Abstract
Restriction-like endonuclease (RLE) bearing non-LTR retrotransposons are site-specific elements that integrate into the genome through a target primed reverse transcription mechanism (TPRT). R2 elements have been used as a model system for investigating non-LTR retrotransposon integration. We previously demonstrated that R2 retrotransposons require two subunits of the element-encoded multifunctional protein to integrate—one subunit bound upstream of the insertion site and one bound downstream. R2 elements have been phylogenetically categorized into four clades: R2-A, B, C, and D, that diverged from a common ancestor more than 850 million years ago. All R2 elements target the same sequence within 28S rDNA. The amino-terminal domain of R2Bm, a R2-D clade element, contains a single zinc finger and a Myb motif that are responsible for binding R2 protein downstream of the insertion site. Target site recognition is of interest as it is the first step in the integration reaction and may help elucidate evolutionary history and integration mechanism. The amino-terminal domain of R2-A clade members contains three zinc fingers and a Myb motif. We show here that R2-Lp, an R2-A clade member, uses its amino-terminal DNA binding motifs to bind upstream of the insertion site. Because the R2-A and R2-D clade elements recognize 28S rDNA differently, we conclude the A- and D-clades represent independent targeting events to the 28S site. Our results also indicate a certain plasticity of insertional mechanics exists between the two clades.
Introduction
Non-LTR retrotransposons transpose through an ordered series of DNA cleavage and polymerization events using encoded endonuclease and polymerase functions.Citation1 Non-LTR retrotransposons have been phylogenetically classified into at least 15 clades to date.Citation2–Citation6 These clades can be classified as either early branching or late branching. Elements belonging to the late branching group tend to have two open reading frames (ORFs) and use an apurinic-apyrimidinic endonuclease (APE) to initiate target primed reverse transcription (TPRT).Citation7–Citation9 These elements typically insert in a non-specific manner and may accumulate to high copy numbers. For example, the human Long Interspersed Element 1 family (L1Hs) inserts into T-A rich sequences and comprises about one sixth of the genome (reviewed in ref. Citation7, Citation10 and Citation11). Multiple elements from the late branching group have been studied and serve as model systems for this group including L1, I factor, Tras, R1 and Tx1L.Citation12–Citation22
The early branching elements tend to encode a single open reading frame and utilize a type IIs restriction-like endonuclease (RLE) to initiate target primed reverse transcription (TPRT).Citation4,Citation23,Citation24 The RLE-bearing elements encode DNA binding motifs, like zinc fingers (ZF) and Myb domains, to target insertion events to specific sites in the genome (although some non-site-specific members exist).Citation4,Citation25–Citation27 Often these specific target sites are within repetitive sequences (e.g., ribosomal genes or another transposon). Targeting a highly repetitive locus is thought to be a useful survival strategy, allowing the transposon to avoid essential host genes. The early branching group has been divided into five clades, Genie, CRE, R4, R2 and NeSL.Citation2–Citation4,Citation6,Citation28 R2 (especially the R2Bm element) is the most well studied of the five clades and serves as the model for all RLE bearing non-LTR retrotransposons—and sometimes for the APE-bearing non-LTR retrotransposons. Indeed, TPRT, the first half of the integration reaction of non-LTR retrotransposons, was first determined for the R2Bm element and later confirmed with L1Hs and other late branching model systems.Citation9,Citation12,Citation23
R2 elements are vertically inherited and insert into a particular sequence within the 28S rDNA gene.Citation4,Citation5,Citation29 The insertion site lies within a patch of highly conserved sequence (). The extreme conservation is likely due to the fact that this sequence forms a major interaction surface with the small ribosomal subunit in the context of the folded ribosome.Citation30 Upon insertion, a full length element generates a 0–5 bp target site deletion.Citation31 R2 elements have been found in a wide range of arthropods (e.g., Bombyx mori, Nasonia vitripennis, Popilla japonica and Limulus polyphemus). R2 element have also been found in Platyhelminthes (e.g., Schistosoma mansoni), chordates (e.g., Ciona intestinalis) and vertebrates (e.g., Danio rerio).Citation4,Citation5 The R2 super-clade has been subdivided into four clades—R2-A, R2-B, R2-C and R2-D—based on reverse transcriptase phylogeny (see ).Citation5
The amino-terminal region of R2 encodes a Myb domain and up to three zinc finger motifs. R2-A clade members have three zinc finger motifs (see ).Citation4,Citation5,Citation32 The first zinc finger is located next to the Myb and has a canonical cysteine and histidine spacing for a zinc finger, C X2C X3FXT/SX2GX3 HX4H. The second zinc finger is a variant with cysteine replacing a histidine, CX2CX12HX3C. The third zinc finger is similar to the first, with a spacing of CX2CX12HX4H. The other clades have subsets of the zinc fingers found in the R2-A clade.Citation4,Citation5,Citation32 The R2-C clade lacks the zinc finger corresponding to R2-A clade ZF2.Citation5 The R2-D clade lacks the zinc fingers corresponding to R2-A clade ZF2 and ZF3. The amino- terminal structure of the R2-B clade has not been determined.Citation5
As the R2-A clade appears to carry a full complement of amino-terminal motifs, and as it is early branching within the R2 superclade, it is reasonable to assume that the ancestral R2 element had three zinc fingers and a Myb motif.Citation5 The R2-C and R2-D clades, then, represent lineages that have lost one or more of the ancestral amino-terminal motifs. That said, it is important to note that each of the R2 clades—R2-A, R2-B, R2-C and R2-D—are themselves highly divergent. Divergence-versus-age analysis is consistent with the clades originating prior to the protostome/deuterostome split.Citation5,Citation29,Citation33 The R2-A and R2-D clades diverged from a common ancestor more than 850 million years ago.Citation5,Citation29,Citation33
Insertion of R2 is often accompanied by concomitant deletions of large segments of the rDNA units—often deleting previous R2 insertions.Citation34 In addition, inter- and intra-chromosomal recombination can increase or decrease the number of rDNA units over time. In one study the number of rDNA units ranged from 140 to 310 in replicate Drosophila melanogaster lines over 400 generations.Citation35 Recombination rates in the rDNA locus appear to be quite high (reviewed in ref. Citation36). Chromosomes with the largest deletions appear to be eliminated from the population.Citation34 The rDNA locus is dynamic; R2 elements must continually transpose or face possible elimination from the locus.Citation34,Citation37,Citation38 Indeed, the genomes of some organisms appear to have purged R2. The extinction of R2 is evident in several arthropod lineages including, the cricket A. domesticus and the mosquito A. gambiae.Citation6,Citation39 Additionally, while present in D. rerio, R2 is not found in the genomes of human, mouse or pufferfish, indicating that R2 extinction likely has occurred several times in vertebrate evolution.Citation40–Citation42 Most genomes that harbor R2 have lost all but a single clade (R2-D clade in the case of B. mori, R2-A clade in the case of L. polyphemus).Citation4,Citation43 Interestingly, a few organisms have been found still harboring multiple clades of R2—C. intestinalis and N. vitripennis have both retained R2-A and R2 D-clade elements.Citation4,Citation6,Citation44
In vitro studies investigating the R2Bm protein and its activities have elucidated much about the insertional mechanism the R2-D clade elements.Citation1,Citation45–Citation47 Two subunits of R2Bm protein, one bound upstream of the insertion site and one bound downstream of the insertion site, effect integration through a series of coordinated DNA cleavage and DNA polymerization events. The upstream subunit binds to the DNA using an undetermined DNA binding motif. This subunit cleaves the target DNA and performs TPRT. The downstream subunit uses the amino-terminal ZF and Myb motifs to bind to DNA. The downstream subunit cleaves the second DNA strand and is hypothesized to perform second strand synthesis. Little is known about how the other R2 clades bind to DNA and integrate. The R2-A clade is of particular interest as it may represent the more ancestral R2 state.
As DNA binding is the first step in the integration reaction and is key to evolving novel integration sites, it is important to understand how non-LTR retrotransposons with different DNA binding motifs and evolutionary histories target their integration sites. In this paper, we show that the R2-A clade member R2Lp uses its amino-terminal DNA binding motifs to bind to sequences upstream of the 28S rDNA insertion site. This finding is in contrast to R2-D clade member R2Bm which uses its amino-terminal DNA binding motifs to bind downstream of the 28S rDNA insertion site.Citation1,Citation25 We conclude that the different binding modes indicate that the R2-A and R2-D clades may have independently targeted the R2 site. Our findings also suggest that a certain plasticity of integration-mechanism wiring exists between the R2 clades.
Results
DNA binding activity of an R2-A clade element's amino-terminal region.
In order to investigate how R2-A clade members target 28S rDNA, bacterial expression constructs were generated by expressing the amino-terminal zinc fingers and Myb domains of the horseshoe crab (L. polyphemus) transposon (R2Lp) (). R2Lp was chosen because L. polyphemus is a deep branching arthropod. The thick black lines under the structure diagrams in denote the portions of R2Lp that were cloned and expressed.
The largest R2-A clade polypeptide we expressed, R2Lp ZF3-Myb, was used in Electrophoretic Mobility Shift Assays (EMSA) to determine if the amino-terminal motifs are used to bind target DNA similar to the R2-D clade elements (). The DNA target in was a 150 bp segment of 28S rDNA containing the insertion site with 80 bp of upstream and 70 bp of downstream sequence. The migration of naked DNA (e.g., the “reference DNA” in lane 1 and the “free DNA” in lane 2) and protein:DNA complexes (i.e., “bound DNA”) are marked. R2Lp ZF3-Myb peptide binds to target DNA ( and lane 2). The protein:DNA complex observed in lane 2 appears to be due to the R2Lp ZF3-Myb peptide and not contaminating E. coli DNA binding proteins in the protein preparation as protein extract isolated from E. coli expressing the negative control construct pGUS did not appreciably bind DNA. A weak band can be observed, but the complex runs slower than the R2Lp ZF3-Myb bound DNA and requires vast excess of protein extract (>10X R2Lp-Myb) to be observed ( and lane 3).
In order to determine whether the R2Lp ZF3-Myb polypeptide bound upstream or downstream of the insertion site, a second EMSA experiment was performed using “half-site” DNA targets (). The 32P labeled target sequences used in this assay corresponded to the 49 bp of DNA immediately upstream of the insertion site (lanes 1 and 2) or the 49 bp of DNA immediately downstream of the insertion site (lanes 3 and 4). The R2Lp ZF3-Myb polypeptide appears to bind upstream DNA in preference to downstream DNA by a factor of >4X, shown in lanes 2 and 4 respectively. Although binding upstream is preferred, it is unclear if the binding is specific or non-specific in nature. It is possible that the greater relative binding to upstream DNA represents a stable and specific interaction while downstream binding is the result of non-specific interactions. If true, this result would be in stark contrast to the binding observed for the R2-D clade element R2Bm. The zinc finger plus Myb motif from R2Bm binds downstream of the insertion site.Citation1,Citation25
Mapping the DNA binding activity of R2Lp Zf3-Myb polypeptide using DNA footprint analysis.
If the binding of the R2Lp ZF3-Myb polypeptide to target DNA is specific, it should form a stable protein:DNA complex at a given DNA sequence, be it to upstream or downstream DNA. A stable complex at a given site should be detectable by DNase I footprinting. Thus, in order to detect and map the site of R2Lp ZF3-Myb polypeptide interaction with DNA, a DNase I protection analysis was performed on the protein:DNA complexes formed on the 150 bp target DNA. DNase I was used to cleave the DNA with one cleavage event per DNA target. Segments of DNA that are bound by protein are protected from cleavage. The unbound-reference-DNA and protein-bound DNA-complexes are fractionated using EMSA and assayed by denaturing gel electrophoresis. The DNase I protection data are presented in , with top strand data on the left and bottom strand data on the right. The lanes marked “bound” and “ref.” are the DNase I treated protein-bound-to-DNA fraction and the DNase I treated naked-DNA-reference fraction, respectively, run on a denaturing polyacrylamide gel. Note that either “free” or “ref.” as defined in can be valid sources of a naked-DNA-reference for this type of experiment so long as the protein being footprinted does not fall off or recycle prior to EMSA fractionation. The ref. DNA used here came from a DNase I reaction lacking the R2Lp ZF3-Myb polypeptide. Next to the bound lane the areas of DNase I protection denoting R2Lp ZF3-Myb binding have been marked with long thick black lines next to the denaturing gel. Short thick black lines denote DNase I hypersensitive sites that were induced by the binding of the R2Lp ZF3-Myb polypeptide. There are three blocks of protection: one centered roughly around the −35 nt region (top strand −40 to −27, bottom strand −40 to −27), one in the −20 region (top strand −20 to −13, bottom strand −24 to −18), and one in the −10 region (top strand −9 to −5, bottom strand −14 to −7). As in , the nucleotide numbering is relative to the R2Bm insertion dyad.
In order to gain a higher resolution map of the R2Lp ZF3-Myb polypeptide bound to target DNA, a missing nucleoside footprint was generated. Missing nucleoside footprinting is a binding interference assay in which random DNA bases are chemically removed from labeled DNA target (one base per target).Citation49 The treated DNA is used in a binding reaction. The protein of interest selects out target DNA that it can bind to. Targets with a base missing at positions that interfere with protein binding preferentially accumulate in the free fraction. For this reason, the “free” and “ref.” DNA are not equivalent. In a missing nucleoside assay, reference (ref.) is the chemically treated “missing nucleoside” DNA. The free and bound fractions are the result of missing nucleoside DNA exposed to protein in a binding reaction followed by EMSA fractionation. The resulting reference, bound and free DNA fractions are analyzed on a denaturing polyacrylamide gel. Bands corresponding to a DNA base that, when missing, interferes with protein binding will be under-represented in the bound fraction and over-represented in the free fraction when compared to the reference DNA. The missing nucleoside data presented in indicated 28S target DNA bases involved in interacting with the R2Lp ZF3-Myb polypeptide are found in the −35 bp region (top strand −40 to −32, bottom strand −38 to −30), the −20 bp region (top strand −22 to −18, bottom strand −23 to −18), and to a lesser extent the −10 bp region (bottom strand −12 to −9) in good agreement with the DNase I footprint data presented in .
Locating the Myb binding site.
In order to ascertain which portion of the R2Lp ZF3-Myb footprint is due to the Myb domain and which portion is due to the ZF motifs, a missing nucleoside footprint of the R2Lp Myb only polypeptide was generated (). The R2Lp Myb footprinted to the −35 region (top strand −40 to −32, bottom strand −38 to −30). For comparison, a missing nucleoside footprint of the Myb domain from the R2-D-clade-member R2Bm is shown bound to the +10 bp region ( and see also ref. Citation25). In R2Bm, the −35 region is bound by an as-yet-undefined DNA binding domain of the R2Bm protein.Citation1,Citation25
The remainder of the R2Lp Zf3-Myb polypeptde's footprint ( and ) must be due to one or more of the R2Lp zinc fingers. It is possible that R2Lp ZF1 binds to the −20 region, and ZF3 may bind to the −10 region. The DNase footprints of the ZF2-Myb and the ZF1-Myb are similar—lacking the −10 region but containing the −35 region and most of the −20 region (see Sup. Data and ). Additional studies would need to be performed to confirm the suggested DNA binding roles of ZF1 and ZF3. Regardless, it is unlikely that all three zinc fingers bind to DNA cooperatively and/or specifically as the ZF2-Myb and ZF1-Myb footprints are similar and as the R2Lp ZF3-1 polypeptide did not bind tightly in EMSA gels (data not shown).
Discussion
shows a summary of the R2Lp amino-terminal-derived polypeptide footprint data overlaid on a linear DNA sequence. The black dotted lines denote R2Lp missing nucleoside data generated from the R2Lp Myb and R2Lp ZF3-Myb protein-DNA complexes (see also and ). The thick black lines denote the R2Lp ZF3-Myb DNase I footprint. The DNase I hypersensitive sites have been left out for clarity. The thick gray lines denote R2Lp ZF2-Myb and ZF1-Myb DNase I footprints (see Sup. data). The location of the Myb binding region (the −35 bp region) and putative zinc finger binding regions (the −20 bp and −10 bp regions) have been labeled “Myb,” “ZF1?” and “ZF3?” respectively. The binding of the the R2Bm (i.e., R2-D clade) Myb is indicated by a gray box (see also ). The results clearly show that R2-A and R2-D clade Mybs are used to target the R2 site differently. The data indicate that an R2-A clade Myb is likely used to target a protein subunit upstream of the insertion site while the R2-D clade Myb is used to target a subunit downstream of the insertion site. There is virtually no sequence similarity between the two binding sites. Interestingly, the sequence where the R2-A clade Myb binds sits within the region (DNase I) footprinted by the R2-D clade upstream subunit—top strand −38 to −10, bottom strand −42 to −16—albeit using an unidentified DNA binding motif(s).Citation50 Similarly, it is not known which conserved protein motif is used to target (or not) a R2-A clade subunit to downstream sequences. Presumably both clades are using the same (re-purposed) previously-hypothesized “second DNA-binding domain.”Citation1,Citation25
Both clades appear to use the Myb associated zinc finger(s) to assist the Myb in targeting the DNA.Citation25 The Myb and the first zinc finger seem to function as a unit (our data and ref. Citation25) It is less clear what role the additional zinc fingers of the R2-A clade elements play. We hypothesize that perhaps the third zinc finger binds DNA in the −10 region, leaving ZF2 performing some other role in integration. It is unlikely that all three R2-A clade zinc fingers bind DNA as the R2Lp ZF3-1 polypeptide did not bind DNA tightly in our reactions (data not shown). While our footprint data is consistent with above zinc finger binding interpretation, additional studies would need to be performed to confirm our proposed ZF assignment.
Because R2-A and R2-D-clades target the R2 site differently, there must be plasticity in how the various functional domains and integration activities are linked together. In R2Bm it was shown that the upstream subunit bound to target through an unidentified protein domain, cleaved the bottom antisense rDNA strand and initiated TPRT. The downstream subunit bound to target DNA through the amino-terminal ZF and Myb, cleaved the sense rDNA strand and (hypothetically) synthesized the second cDNA strand.Citation1,Citation25,Citation46 In R2Lp some or all of the links between DNA binding domains, DNA cleavage and polymerization functions have to have been remapped with regard to how the upstream and downstream subunits function in R2Bm.
In addition, because R2Lp and R2Bm target the 28S rDNA differently, we conclude that the R2-A and the R2-D clades must have targeted the R2 site independently of each other at some stage. Assuming the R2 site is the ancestral site, ether the R2-A or the R2-D clade must have derived a new way to bind upstream and downstream of the site. The new way of binding may have allowed the new element to flourish and compete for the the canonical site. Perhaps the re-engineering event(s) occurred as a result of the R2-D clade losing two of the (presumably) three ancestral zinc fingers. The “re-targeting” could have happened while still at the canonical R2 site. Alternatively, there may have been pressure to escape the 28S or the ribosomal locus at some point. For example, existing competition for the canonical site or an overall paucity of ribosomal repeats could have occurred. A re-engineered ancestral element may have evolved to target elsewhere in the genome then reacquired the canonical site at a later point. In either case, it appears that R2 elements have the ability to adapt and to change how they interact with target DNA. Indeed, R2-like elements have been discovered that phylogenetically clade with R2 but are not targeted to the R2 insertion site. R8, an element found in hydra, groups with the R2-A clade but targets the 18S rDNA.Citation52 R9, an element found in bdelloid rotifers, groups with the R2-A clade and targets a site in the 28S rDNA approximately 1,436 bp upstream of the standard R2 site.Citation53 R9 is also unique in that it generates a 126 bp target site duplication.Citation53 It will be interesting to see how R8 and R9 target their respective sites. R9 is also interesting in light of the large target site duplication which is very atypical for RLE bearing non-LTR retrotransposons. Identifying how other RLE bearing non-LTR retrotransposons bind to their target may lead to the engineering of R2 elements to target sites of interest (e.g., for use as site specific gene targeting vectors).
Materials and Methods
Generating expression constructs.
Constructs containing a varying number of R2 derived amino terminal zinc finger motifs were generated and named as follows: R2Lp ZF3-Myb corresponds to codons 67–280 of the L. polyphemus R2 transposon, R2Lp ZF2-Myb to codons 116–280, R2Lp ZF1-Myb to codons 154–280, R2Lp ZF3-1 to codons 67–183, R2Lp Myb to codons 199–280 and R2Bm Myb to codons 138–228 of the B. mori R2 transposon (). Polymerase chain reaction (PCR) primers used to amplify the regions of interest from L. polyphemus genomic DNA or a codon optimized R2Bm gene are listed in .
The above regions of interest were cloned into the bacterial expression vector pET28a (Novagen 69864-3), the Gateway destination vector pDEST 17 (Invitrogen 11803-012), or the Gateway compatible destination vector pDESTTAP (see below). Initial ligation and recombination reactions were transformed into electroporation competent XL-1 Blue Escherichia coli (Agilent 200259) or chemically competent Oneshot Top10 E. coli (Invitrogen C4040-10) for screening purposes. Resulting colonies were screened by PCR and sequenced (Big Dye, Applied Biosystems 4337455).
The R2Lp ZF3-Myb fragment was cloned into the NdeI and BamHI sites of pET28a with T4 ligase. All other fragments were first recombined into the Gateway donor vector pENTR/TEV/D-TOPO (Invitrogen K2535-20) using the manufacturer's protocol. Inserts from confirmed Gateway entry clones were then recombined into destination vectors. R2Lp ZF2-Myb and R2Lp ZF1-Myb went into pDEST17 while R2Lp Myb went into pDESTTAP. Additionally a pDESTTAP version of the Gateway LR-clonase control-reaction-gene GUS was generated for use as a negative control.
The pET28a and the pDEST17 constructs used the amino-terminal 6X His tag engineered into those vectors. The Gateway-compatible destination vector pDESTTAP is a low-copy-number plasmid with amino terminal 6X His and malE tags. The pDESTTAP plasmid was constructed by cloning the malE gene from pMALc4x (New England Biolabs n81085) and the Gateway destination cassette from pDEST17 (Invitrogen 11803-102) into KpnI and XhoI sites of the pET45b plasmid (Novagen 71327-3). The PCR primers used to amplify the destination cassette and malE are listed in .
Protein expression and purification.
Sequence confirmed clones were transformed into ArcticExpress DE3 or ArcticExpress DE3 RIL E. coli (Stratagene 230192, 230193) for expression. Two-hundred mL of Luria Broth was inoculated with 240 µL of a saturated starter culture and grown to an optical density (A600) of 0.6 in a 37°C incubator shaker. Cultures were then briefly cooled to 12°C prior to induction with 0.1 mM IPTG in a 12°C incubator shaker for 24 hours. Cells from the induced culture were harvested with centrifugation at 4,000x g for 20 minutes at 4°C. Cell pellets were rinsed with 10 mM Tris HCl pH 7.5 and stored at −80°C.
Frozen pellets were resuspended in 2.5 mL of a 50% glycerol, 100 mM Hepes pH 7.5, 5 mM beta-mercaptoethanol solution containing 2 mg/mL of hen egg white lysozyme (Amresco 0663) for 15 min at room temperature. Thirteen mL of lysis solution (100 mM Hepes pH 7.5, 1 M NaCl, 5 mM beta-mercaptoethanol and 0.2% triton X-100) was added to the resuspension and inverted several times to mix. The cell lysate was centrifuged at 33,000 rpm (69,888x g) in a T 865 rotor at 2°C for 20 hours. Supernatant was decanted and allowed to gravity flow through a prewashed (3 mL of 50 mM Hepes pH 7.5, 500 mM NaCl, 0.02% triton X-100, 5 mM imidazole pH 7.5 and 2 mM beta-mercaptoethanol) Talon affinity resin (Clontech 635501) column. Resin bound protein was washed with increasingly stringent solutions of column buffer, wash one (1.2 mL of prewash with 10 mM imidazole), wash two (1.2 mL of prewash with 20 mM imidazole), wash three (one milliliter of prewash with 300 mM NaCL and 30 mM imidazole), wash 4 (600 µL of prewash with 300 mM NaCl and 40 mM imidazole) and eluted (300 µL of prewash with 300 mM NaCl and 60 mM imidazole). Elutant was diluted to 0.5x with 100% glycerol (for final concentrations of 25 mM Hepes pH 7.5, 150 mM NaCl, 0.01% triton X-100, 30 mM imidazole pH 7.5, 1 mM beta-mercaptoethanol and 50% glycerol) and stored at −20°C.
Protein concentration was determined using a BSA standard (Biorad 500-0202) run along with sample on a SDS PAGE gel stained with Sypro Orange (Biorad 170-3120) or Comassie Blue R-250 (Amresco 0472-10G). ImageJ and linear regression analysis was used to determine sample protein concentrations from gel image.Citation48 Apparent purities were greater than 80%.
Electrophoretic mobility shift assays and footprints.
Three target DNAs were used in this study: a 150 bp sequence flanking the insertion site with 80 bp of upstream sequence and 70 bp of downstream sequence, 49 bp of upstream sequence ending at the insertion site, and 49 of downstream sequence starting at the insertion site. The 150 bp target was generated by PCR and used in Electrophoretic Mobility Shift Assays (EMSA) and DNA footprints. The shorter targets were generated by annealing complementary oligonucleotides and were used in EMSA reactions. See for a list of the oligonucleotides and primers used to make target DNA. Radioactive labeling of DNA, EMSA and missing nucleoside footprints were as previously described in references Citation25, Citation49 and Citation50. DNase I footprints were also as previously described except that binding and cleaving reactions were performed in 10 mM Tris HCl pH 7.5, 75 mM NaCl, 3 mM MgCl2, 0.5 mM CaCl2, 0.01% triton X-100, 1 mM DTT and 1.65% glycerol.Citation25,Citation50,Citation51 Post binding and cleavage the glycerol concentration was adjusted to 12% to facilitate loading onto EMSA gels. All EMSA and footprint analyses were repeated at least twice.
Abbreviations
| LTR | = | long terminal repeat |
| TPRT | = | target primed reverse transcription |
| TE | = | transposable element |
| RLE | = | restriction like endonuclease |
| PCR | = | polymerase chain reaction |
| EMSA | = | electrophoretic mobility shift assay |
| ZF | = | zinc finger |
| Bm | = | Bombyx mori |
| Lp | = | Limulus polyphemus |
| bp | = | base pair |
| nt | = | nucleotide |
| APE | = | apurinic-apyrimidinic endonuclease |
Figures and Tables
Table 1 Primers used in cloning and target DNA preparation
Figure 1 R2 clades and ORF structure. (A) The phylogeny represents the relationships between the known R2 elements based on the reverse transcriptase sequence.Citation5 R2 elements have been divided into four clades—R2-A, B, C and D. The amino-terminal domain structure differs between the clades along phylogenic lines. To date, no amino terminal sequence information is available for the R2-B clade. Key: ZF denotes a zinc finger domain, Myb denotes a Myb domain, RT is the reverse transcriptase, cchc denotes the c-terminal cysteine and histidine motif, and RLE denotes the restriction-like endonuclease. The rectangle represents the element's ORF. Lines projecting out from the rectangle represent the 5′ and 3′ untranslated regions. Drawings are not to scale. (B) Alignment of R2 insertion site from representative organisms along with what clades are found in each organism.Citation4–Citation6 The numbering is centered on the insertion site of R2 with upstream flanking DNA given in negative numbers and downstream flanking DNA in positive numbers.
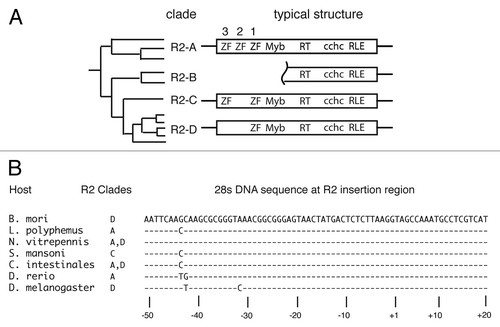
Figure 2 Expression constructs. Thick solid lines denote the segment of the R2Lp and R2Bm ORFs that were expressed in bacteria and purified using engineered 6x His tags. The clone names are to the right of the solid lines. See Materials and Methods for additional information.
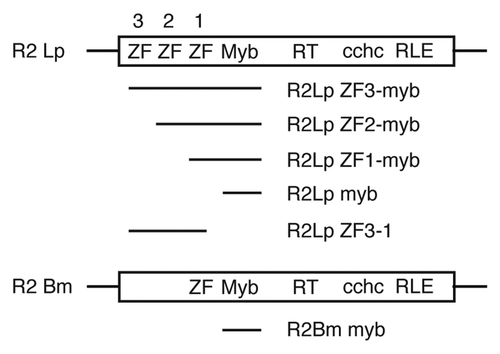
Figure 3 Electrophoretic mobility shift assay. (A) R2Lp ZF3-Myb forms a protein:DNA complex in the presence of 150 bp 28S rDNA target sequence. Lane 1 is the reference lane for DNA migration in the absence of protein (ref.). Lane 2 shows a mobility shift of the DNA in the presence of R2Lp ZF3-Myb protein (bound vs. free). Lane 3 is in the presence of the negative control pGUS protein. Greater that 10X more pGus protein extract was used than R2Lp ZF3-Myb. Lanes 1 and 2 came from the different parts of a gel that smiled during running. Lane 3 came from a separate gel. (B) R2Lp ZF3-Myb bound to the 32P-labeled 49 bp half site target sequences. The efficiency of 32P-labeling was greater for the upstream DNA. Equal amounts of DNA used in lanes 1–4. Lanes 1 and 2 are upstream DNA. Lanes 3 and 4 are downstream DNA. Lanes 1 and 3 are reference lanes for DNA migration in the absence of protein. Lanes 2 and 4 are in the presence of R2Lp ZF3-Myb protein. An equal amount of protein was used in lanes 2 and 4. All lanes came from different parts of the same gel.
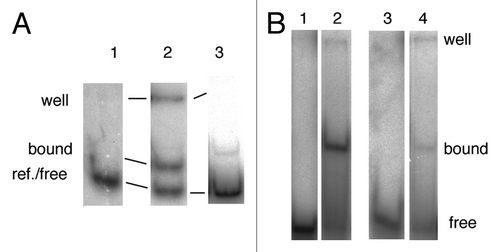
Figure 4 DNase I footprint of the R2Lp ZF3-Myb polypeptide. The denaturing polyacrylamide gel used to assay the DNase I footprint of the R2Lp ZF3-Myb polypeptide bound to top or bottom strand 32P end labeled 150 bp target DNA substrate is presented. Top strand labeled data is on the left and bottom strand labeled data is on the right. The lanes marked A+G are adenosine plus guanosine cleaved target DNAs that are used as linear guides to determine location along the target DNA. The lanes marked “bound” and “ref.” are protein:DNA complexes treated with DNase I and naked-DNA treated with DNase I, respectively, that were fractionated by EMSA and analyzed by denaturing gel electrophoresis. Regions of DNA that are protected from DNase I degradation by the presence of the R2Lp ZF3-Myb polypeptide are marked with thick black lines. Short thick black lines mark polypeptide binding induced DNase I hypersensitive sites.
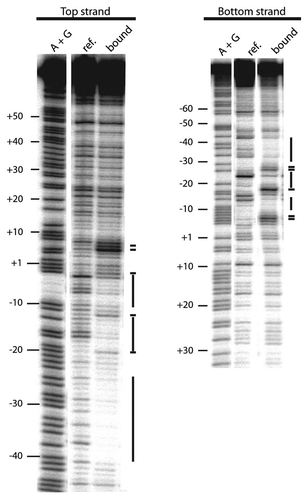
Figure 5 Missing nucleoside footprint of the R2Lp ZF3-Myb polypeptide. The denaturing polyacrylamide gel used to assay the missing nucleoside footprint of the R2Lp ZF3-Myb polypeptide bound to the 150 bp target DNA substrate is presented. Top strand labeled data is on the left and bottom strand labeled data is on the right. The lanes marked A+G are adenosine plus guanosine cleaved target DNAs that are used linear guides to determine location along the target DNA. The lanes marked “ref.” is the hydroxyradical treated DNA (i.e., the “missing nucleoside” DNA). The lanes marked “free” and “bound” are missing nucleoside DNA that was exposed to protein in a binding reaction in which about 50% of the DNA substrate was bound by protein. The bound and free DNA fractions in the binding reaction were fractionated by EMSA prior to denaturing gel electrophoresis. Nucleotides involved in DNA binding are marked with a black dotted line.
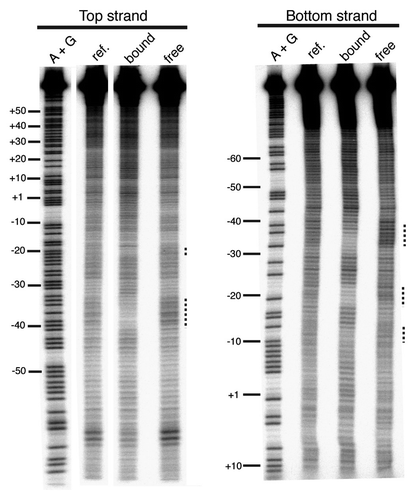
Figure 6 Missing nucleoside footprint of R2-A and R2-D clade Myb polypeptides. Part A is the R2Lp Myb footprint data. Part B is the R2Bm Myb footprint data. Lanes and markings are as in . The R2Bm data is in agreement with reference Citation25.
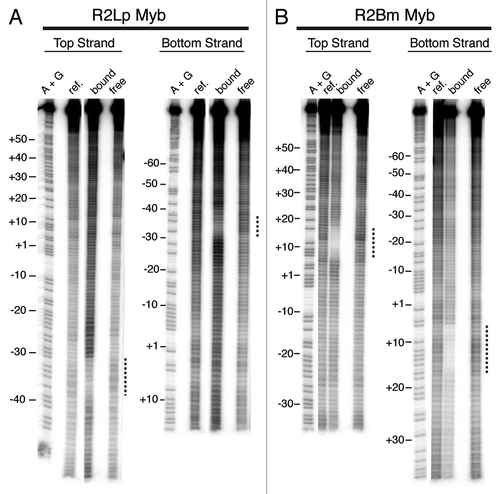
Figure 7 Summary of R2Lp footprint data. The footprint data generated for each polypeptide examined is overlaid on a linear DNA sequence. The R2Lp polypeptide that generated a given footprint is listed to the left. Black dotted lines denote missing nucleoside footprint data from and . Thick black and thick gray lines denote DNase I footprint data from and the Supplemental Figure respectively. For clarity, the DNase I hypersensitive sites have been left out. The R2-A clade Myb binding site (Myb) is marked as are the hypothesized zinc finger (ZF) binding sites (deduced from , and the Sup. Fig.). The binding of the R2-D clade Myb (see ) is indicated by a gray box.
Acknowledgments
The authors would like to thank Dillion Cawley for generating the R2Lp ZF3-Myb clone and for the preliminary footprint trial. The authors would also like to thank Kimberly Bowles, Ph.D., Brad Reveal, Ph.D., and Haridha Shivram for critical reading of the manuscript and Dr. Raymond Jones for helpful advice. Funding generously provided by NSF.
References
- Christensen SM, Eickbush TH. R2 target-primed reverse transcription: ordered cleavage and polymerization steps by protein subunits asymmetrically bound to the target DNA. Mol Cell Biol 2005; 25:6617 - 6628
- Burke WD, Malik HS, Rich SM, Eickbush TH. Ancient lineages of non-LTR retrotransposons in the primitive eukaryote, Giardia lamblia. Mol Biol Evol 2002; 19:619 - 630
- Malik HS, Eickbush TH. NeSL-1, an ancient lineage of site-specific non-LTR retrotransposons from Caenorhabditis elegans. Genetics 2000; 154:193 - 203
- Burke WD, Malik HS, Jones JP, Eickbush TH. The domain structure and retrotransposition mechanism of R2 elements are conserved throughout arthropods. Mol Biol Evol 1999; 16:502 - 511
- Kojima KK, Fujiwara H. Long-term inheritance of the 28S rDNA-specific retrotransposon R2. Mol Biol Evol 2005; 22:2157 - 2165
- Kojima KK, Fujiwara H. Cross-genome screening of novel sequence-specific non-LTR retrotransposons: various multicopy RNA genes and microsatellites are selected as targets. Mol Biol Evol 2004; 21:207 - 217
- Moran JV, Gilbert N. Craig NL, Craigie R, Gellert M, Lambowitz AM. Mammalian LINE-1 Retrotransposons and Related Elements. Mobile DNA II 2002; Washington, DC ASM Press 836 - 869
- Feng Q, Moran JV, Kazazian HHJ, Boeke JD. Human L1 retrotransposon encodes a conserved endonuclease required for retrotransposition. Cell 1996; 87:905 - 916
- Cost GJ, Feng Q, Jacquier A, Boeke JD. Human L1 element target-primed reverse transcription in vitro. EMBO J 2002; 21:5899 - 5910
- Konkel MK, Batzer MA. A mobile threat to genome stability: The impact of non-LTR retrotransposons upon the human genome. Semin Cancer Biol 2010;
- Deininger PL, Batzer MA. Mammalian retroelements. Genome Res 2002; 12:1455 - 1465
- Moran JV, Holmes SE, Naas TP, DeBerardinis RJ, Boeke JD, Kazazian HHJ. High frequency retrotransposition in cultured mammalian cells. Cell 1996; 87:917 - 927
- Takahashi H, Fujiwara H. Transplantation of target site specificity by swapping the endonuclease domains of two LINEs. EMBO J 2002; 21:408 - 417
- Zingler N, Weichenrieder O, Schumann GG. APE-type non-LTR retrotransposons: determinants involved in target site recognition. Cytogenet Genome Res 2005; 110:250 - 268
- Seleme MC, Disson O, Robin S, Brun C, Teninges D, Bucheton A. In vivo RNA localization of I factor, a non-LTR retrotransposon, requires a cis-acting signal in ORF2 and ORF1 protein. Nucleic Acids Res 2005; 33:776 - 785
- Maita N, Aoyagi H, Osanai M, Shirakawa M, Fujiwara H. Characterization of the sequence specificity of the R1Bm endonuclease domain by structural and biochemical studies. Nucleic Acids Res 2007; 35:3918 - 3927
- Gasior SL, Roy-Engel AM, Deininger PL. ERCC1/XPF limits L1 retrotransposition. DNA Repair (Amst) 2008; 7:983 - 989
- Khazina E, Weichenrieder O. Non-LTR retrotransposons encode noncanonical RRM domains in their first open reading frame. Proc Natl Acad Sci USA 2009; 106:731 - 736
- Yoshitake K, Aoyagi H, Fujiwara H. Creation of a novel telomere-cutting endonuclease based on the EN domain of telomere-specific non-long terminal repeat retrotransposon, TRAS1. Mob DNA 2010; 1:13
- Beck CR, Collier P, Macfarlane C, Malig M, Kidd JM, Eichler EE, et al. LINE-1 retrotransposition activity in human genomes. Cell 2010; 141:1159 - 1170
- Martin SL. Nucleic acid chaperone properties of ORF1p from the non-LTR retrotransposon, LINE-1. RNA Biol 2010; 7:67 - 72
- Doucet AJ, Hulme AE, Sahinovic E, Kulpa DA, Moldovan JB, Kopera HC, et al. Characterization of LINE-1 ribonucleoprotein particles. PLoS Genet 2010; 6:e10001150
- Luan DD, Korman MH, Jakubczak JL, Eickbush TH. Reverse transcription of R2Bm RNA is primed by a nick at the chromosomal target site: a mechanism for non-LTR retrotransposition. Cell 1993; 72:595 - 605
- Yang J, Malik HS, Eickbush TH. Identification of the endonuclease domain encoded by R2 and other site-specific, non-long terminal repeat retrotransposable elements. Proc Natl Acad Sci USA 1999; 96:7847 - 7852
- Christensen SM, Bibillo A, Eickbush TH. Role of the Bombyx mori R2 element N-terminal domain in the target-primed reverse transcription (TPRT) reaction. Nucleic Acids Res 2005; 33:6461 - 6468
- Volff JN, Korting C, Froschauer A, Sweeney K, Schartl M. Non-LTR retrotransposons encoding a restriction enzyme-like endonuclease in vertebrates. J Mol Evol 2001; 52:351 - 360
- Mandal PK, Bagchi A, Bhattacharya A, Bhattacharya S. An Entamoeba histolytica LINE/SINE pair inserts at common target sites cleaved by the restriction enzyme-like LINE-encoded endonuclease. Eukaryot Cell 2004; 3:170 - 179
- Eickbush TH, Malik HS. Craig NL, Craigie R, Gellert M, Lambowitz AM. Origins and Evolution of Retrotransposons. Mobile DNA II 2002; Washington, DC ASM Press 1111 - 1146
- Malik HS, Burke WD, Eickbush TH. The age and evolution of non-LTR retrotransposable elements. Mol Biol Evol 1999; 16:793 - 805
- Ban N, Nissen P, Hansen J, Moore PB, Steitz TA. The complete atomic structure of the large ribosomal subunit at 2.4 A resolution. Science 2000; 289:905 - 920
- Stage DE, Eickbush TH. Origin of nascent lineages and the mechanisms used to prime second-strand DNA synthesis in the R1 and R2 retrotransposons of Drosophila. Genome Biol 2009; 10:49
- Eickbush TH. Craig NL, Craigie R, Gellert M, Lambowitz AM. R2 and Related Site-Specific Non-Long Terminal Repeat Retrotransposons. Mobile DNA II 2002; Washington, DC ASM Press 813 - 835
- Burke WD, Malik HS, Lathe WCr, Eickbush TH. Are retrotransposons long-term hitchhikers?. Nature 1998; 392:141 - 142
- Zhang X, Zhou J, Eickbush TH. Rapid R2 retrotransposition leads to the loss of previously inserted copies via large deletions of the rDNA locus. Mol Biol Evol 2008; 25:229 - 237
- Averbeck KT, Eickbush TH. Monitoring the mode and tempo of concerted evolution in the Drosophila melanogaster rDNA locus. Genetics 2005; 171:1837 - 1846
- Eickbush TH, Eickbush DG. Finely orchestrated movements: evolution of the ribosomal RNA genes. Genetics 2007; 175:477 - 485
- Perez-Gonzalez CE, Eickbush TH. Rates of R1 and R2 retrotransposition and elimination from the rDNA locus of Drosophila melanogaster. Genetics 2002; 162:799 - 811
- Zhang X, Eickbush TH. Characterization of active R2 retrotransposition in the rDNA locus of Drosophila simulans. Genetics 2005; 170:195 - 205
- Jakubczak JL, Burke WD, Eickbush TH. Retrotransposable elements R1 and R2 interrupt the rRNA genes of most insects. Proc Natl Acad Sci USA 1991; 88:3295 - 3299
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, et al. Initial sequencing and analysis of the human genome. Nature 2001; 409:860 - 921
- Aparicio S, Chapman J, Stupka E, Putnam N, Chia JM, Dehal P, et al. Whole-genome shotgun assembly and analysis of the genome of Fugu rubripes. Science 2002; 297:1301 - 1310
- Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, Agarwal P, et al. Initial sequencing and comparative analysis of the mouse genome. Nature 2002; 420:520 - 562
- Burke WD, Calalang CC, Eickbush TH. The site-specific ribosomal insertion element type II of Bombyx mori (R2Bm) contains the coding sequence for a reverse transcriptase-like enzyme. Mol Cell Biol 1987; 7:2221 - 2230
- Stage DE, Eickbush TH. Maintenance of multiple lineages of R1 and R2 retrotransposable elements in the ribosomal RNA gene loci of Nasonia. Insect Mol Biol 2010; 19:37 - 48
- Kurzynska-Kokorniak A, Jamburuthugoda VK, Bibillo A, Eickbush TH. DNA-directed DNA polymerase and strand displacement activity of the reverse transcriptase encoded by the R2 retrotransposon. J Mol Biol 2007; 374:322 - 333
- Christensen SM, Ye J, Eickbush TH. RNA from the 5′ end of the R2 retrotransposon controls R2 protein binding to and cleavage of its DNA target site. Proc Natl Acad Sci USA 2006; 103:17602 - 17607
- Bibillo A, Eickbush TH. End-to-end template jumping by the reverse transcriptase encoded by the R2 retrotransposon. J Biol Chem 2004; 279:14945 - 14953
- MD A, PJ M, SJ R. Image Processing with ImageJ. Biophotonics International 2004; 11:36 - 42
- Hayes JJ, Tullius TD. The missing nucleoside experiment: a new technique to study recognition of DNA by protein. Biochemistry 1989; 28:9521 - 9527
- Christensen S, Eickbush TH. Footprint of the retrotransposon R2Bm protein on its target site before and after cleavage. J Mol Biol 2004; 336:1035 - 1045
- Brenowitz B, Senear DF, Kingston RE. Ausubel FM. DNase I Footprint Analysis of Protein-DNA Binding. Current Protocols in Molecular Biology 2003; 12:Hoboken, NJ John Wiley and Sons, Inc. 4 - 5
- Kojima KK, Kuma K, Toh H, Fujiwara H. Identification of rDNA-specific non-LTR retrotransposons in Cnidaria. Mol Biol Evol 2006; 23:1984 - 1993
- Gladyshev EA, Arkhipova IR. Rotifer rDNA-specific R9 retrotransposable elements generate an exceptionally long target site duplication upon insertion. Gene 2009; 448:145 - 150