the regulatory network might well be retained across evolutionary time long after the elements from which they arose had been lost from the host genome. Given the documented role of miRs as regulators in both abiotic stress and nutrient deprivation, we postulate that the enigmatic origins of some of the more ancient, conserved miR loci might be explained via TE derivation. Under such a model, selective pressure to maintain only the components essential to transcriptional regulation, target recognition and hairpin structure could account for a decay of nonessential components resulting in the identification of TE-derived miRs and miR regulatory networks becoming more difficult to ascertain.
In contrast, we find the genomic origins of recently established miR-loci readily definable, and we also describe numerous examples of taxon-specific miR expansions coupled to taxon-specific, actively mobilizing TE repertoires. For instance, the miR-466 and -467 families initially formed from a mouse-specific CR1 non-LTR retrotransposon account for 50 of the 672 currently annotated mouse miR loci. Similarly, 70 of the 1,048 currently annotated human miR genomic loci were formed from primate-specific Alu repeats. As Alu repeats have formed numerous human miR loci,Citation19,Citation23 and mobilize regulatory elements, it is tempting to speculate that the ongoing primate-specific Alu expansion does not represent a failure of the RNAi machinery to constrain Alu transposition, but instead a fortuitous genetic symbiosis in which insertion of Alu elements into noncoding regions of transcripts has resulted in slight perturbations of gene expression ultimately giving way to an enhanced adaptation rate for the human genome.
Importantly, we stress that the results presented here do not argue against other known mechanisms that generate miR loci. Previous reports have indicated some miR loci being derived from regional duplications and/or processed antisense pseudogene transcripts.Citation44,Citation45 In addition, while we find many miR loci were initially formed by the chance integrations of transposable elements (TEs) into positions immediately adjacent to related TEs on the opposing strand ( and B), we find several loci apparently formed de novo from internal SINE sequences (). Importantly, families of viruses and retrotransposons commonly utilize tRNAs as primers for the initiation of transposition.Citation35 Models describing the origins of tRNA-derived SINEs from the genomic insertion of these intermediates have been previously described in references Citation38–Citation40. Conceivably, point mutations in the tRNAs involved in these processes could lead to the formation of stable hairpin structures, which if processed by the host RNAi defense machinery would allow the regulation of progenitor moieties as well as any additional host transcripts previously subject to their translocation. In either scenario, transposon mediated interactions may establish a mechanism whereby TE-derived miRs could integrate into and “experiment” with existing primitive gene networks. Stress-induced expression patterns of TEs are well documented with examples of their physiological induction in plants and animals.Citation46–Citation49 Given these data, we propose that the miR-based system of regulation may have arisen via selective subfunctionalization created by the associated benefit of regulating host genes containing portions of TEs (). Now identified in algal and protozoan genomes, miR regulatory networks predate the origins of multicellularity, and may have facilitated the evolution of complex developmental networks. A possible driver for the subfunctionalization may have been physiological stress resulting in the propagation of TEs that aided in the initial establishment of these regulatory networks.
Finally, there is currently no clear strategy for accurate miR target prediction. The recurrent observation of perfect complementarity between a seed and seed match () in the few characterized miR:target interactions has resulted in algorithms that base target searches on identification of perfect seed matches after which programs differ predominantly through the significance assigned to multiple seed matches within a given mRNA, the degree of complementarity between the remainder of a miR and proposed target, and seed match conservation across species.Citation11–Citation18 Our work strongly suggests that seed match conservation may hinder target recognition because of ongoing TE propagations after miR-locus formation, as well as the prevalence of taxon-specific miR expansions. In all probability, a uniform description of miR target interaction has not yet been identified because there is no uniform description of miR target interaction, which is due to various contributing factors such as GU base-pairing, nucleotide editing, local secondary structure/target accessibility, position effects due to nucleotide composition and RNA-binding protein availability.Citation8 This work, however, corroborated by several previous reports in references Citation23–Citation27, describes a molecular origin for many miRs that may help circumvent many of the difficulties in accurate target prediction.Citation8 Since active TEs are present in multiple copies across the genomeCitation33 and because miRs target sequences through complementary basepairing, requiring that a miR target site occur in a TE related to those from which a miR was initially formed represents a logical and (more importantly) simplifying addition to current informatic strategies ().
Materials and Methods
Retrieving miR and human 3′UTR sequences.
Single FASTA files containing the full sets of miR mature and stem loop sequences (from all species) were downloaded from the miR Registry housed at SangerCitation37 (http://www.mirbase.org/). Flanking genomic sequences were obtained for miRs corresponding to genomes currently available in EnsemblCitation52 by altering the nucleotide positions provided by RFAM as required then extracting the full sequences using the Biomart utilityCitation53 (www.ensembl.org/biomart/martview). The full set of ENSEMBL human 3′UTR sequences were also compiled in and retrieved using the Biomart mining utility.
Screening miR loci for repetitive elements.
Importantly, all alignment analyses and annotations were identically run in parallel by two independent research teams then merged for verification. FASTA files containing all miR stem loops in isolation and with 500 nt of flanking sequence both 5′ and 3′ (when available) were analyzed using the Genetic Information Research Institute online censor utility, “Censor Server”Citation33 (http://www.girinst.org/censor/index.php). All loci were also aligned against the full “all species” set of RepBaseCitation31 annotated repetitive elements, the publically available RFAM collectionCitation32 (www.sanger.ac.uk/Software/Rfam), and the tRNAscan-SE databaseCitation56 created by Todd Lowe at lowelab.cse.ucsc.edu/GtRNAdb using an in-house, stand-alone BLAST (BLASTN 2.2.15 with -FF, -r2, -W7, -e.1 flags). Significant alignments were strictly defined as ≥80% identity to at least 40 nt or ≥70% identity to at least 50 nt of an individual pre-miR. The highest p-scored alignment for each miR hairpin (averaging 82.9% identity over 85.7 nts) was utilized to define miR origins. Following sequence based annotation all miRs were separated into familial clusters based on miRBase nomenclature.Citation37 Common familial origin was defined as: (1) having the same TE produce the highest p-scored alignment to multiple members of a miR family and (2) no other family members producing significant alignments to a different TE.
MiR target prediction.
Six distinct sequences were assembled for each human miR locus whose progenitor TE(s) had been identified: the entire miR stem loop, the stem loop plus 100 nt 5′ and 100 nt 3′, the mature miR plus 20 nt 5′, the mature plus 20 3′ nt, the mature plus 10 nt 5′ and the mature plus 10 nt 3′. Each of these sequences (all containing both a mature miR and some amount of flanking sequence) were screened against the full set of human 3′ UTRs currently available in Ensembl (>18,000 sequences) using BLASTN 2.2.15 with -FF, -r2, -S2, -W7 flags. Putative target “hits” were required to (1) contain a perfect miR seed match (2) bear >50% identity to flanking sequences and (3) be located within 3′UTR sequences annotated as a particular miR's progenitor TE by the Genetic Information Research Institute online censor utility.Citation33
Abbreviations
| bp | = | basepair |
| LINE | = | long interspersed repeated element |
| LTR | = | long terminal repeat |
| miR | = | microRNA |
| mRNA | = | messenger RNA |
| nt | = | nucleotide |
| ORF | = | open reading frame |
| Pol III | = | RNA polymerase III |
| RNAi | = | RNA interference |
| scaRNA | = | small cajal body-specific RNA |
| SINE | = | short interspersed repeated elements |
| siRNA | = | small interfering RNA |
| snoRNA | = | small nucleolar RNA |
| SRP | = | signal recognition particle |
| TE | = | transposable element |
| tRNA | = | transfer RNA |
| UTR | = | untranslated region |
Figures and Tables
Figure 1 MiR biology. (A) MiR production. MiRs occur inter- and intragenically and can be transcribed by RNA Polymerase II or III (Pol-II or Pol-III).Citation23 Prior to nuclear export, the “pri-miR” hairpin is excised from the initial transcript by Drosha. Following nuclear export, the hairpin is processed by Dicer to produce the ∼20 nt mature miR. Image adapted from Bartel et al.Citation54 (B) MiR seeds and seed matches. Cartoon depicting a perfect seed match between a mature miR (top) and a target mRNA (bottom). The miR nucleotides commonly referred to as a “seed” (basepairs 2 through 8) and a perfect seed match in the mRNA are shown in red. Vertical lines indicate basepairing.
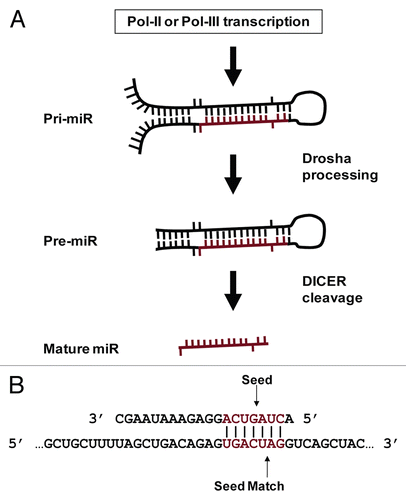
Figure 2 MiRs commonly occur at the intersection of related, converging TEs. (A) Cartoon depicting the theoretical origin of numerous miRs. A pri-miR is depicted just above an arrow indicating read through transcription from a positive strand LINE1 (L1) element into an adjacent negative strand L1. This relationship suggests a likely series of events leading to the creation of a potential miR hairpin in which an L1 is inserted immediately adjacent to a related L1 on the opposite strand creating the convergent or “tail to tail” organization illustrated. Next, transcriptional read through would result in an imperfect RNA hairpin being produced potentially recognized and processed by the RNAi machinery with each stem corresponding to the terminal nucleotides of the contributing LINEs. (B) Examples of human miR loci alignments to the RepBase dataset. Importantly, all pre-miRs significantly aligning with a Censor Server repetitive element annotation have been reported irrespective of agreement with the scenario portrayed in (A)—while we find numerous loci arising by this mechanism, we find others (like miR-640) do not. Entirely contained within an THER1 SINE, we propose an additional mechanism (point mutation(s) resulting in an alteration of normal SINE secondary structure gave rise to pre-miR-640). All repetitive elements (grey rectangles) occurring within 500 bp (5′ and 3′) have been included in the scale diagrams for uniformity. The RepBase repetitive element annotations found in these diagrams are described immediately beneath each locus as “Element 1, Element 2, etc.,” as they occur 5′ to 3′. “Base Positions” refers to the basepairs occupied by a miR hairpin (in the current Ensembl assembly). All loci have been diagrammed with respect to the Watson strand and the orientation of internal elements indicated by position above (5′ to 3′) or below (3′ to 5′) the center line. Element basepair positions are in respect to distance (±) from the 1st nucleotide of the pre-miR (as occurring on the Watson strand). *previously described origin.Citation21,Citation23 Figures adapted from references Citation21 and Citation23.
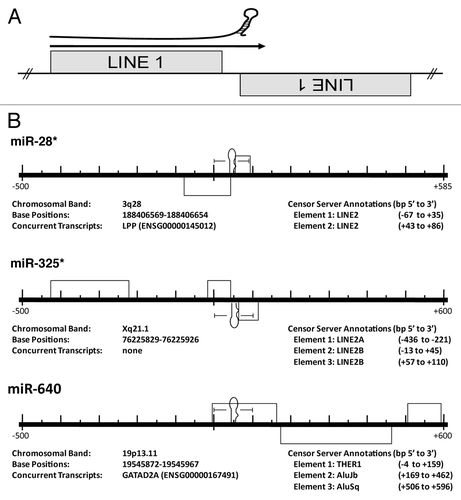
Figure 3 MiR-284 familial alignment. Alignment of the 12 miR-284 hairpins. Individual hairpin sequences along with species (right) and miRBase identifier (left) are shown. *indicates 100% nucleotide conservation. Grey highlight indicates specific miR hairpins annotated as bearing significant sequence complementarity to Mermite-35.

Figure 4 MiR-28 alignments with predicted targets. Alignments between three predicted miR-28 target 3′UTRs (top), a consensus L2 LINE (middle) and the miR-28 genomic sequence reverse complemented (bottom) are illustrated. Mature miR-28 is highlighted in grey. Open boxes indicate perfect seed matches. To qualify as a 3′UTR “hit”, alignments were required to (1) contain a perfect seed match, (2) match .50% of the flanking sequence used in the target query and (3) occur within a 3′UTR sequence annotated as an L2 sequence by Censor Server. Vertical lines indicate base identity with the L2 consensus sequence. Dotted lines indicate purine/pyrimidine conservation. LYPD3, “LY6/PLAUR domain containing 3”; E2F6, “E2F transcription factor 6”; CXCL9, “chemokine (C-X-C motif) ligand 9”.
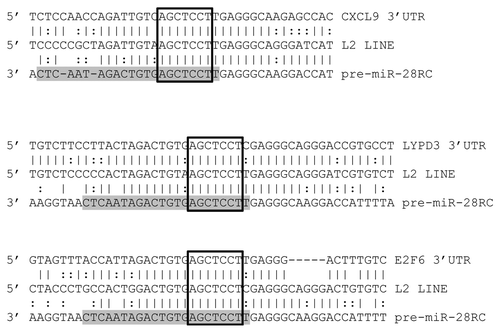
Figure 5 Molecular events responsible for miR establishment. MiRs “arise” when an advantageous regulatory niche has developed out of a series of random TE insertions after which the fortuitous formation of a TE juxtaposition (shown, ) and subsequent processing by RISC can lead to miR establishment if the resulting small RNAs confer some regulatory advantage in order to be selected for (e.g., improved cell tolerance to apoptotic stimulus due to delayed response accompanying translational repression). Numbers indicate the sequential steps necessary for miR establishment. Thick lines indicate genomic DNA and thin lines denote RNA.
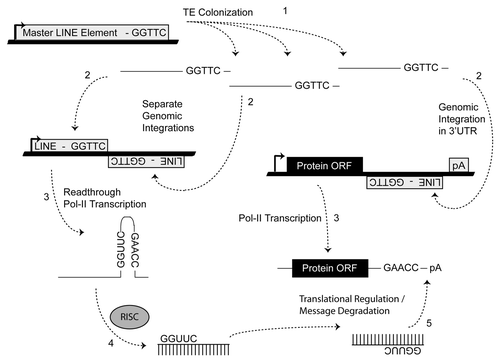
Table 1 Summary of MiR loci progenitor transposable elements
Table 2 Summary of familial inclusions
Additional material
Download Zip (658.8 KB)Acknowledgements
This work was funded by the School of Biological Sciences, the College of Arts and Sciences at Illinois State University, and National Institutes of Health Grant 1 R15 CA137608.
References
- Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993; 75:843 - 854
- Lee RC, Ambros V. An extensive class of small RNAs in Caenorhabditis elegans. Science 2001; 294:862 - 864
- Lau NC, Lim LP, Weinstein EG, Bartel DP. An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science 2001; 294:858 - 862
- Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998; 391:806 - 811
- Cai X, Hagedorn CH, Cullen BR. Human microRNAs are processed from capped, polyadenylated transcripts that can also function as mRNAs. Rna 2004; 10:1957 - 1966
- Hutvagner G, Zamore PD. A microRNA in a multiple-turnover RNAi enzyme complex. Science 2002; 297:2056 - 2060
- Farazi TA, Spitzer JI, Morozov P, Tuschl T. miRNAs in human cancer. J Pathol 2011; 223:102 - 115
- Smalheiser NR, Torvik VI. Complications in mammalian microRNA target prediction. Methods Mol Biol 2006; 342:115 - 127
- Zeng Y, Wagner EJ, Cullen BR. Both natural and designed micro RNAs can inhibit the expression of cognate mRNAs when expressed in human cells. Mol Cell 2002; 9:1327 - 1333
- Lai EC. Micro RNAs are complementary to 3′ UTR sequence motifs that mediate negative post-transcriptional regulation. Nat Genet 2002; 30:363 - 364
- Burgler C, Macdonald PM. Prediction and verification of microRNA targets by MovingTargets, a highly adaptable prediction method. BMC Genomics 2005; 6:88
- Kruger J, Rehmsmeier M. RNAhybrid: microRNA target prediction easy, fast and flexible. Nucleic Acids Res 2006; 34:451 - 454
- Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP, Burge CB. Prediction of mammalian microRNA targets. Cell 2003; 115:787 - 798
- Nam JW, Shin KR, Han J, Lee Y, Kim VN, Zhang BT. Human microRNA prediction through a probabilistic co-learning model of sequence and structure. Nucleic Acids Res 2005; 33:3570 - 3581
- Rehmsmeier M, Steffen P, Hochsmann M, Giegerich R. Fast and effective prediction of microRNA/target duplexes. RNA 2004; 10:1507 - 1517
- Rhoades MW, Reinhart BJ, Lim LP, Burge CB, Bartel B, Bartel DP. Prediction of plant microRNA targets. Cell 2002; 110:513 - 520
- Saetrom O, Snove O Jr, Saetrom P. Weighted sequence motifs as an improved seeding step in microRNA target prediction algorithms. RNA 2005; 11:995 - 1003
- Wang X. Systematic identification of microRNA functions by combining target prediction and expression profiling. Nucleic Acids Res 2006; 34:1646 - 1652
- Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, et al. The sequence of the human genome. Science 2001; 291:1304 - 1351
- Diao XM, Lisch D. Mutator transposon in maize and MULEs in the plant genome. Yi Chuan Xue Bao 2006; 33:477 - 487
- Smalheiser NR, Torvik VI. Mammalian microRNAs derived from genomic repeats. Trends Genet 2005; 21:322 - 326
- Tempel S, Jurka M, Jurka J. Visual-Repbase: an interface for the study of occurrences of transposable element families. BMC Bioinformatics 2008; 9:345
- Borchert GM, Lanier W, Davidson BL. RNA polymerase III transcribes human microRNAs. Nat Struct Mol Biol 2006; 13:1097 - 1101
- Yao C, Zhao B, Li W, Li Y, Qin W, Huang B, et al. Cloning of novel repeat-associated small RNAs derived from hairpin precursors in Oryza sativa. Acta Biochim Biophys Sin (Shanghai) 2007; 39:829 - 834
- Yan Y, Zhang Y, Yang K, Sun Z, Fu Y, Chen X, et al. Small RNAs from MITE-derived stem-loop precursors regulate abscisic acid signaling and abiotic stress responses in rice. Plant J 2011; 65:820 - 828
- Piriyapongsa J, Jordan IK. A family of human microRNA genes from miniature inverted-repeat transposable elements. PLoS One 2007; 2:203
- Devor EJ, Peek AS, Lanier W, Samollow PB. Marsupial-specific microRNAs evolved from marsupial-specific transposable elements. Gene 2009; 448:187 - 191
- Agrawal A, Eastman QM, Schatz DG. Transposition mediated by RAG1 and RAG2 and its implications for the evolution of the immune system. Nature 1998; 394:744 - 751
- Allen TA, Von Kaenel S, Goodrich JA, Kugel JF. The SINE-encoded mouse B2 RNA represses mRNA transcription in response to heat shock. Nat Struct Mol Biol 2004; 11:816 - 821
- Espinoza CA, Allen TA, Hieb AR, Kugel JF, Goodrich JA. B2 RNA binds directly to RNA polymerase II to repress transcript synthesis. Nat Struct Mol Biol 2004; 11:822 - 829
- Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O, Walichiewicz J. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet Genome Res 2005; 110:462 - 467
- Griffiths-Jones S, Moxon S, Marshall M, Khanna A, Eddy SR, Bateman A. Rfam: annotating non-coding RNAs in complete genomes. Nucleic Acids Res 2005; 33:121 - 124
- Kohany O, Gentles AJ, Hankus L, Jurka J. Annotation, submission and screening of repetitive elements in Repbase: RepbaseSubmitter and Censor. BMC Bioinformatics 2006; 7:474
- Konkel MK, Batzer MA. A mobile threat to genome stability: The impact of non-LTR retrotransposons upon the human genome. Semin Cancer Biol 2010; 20:211 - 221
- Bushman FD. Targeting survival: integration site selection by retroviruses and LTR-retrotransposons. Cell 2003; 115:135 - 138
- Ni J, Clark KJ, Fahrenkrug SC, Ekker SC. Transposon tools hopping in vertebrates. Brief Funct Genomic Proteomic 2008; 7:444 - 453
- Kozomara A, Griffiths-Jones S. miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res 2011; 39:152 - 157
- Ponicsan SL, Kugel JF, Goodrich JA. Genomic gems: SINE RNAs regulate mRNA production. Curr Opin Genet Dev 2010; 20:149 - 155
- Ohshima K, Okada N. SINEs and LINEs: symbionts of eukaryotic genomes with a common tail. Cytogenet Genome Res 2005; 110:475 - 490
- Dewannieux M, Heidmann T. LINEs, SINEs and processed pseudogenes: parasitic strategies for genome modeling. Cytogenet Genome Res 2005; 110:35 - 48
- Berezikov E, van Tetering G, Verheul M, van de Belt J, van Laake L, Vos J, et al. Many novel mammalian microRNA candidates identified by extensive cloning and RAKE analysis. Genome Res 2006; 16:1289 - 1298
- Bentwich I, Avniel A, Karov Y, Aharonov R, Gilad S, Barad O, et al. Identification of hundreds of conserved and nonconserved human microRNAs. Nat Genet 2005; 37:766 - 770
- Jurka J. Evolutionary impact of human Alu repetitive elements. Curr Opin Genet Dev 2004; 14:603 - 608
- Voinnet O. Origin, biogenesis and activity of plant microRNAs. Cell 2009; 136:669 - 687
- Megraw M, Sethupathy P, Corda B, Hatzigeorgiou AG. miRGen: a database for the study of animal microRNA genomic organization and function. Nucleic Acids Res 2007; 35:149 - 155
- Hirayama T, Shinozaki K. Research on plant abiotic stress responses in the post-genome era: past, present and future. Plant J 2010; 61:1041 - 1052
- Beauregard A, Curcio MJ, Belfort M. The take and give between retrotransposable elements and their hosts. Annu Rev Genet 2008; 42:587 - 617
- Cho K, Lee YK, Greenhalgh DG. Endogenous retroviruses in systemic response to stress signals. Shock 2008; 30:105 - 116
- Ebina H, Levin HL. Stress management: how cells take control of their transposons. Mol Cell 2007; 27:180 - 181
- Griffiths-Jones S, Bateman A, Marshall M, Khanna A, Eddy SR. Rfam: an RNA family database. Nucleic Acids Res 2003; 31:439 - 441
- Griffiths-Jones S, Moxon S, Marshall M, Khanna A, Eddy SR, Bateman A. Rfam: annotating non-coding RNAs in complete genomes. Nucleic Acids Res 2005; 33:121 - 124
- Birney E, Andrews D, Caccamo M, Chen Y, Clarke L, Coates G, et al. Ensembl 2006. Nucleic Acids Res 2006; 34:556 - 561
- Durinck S, Moreau Y, Kasprzyk A, Davis S, De Moor B, Brazma A, et al. BioMart and Bioconductor: a powerful link between biological databases and microarray data analysis. Bioinformatics 2005; 21:3439 - 3440
- Bartel B, Bartel DP. MicroRNAs: at the root of plant development?. Plant Physiol 2003; 132:709 - 717
- Griffiths-Jones S, Bateman A, Marshall M, Khanna A, Eddy SR. Rfam: an RNA family database. Nucleic Acids Res 2003; 31:439 - 441
- Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 1997; 25:955 - 964