Abstract
Restriction-like endonuclease (RLE) bearing non-LTR retrotransposons are site-specific elements that integrate into the genome through target primed reverse transcription (TPRT). RLE-bearing elements have been used as a model system for investigating non-LTR retrotransposon integration. R2 elements target a specific site in the 28S rDNA gene. We previously demonstrated that the two major sub-classes of R2 (R2-A and R2-D) target the R2 insertion site in an opposing manner with regard to the pairing of known DNA binding domains and bound sequences-indicating that the A- and D-clades represent independently derived modes of targeting that site. Elements have been discovered that group phylogenetically with R2 but do not target the canonical R2 site. Here we extend our earlier studies to show that a separate R2-A clade element, which targets a site other than the canonical R2 site, does so by using the amino-terminal zinc fingers and Myb motifs. We further extend our targeting studies beyond R2 clade elements by investigating the ability of the amino-terminal zinc fingers from the nematode NeSL-1 element to target its integration site. Our data are consistent with the use of an amino-terminal DNA binding domain as one of the major targeting determinants used by RLE-bearing non-LTR retrotransposons to secure a protein subunit near the insertion site. This amino-terminal DNA binding domain can undergo modifications, allowing the element to target novel sites. The binding orientation of the amino-terminal domain relative to the insertion site is quite variable.
Introduction
Non-long-terminal-repeat (non-LTR) retrotransposons are a major class of eukaryotic transposable elements. These elements are vertically inherited and can impact the evolution of their host's genome in many ways.Citation1–Citation5 Non-LTR retrotransposons replicate through an ordered series of DNA cleavage and polymerization events using encoded nucleic acid binding, endonuclease, and polymerase functions.Citation6 The element encoded protein(s), once translated, form a ribonucleoprotein (RNP) particle with the transcript from which they were translated—a process called cis-preference. The RNP binds to the target DNA, cuts one of the DNA strands, and uses the target site's exposed 3′-OH to prime reverse transcription of the element RNA into cDNA (cDNA)—a process called target primed reverse transcription (TPRT).Citation7 The opposing target DNA strand is then cleaved.Citation6 The cDNA is turned into double stranded DNA, completing the integration event in a process that is not yet well understood.Citation6
Non-LTR retrotransposons can be grouped into those elements that harbor a restriction-like endonuclease (RLE) to initiate TPRT and those that harbor an apurinic-apyrimidinic endonuclease (APE) to initiate TPRT (reviewed in refs. Citation8–Citation10).Citation7,Citation11–Citation14 The RLE-bearing elements tend to be site specific (i.e., inserting into a given sequence within a genome) while the APE-bearing elements tend to be nonspecific—although examples of both site-specific and nonspecific targeting can be found within each group.Citation1,Citation11,Citation15,Citation16 Phylogenetic analysis of the reverse transcriptase domain from RLE and APE bearing elements indicates that the RLE-bearing elements are likely the earlier branching group (reviewed in ref. Citation10).
The site specificity of RLE-bearing non-LTR retrotransposons makes them attractive systems with which to study the TPRT integration reaction. In addition, once it is understood how RLE-bearing elements target DNA, it may be possible to engineer these elements for use as site specific gene targeting vehicles. We are conducting a systematic study of the DNA targeting functions of RLE-bearing non-LTR retrotransposons.Citation6,Citation17–Citation19 DNA recognition is the first step in the integration reaction and is therefore the step that must undergo modification when an element targets a novel site. Target site recognition is believed to be achieved primarily through distinct DNA binding motifs located within the coding region of the element, as opposed to any inherent specificity of the RLE. The endonuclease is believed to be largely nonspecific while the DNA binding domains target insertion events to specific sites in the genome.Citation6,Citation12,Citation16,Citation18,Citation20
RLE-bearing non-LTR retrotransposons encode a single multifunctional protein with RNA binding, DNA binding, DNA endonuclease, and reverse transcriptase activities. These retrotransposons have been phylogenetically classified into at least five clades based upon sequence comparisons of the reverse transcriptase domain: R2, R4, Genie, CRE, and NeSL ().Citation12,Citation21–Citation24 All of these clades share a similar basic ORF structure with a central reverse transcriptase domain (RT), a carboxyl-terminal cysteine-histidine motif (cchc), and a carboxyl-terminal restriction-like endonuclease. The major differences between the clades resides within the N-terminal domain. Elements belonging to the Genie, CRE, and NeSL clades typically contain two N-terminal zinc fingers (ZFs), while R2 clade elements contain a Myb motif and a variable number of ZFs. The R4 clade appears to lack both ZFs and Myb motifs. Within each clade N-terminal structural variants exist (e.g., a variable number of ZFs). In addition, the name-sake of the NeSL clade, NeSL-1, contains a cysteine protease domain (PRO) of unknown function.Citation21
R2 is the most well studied of the five clades. The R2 designation is actually a double criteria designation—a phylogenetic grouping in conjunction with an insertion site designation. R2 elements insert into a particular sequence within the 28S rDNA gene ( and ).Citation1,Citation12,Citation25 R2 elements are further subdivided into the R2-A, R2-B, R2-C, and R2-D groups based on reverse transcriptase phylogeny.Citation25 Each R2 group has a different configuration of ZFs and Myb motifs in the N-terminal domain of the encoded R2 protein. The two major subdivisions are the R2-A and the R2-D groups (). The R2-D group, exemplified by the well studied Bombyx mori R2 element (R2Bm), has a single N-terminal ZF and a Myb motif. R2Bm uses two protein subunits to integrate.Citation6 These two subunits take on different conformations and roles in the integration reaction ().Citation6,Citation26 One protein subunit is bound upstream of the insertion site, and the other one is bound downstream of the insertion site. The upstream subunit binds to the DNA using an undetermined DNA binding motif, cleaves the target DNA, and performs TPRT.Citation6 The downstream subunit uses the N-terminal ZF and Myb motifs to bind to the DNA.Citation18 The downstream subunit cleaves the second DNA strand and is hypothesized to perform second strand synthesis. The 3′ untranslated region (UTR) of the R2 RNA is bound to the upstream subunit, and the 5′ UTR is bound to the downstream subunit.Citation26 The R2-A group binds the target site differently than the R2-D group. The R2-A group elements have three N-terminal zinc fingers as well as a Myb motif and are thought to be the more ancestral R2 group. The R2-A group element from Limulus polyphemus (Lp), R2Lp, uses the N-terminal ZFs and a Myb to bind to the upstream DNA sequences as opposed to downstream sequences.Citation17 The different DNA binding modes indicate R2-A and R2-D are independent targeting (or retargeting) events to the canonical R2 site. The distinct DNA binding modes also suggest that the R2-A and R2-D elements use a different linkage configuration between the various nucleic acid binding domains and catalytic activities involved in the integration reaction.Citation17
Recently, elements that phylogenetically group with the R2-A group have been discovered that do not target the canonical R2 site. R9 from Adineta vaga (R9Av) targets a site within the 28S about 1436 bp upstream of the R2 site, and R8 from Hydra magnipapillata (R8Hm) targets a sequence in the 18S rDNA ().Citation27,Citation28 The R2 site is thought to be the more ancestral site as most species that have retained R2 clade elements have done so at the R2 site. Given this interpretation, the R8 and R9 sites are instances of R2 clade elements acquiring novel site specificity.Citation27 In this paper we investigate the role that the N-terminal ZF and Myb motifs have in targeting an R2 clade element to a novel genomic site. We show that the N-terminal domain of R9Av has been modified so as to target the R9 site and not the R2 site. Interestingly, the orientation of the binding of the ZFs and Myb motif differs from the R2-A and the R2-D elements that target the R2 site. We also extended our DNA targeting studies to elements beyond the R2 clade. Of the five clades—R2, R4, Genie, CRE, and NeSL—only R2 has a Myb motif in the N-terminal region (). That Myb motif is a major contributor to the specificity observed in the R2 clade elements, with the ZFs providing fewer DNA contacts as a whole than the Myb motif.Citation17,Citation18 Genie, CRE, and NeSL clades only have ZFs—typically two ZFs—and no Myb motifs. In order to ascertain how Genie, CRE, and NeSL clade elements target DNA, we examined the DNA binding potential of a NeSL clade element. We show that the NeSL-1 element uses its two N-terminal ZFs to target DNA. Along with our previously reported study, our results indicated that the N-terminal ZFs (and Myb motif if present) may represent a universal targeting module for all site specific RLE-bearing non-LTR retrotransposons that contain these motifs. The Myb and ZFs can undergo modification, allowing novel sites to be targeted. During modification, individual ZF and Myb motifs can be acquired or lost. In addition, the physical/temporal linkage configurations between the various nucleic acid binding activities (5′ UTR RNA binding, 3′ UTR RNA binding, upstream DNA binding, and downstream DNA binding) and catalytic activities (first strand cleavage, TPRT, second strand cleavage, and second strand synthesis) may get reconfigured as elements transition to target new sites in the genome.
Results and Discussion
DNA binding activity of an R2 element directed to a noncanonical target site: Mapping the DNA binding activity of R9Av.
R2-D group elements that target the R2 site (e.g., R2Bm) use the N-terminal ZF and Myb motif to secure an R2 protein subunit downstream of the insertion site.Citation6,Citation18 R2-A group elements that target the R2 site (e.g., R2Lp) use their N-terminal ZFs and Myb motif to secure an R2 protein subunit upstream of the insertion site.Citation17 Although both R2-D and R2-A group elements target the same site, they do so differently. To better understand R2-clade mechanistic plasticity and site specificity, R9Av was examined. R9Av is an R2-A group element that does not target the canonical insertion site (). R9Av is also interesting as it generates a 126 bp target site duplication (TSD) upon insertion as opposed to the blunt or small 1–10 bp deletion observed for most R2 insertions.Citation29
Full-length non-LTR retrotransposon proteins are very difficult to express and purify in a soluble and active form. For this reason, N-terminal derived polypeptides containing the ZFs and Myb motifs expected to be involved in targeting were expressed and purified in order to ascertain if the R9Av ZF and Myb motifs have modified so as to direct the R2 clade element to the R9 site instead of the R2. Initially, four target DNAs were used in electrophoretic mobility shift assays (EMSA) to identify if and where the R9Av N-terminal ZF and Myb bind (). The R9Av polypeptide spanned from ZF3 through the Myb motif and was given the name R9Av ZF3-Myb (). A diagram of the four target site DNAs used in the EMSA reactions are shown in . Target 1 consisted of the segment of the 28S that becomes duplicated upon R9 insertion (126 bp TSD region) along with 112 bp of upstream flanking sequence (net 238 bp). Target 2 was the 126 bp TSD region along with 101 bp of downstream flanking sequence (net 227bp). Target 3 was the 112 bp of upstream flanking sequence. Target 4 was the 101 bp of downstream flanking sequence. Each target DNA was end-labeled with 32P and put into binding reactions with varying amounts of R9Av ZF3-Myb polypeptide. The polypeptide concentrations ranged from 240 nM to 9 nM in 3-fold increments. The binding reactions contained 9 ng of DNA, which translates to around 9.6 nM for Targets 1 and 2, and around 21 nM for Targets 3 and 4.
The R9Av ZF3-Myb polypeptide bound best to Target 1, with observable binding occurring in the presence of 9 nM of polypeptide—roughly a 1:1 molar ratio of protein to DNA (, Lane 5). Greater than 50% of Target 1 was bound in the presence of 27 nM polypeptide (, Lane 4), and 100% of target bound at 80 nM—a 8.3:1 protein to DNA molar ratio (, Lane 3). At the higher protein to DNA ratios, additional protein- DNA complexes occur (, Lanes 2–4). Presumably the fastest migrating protein-DNA complex represents a single polypeptide bound to DNA (hereafter called a monomer) as it appears in the lower protein concentration samples. The slower migrating protein-DNA complexes, then, may represent higher order protein-DNA complexes (e.g., dimer, trimer, etc.). Target 3 was also bound efficiently by the R9Av ZF3-Myb polypeptide in that protein-DNA complexes were observed at low protein concentrations (e.g., , Lane 5, a 1:2.3 molar ratio of protein to DNA). Target 2 was bound much less efficiently (at least 9× less efficient) than Target 1. Observable R9Av ZF3-Myb polypeptide binding to Target 2 did not occur until the 80 nM of polypeptide level—a 8.3:1 protein to DNA molar ratio (, Lane 3). The R9Av ZF3-Myb polypeptide did not bind appreciably to Target 4 ().
The EMSA reactions involving the R9Av ZF3-Myb polypeptide indicate that this region of the R9 protein is involved in DNA targeting. Because Target 1 and Target 3, which have the upstream DNA in common, were bound most efficiently by the R9Av ZF3-Myb polypeptide (respectively), it appears that the protein motifs contained in the polypeptide are responsible for securing an R9 protein subunit to DNA sequences upstream of the TSD region. However, as Target 2, which contains the TSD region, bound the R9Av ZF3-Myb polypeptide better than Target 4, it is possible that some base-specific interactions may occur within the TSD region in addition to the upstream sequence. Any presumed association of the polypeptide with the TSD region could not be due to the action of either ZF3 or ZF2 as the binding profile of the R9Av ZF1-Myb polypeptide mirrored the binding of the R9Av ZF3-Myb polypeptide on the four targets used in (data not shown). Binding to Target 1 was the most robust, followed by Target 3, then Target 2, and lastly Target 4. It is possible that the greater association of protein with Target 2 relative to Target 4 is related to local DNA structure (e.g., bent DNA for positioning a nucleosome) causing increased association of the polypeptide.
To better map the site of interaction between the R9Av ZF3-Myb polypeptide and the target DNA, a DNase I protection based DNA footprint analysis was performed. Assuming the binding of the R9Av ZF3-Myb polypeptide to target DNA is specific to the upstream sequence and perhaps the TSD region, as indicated by the EMSA results, it should be possible to precisely map the site (or sites) of interaction by DNase footprinting. A footprint signal is indicative of specific binding. So as not to unduly skew the mapping results, the footprint analysis was done using Target 1.
The R9Av ZF3-Myb polypeptide was bound to target DNA that had been end-labeled on either the top strand or the bottom strand, respectively. The R9Av ZF3-Myb polypeptide:DNA ratios were adjusted so as to form primarily monomers or monomers and dimers similar to that seen in , Lanes 4 and 5. The binding reactions were subsequently subjected to DNase I treatment under conditions that yield one cleavage event per DNA target. The DNase I treated protein-DNA complexes were fractionated into bound and free/reference fractions by EMSA (data not shown) prior to analysis on a denaturing polyacrylamide gel (). Segments of DNA that show protection from DNase I cleavage when compared with reference DNA indicate areas of DNA bound by the R9Av ZF3-Myb polypeptide. Of the bound fractions, , Lane 3 are from the dimer and , Lane 4 are from the monomer protein complex. The areas of DNase I protection denoting R9Av ZF3-Myb binding have been marked with long thick black lines next to the denaturing gel. Short thin black lines denote DNase I hypersensitive sites that were induced by the binding of the R9Av ZF3-Myb polypeptide.
The footprinted region for a single polypeptide monomer is restricted to sequences upstream of the TSD region, specifically from base pair position −47 to −27 on the top strand and from base pair position −46 to −27 on the bottom strand (, Lane 4). The base pair numbering is relative to the presumptive site of bottom strand cleavage and TPRT. The TPRT site is inferred from the orientation of inserted R9Av elements as well as the DNA cleavage sites required to generate the observed TSD. Negative numbers represent base pair positions upstream of the site of TPRT, and positive numbers represent base pairs positions downstream of the TPRT site. No obvious additional footprint signal was observed in any higher order complexes (e.g., the dimer complex, , Lane 3) beyond that defined for the monomer (, Lane 4). There is a DNase I hypersensitive site on the bottom strand at base pair position −46 induced by binding of a single polypeptide unit. Additional DNase I hypersensitive sites are observed in the presence of additional polypeptide units being bound (see top strand , Lane 3, positions +16 and +74).
In order to ascertain the binding orientation of the R9 Myb relative to the ZFs and to gain a higher resolution footprint, a missing nucleoside footprint analysis was performed on the R9Av ZF3-Myb polypeptide.Citation30 A second missing nucleoside footprint was performed on the truncated polypeptide, R9Av ZF1-Myb, which is missing ZF3 and ZF2. DNA with random abasic sites (i.e., missing nucleoside DNA) was exposed to protein in a binding reaction followed by EMSA fractionation. The resulting DNA fractions were analyzed on a denaturing polyacrylamide gel. Bands corresponding to a DNA base that, when missing, interfered with protein binding yielded a footprint signal. The missing nucleoside data for the R9Av ZF3-Myb polypeptide bound to DNA are presented in . Only the fastest migrating protein-DNA complex (i.e., the monomer) was analyzed. The DNA bases that were found to interact with the R9Av ZF3-Myb polypeptide are located from −43 to −31 on the top strand and from −46 to −31 on the bottom strand, in good agreement with DNase I footprint data. The missing nucleoside data for the R9Av ZF1-Myb are presented in .
The missing nucleoside data have been summarized for both polypeptides, along with the DNase I data, in . Both DNase I and missing nucleoside footprint data confirm that the R9Av N-terminal DNA binding region, ZF3-Myb, binds to DNA sequences upstream of the 126 bp TSD region. If a specific interaction exists between the ZF3-Myb polypeptide and the TSD region, the interaction is not stable enough to footprint in our reactions. In addition, no additional footprint signal was detected in any of the higher order complexes, indicating a single specific binding site for the polypeptide on the target DNA is likely. The shorter R9 polypeptide, R9 ZF1-Myb, made contacts on both strands in the region of −40 to −31, but just on the top strand in the region spanning −43 to −41. The lack of footprinting on the bottom strand in the region of −46 to −40 with the shorter polypeptide indicated that either, or both, ZF3 and ZF2 associate with the bottom strand in this region. The region from −40 to −31, which footprinted on both strands with both polypeptides, is likely where the Myb motif binds. ZF1 binding may then account for the top strand signal from −43 to −41. This interpretation is consistent with other R2 elements where the Myb motif contacted both DNA strands over roughly a 10 bp region and ZF1 contacted at least one strand over a 3–5 bp region.Citation17,Citation18 Additional studies would have to be done to confirm the ZF assignments in R9Av; however, it is clear that the Myb motif binds closest to the insertion site and the ZFs farther away.
R9Av and R2Lp are both R2-A group elements that use their N-terminal ZFs and Myb to target different sites in the genome. R9Av targets the R9 site, and R2Lp targets the R2 site. Both elements use their respective N-terminal DNA binding module to bind a protein subunit upstream of the insertion site.Citation17 Interestingly, R9Av and R2Lp bind in opposite orientation to each other with respect to the binding order of the Myb motif and ZFs relative to the insertion site. In R2Lp, the ZFs are closest to the insertion site, while in R9Av the Myb motif binds closest to the insertion site. In the R2-D clade element R2Bm, the Myb motif and ZF bind downstream of the insertion site, with the ZF being closest to the insertion site.Citation18
DNA targeting by a non-R2 RLE-bearing non-LTR retrotransposon.
For both R2-A and R2-D group elements, the Myb domain appears to account for the largest continuous swath of base specific contacts for the subunit using the N-terminal motifs to bind to DNA.Citation17,Citation18 Indeed, we have been unsuccessful in getting the ZFs of R2-A and R2-D group transposons to bind tightly enough to be footprinted in the absence of the Myb motif (R9Av data not shown and refs. Citation17 and Citation18). Of the known RLE-bearing non-LTR retrotransposons, only R2 clade elements contain a Myb motif. In this aspect at least, R2 is not representative of other RLE-bearing non-LTR retrotransposons. N-terminal ZFs (typically two ZFs) have been identified in elements belonging to the NeSL, CRE, and Genie clades (). In order to extend our studies of target site recognition beyond R2, the NeSL clade element NeSL-1Ce was examined. NeSL-1Ce contains two N-terminal ZFs and targets the spliced leader-1 gene of Caenorhabditis elegans (). In order to test if the NeSL-1Ce ZFs function in target recognition similar to R2's Myb plus ZF(s) pairing, a polypeptide containing the NeSL-1Ce N-terminal ZFs (NeSL-1 ZFs) was cloned, expressed, and purified. The purified polypeptide was assayed for DNA binding activity against a 125 bp target DNA containing the NeSL-1 insertion site. EMSA analysis showed a slower migrating complex consistent with the NeSL-1 ZFs polypeptide binding to target DNA (). DNase I footprint analysis () was used to determine if the binding was specific. Regions of DNA protected from DNase I degradation in the presence of bound polypeptide were localized to two closely spaced regions: (1) top strand base pair positions −21 to −19, bottom strand −20 to −17; (2) top strand −9 to −7, bottom strand −7 to −5. Base pair positions are relative to the NeSL insertion site (i.e., TPRT site), with negative integers representing base pair positions upstream of the TPRT site and positive integers representing base pair positions downstream of the TPRT site. The DNase I footprint has been overlaid on the insertion site sequence in . The NeSL-1 ZF polypeptide was found to bind to DNA sequences upstream of the insertion site (i.e., within the spliced leader exon). The two zones of DNase I protection observed likely correspond to the binding of the two ZFs, respectively. The binding orientation of the two ZFs is unknown.
Summary of RLE-bearing non-LTR retrotransposon DNA binding modes and implications for the integration model.
The insertion model posited for R2 elements, and by analogy all RLE-bearing non-LTR retrotransposons, requires two subunits of protein to affect element insertion, one subunit bound to each side of the insertion site. The catalytic domains of the two subunits must be in opposite orientation to each other in order to carry out the two half reactions required for insertion (see for more information derived from R2Bm). Amino acid alignments of R2 elements would appear to argue that there is tight integration between the RT and the carboxyl-terminal domain. There is not much room for flexible linkers between the highly conserved domains in the carboxyl terminal domain, unlike the N-terminal domain where there is variability in the number and makeup of the conserved motifs as well as variable spacing between some of the conserved regions.Citation12 Examinations of 5′ junctions of native R2 insertions in various Drosophila species indicate that the processes of second strand cleavage and second strand synthesis are rapidly evolving.Citation29 In R2Bm, where most of the biochemistry has been done, the subunit that performs second strand cleavage (and presumably second strand synthesis) interacts with the 5′ RNA and binds to the target DNA downstream of the insertion site using the N-terminal ZF and Myb ( and ). The upstream subunit binds through an (as yet) unidentified protein domain (indicated by the single question mark in ). In R2Bm, the subunit that uses the unidentified DNA binding domain binds the 3′ RNA and performs first strand cleavage and TPRT. The unidentified DNA binding domain has been hypothesized to be located in the carboxyl-terminal domain.Citation6,Citation12
Collectively, our N-terminal DNA binding data on R2 and NeSL elements indicate that most site-specific RLE-bearing non-LTR retrotransposons likely use their N-terminally located DNA binding motifs to load a transposon subunit onto the target site. There appears to be variability in how the ZFs and Myb motifs bind target DNA (). In some cases the N-terminal motifs are used to secure the upstream subunit and in other cases the downstream subunit. In some cases the ZFs are closest to the insertion site, and in other cases the Myb is closest to the insertion site. This variability may indicate plasticity in how the binding and catalytic domain functions are wired into the overall insertion mechanism. The orientation changes may relate to the absolute orientation of the RLE and RT catalytic domains relative to the insertion site, although flexible linkers could either decouple binding orientation from catalytic orientation or even re-wire the linkage to be opposite of what is known for R2Bm.
In R2Lp the upstream and downstream subunits appear to be swapped relative to R2Bm.Citation17 However, orientation of the ZFs and Myb motif relative to the insertion site are identical to R2Bm (i.e., the ZFs are nearest the insertion site).Citation17 The R2Lp downstream subunit is hypothetical and is based upon the R2Bm model of insertion. The downstream R2Lp subunit is marked with two question marks in , one question mark to signify the presence of the subunit being hypothetical and one question mark to signify that, if present, the subunit would be expected to bind to DNA using the same unidentified (carboxyl-terminal?) protein domain that secures the R2Bm upstream subunit to target DNA. In the case of R9Av, the N-terminal ZFs and Myb bind upstream of the insertion site as in R2Lp, but the binding orientation of the ZFs and Myb appear flipped compared with R2Lp (). The two R9Av subunits would be expected to be near each other in space, assuming the 146 bp TSD region was wrapped around a nucleosome and the downstream subunit bound just downstream of the TSD.Citation28 As in R2Lp, the R9Av downstream subunit would be targeted via the unidentified DNA binding domain. In the case of NeSL-1, the upstream subunit is again bound using the N-terminal ZFs (orientation unknown), and the hypothetical downstream subunit would be bound to target DNA using the unidentified DNA binding domain. In each case, it is tempting to speculate that the subunit that binds the 3′ UTR RNA binds to DNA using the hypothetical carboxyl-terminal DNA binding domain (or the RLE) and performs TPRT as the RT and conserved carboxyl-terminal motifs appear to be more tightly linked. The variable N-terminal domain is attached to the RT through a variable length spacer.Citation12 The subunit that performs the rapidly evolving second strand cleavage and second strand synthesis reactions would be bound to target DNA using the N-terminal ZFs (and Myb). If this model continues to hold, it may be possible to engineer an RLE-bearing element (e.g., R2Bm) to target elsewhere in the genome by swapping out the DNA binding motifs. For RLE-bearing retrotransposons to be used as site-specific gene targeting vehicles, however, the unidentified DNA binding domain will need to be identified. In addition, a greater knowledge of the 3D and globular domain structure is of great interest.
Materials and Methods
Generating expression constructs.
Constructs containing R9 and NeSL-1 derived N-terminal putative DNA binding motifs were generated and named as follows: R9Av ZF3-Myb corresponds to codons 54–295 of the extended ORF (stop codon to stop codon) from R9 Adineta vaga transposon (R9Av) GenBank GQ398057.1, R9Av ZF1-Myb corresponds to codons 154–295 of R9Av, and NeSL-1 ZFs corresponds to codons 110–261 of the Caenorhabditis elegans NeSL-1transposon extended ORF (stop codon to stop codon). The polymerase chain reaction (PCR) primers used to amplify the above regions of interest from A. vaga and C. elegans genomic DNA are listed in . A. vaga genomic DNA was a gift from Irina Arkhipova (Josephine Bay Paul Center for Comparative Molecular Biology and Evolution). C. elegans genomic DNA was a gift from Andre Pires d Silva (University of Texas Arlington). The R9 fragments were cloned into the Gateway® donor vector pENTR/TEV/D-TOPO (Invitrogen, K2535,20) using the manufacturer's protocol and then recombined into the Gateway®-compatible bacterial-expression destination vector pDESTTAP.Citation17 The NeSL-1 ZFs were cloned into the NdeI and BamHI sites of the bacterial expression vector pET28a (Novagen, 69864-3). Initial ligation and recombination reactions were transformed into electroporation competent XL-1 Blue Escherichia coli (Agilent, 200259) or chemically competent Oneshot Top10 E. coli (Invitrogen, C4040-10) for screening purposes. Resulting colonies were screened by PCR and sequenced (Big Dye, Applied Biosystems, 4337455). The expression constructs were maintained in Arctic Express RIL DE3 E. coli cells (Stratagene, 230193) for expression.
Protein expression and purification.
Cells were grown in 200 mL of Luria Bertani medium to an A600 of 0.6–0.7 at 37°C in an incubator shaker. The culture was then cooled to 12°C. Isopropyl-β-D-Galactoside (IPTG) was added to the cooled culture at a final concentration of 1 mM for the R9Av clones and 0.1 mM for the NeSL-1 clone. After the addition of IPTG, the cultures were further incubated for 24 h at 12°C in an incubator shaker. The cultures were centrifuged at 4,000× g for 20 min at 4°C. The pellets were washed with cold 10 mM TRIS-HCl pH 7.5 and were either used directly or stored at −80°C.
The R9Av pellets were resuspended in 2.5 mL of Solution A [50% glycerol, 100 mM HEPES, 5 mM β-mercaptoethanol, and 2 mg/mL of lysozyme (Amresco, 0663)] and incubated on ice for 15 min and at room temperature for 15 min. The resuspended cells were lysed by adding 13.2 ml of solution B (100 mM HEPES, 1 M NaCl, 5 mM β-mercaptoethanol, and 0.1% Triton X-100) and incubating on ice for 30 min. The resuspended pellet was then centrifuged for 20 h at 69,888× g at 2°C. The supernatant was mixed with the Talon resin (Clontech, 635501) that had been prewashed with 10 ml of Talon column buffer (50 mM HEPES pH 7.5, 500 mM NaCl, 0.2% triton X-100, 5 mM imidazole pH 7.5). The resin bound protein was washed with 20 mL of column buffer containing 10 mM imidazole and eluted with 300 ul of column buffer containing 300 mM NaCl and 150 mM imidazole. An equal volume 100% glycerol was added to the eluate for storage at −20°C.
The NeSL-1 pellets were resuspended in 8 mL of lysis buffer (100 mM Hepes pH 7.5, 1 M NaCl, 1 mM β-mercaptoethanol and 0.2% triton X-100, 10 units of DNase I) and passed through a French press two times. The cell lysates were centrifuged in an Eppendorf centrifuge at 12,000 RPM for 10 min at 4°C. The supernatant was adjusted to 10% glycerol and passed over the Talon resin columns by gravity flow, as previously described.Citation17
Protein concentrations were determined by samples run on a SDS 6% PAGE along with a bovine serum albumin standard. SDS PAGE were stained with Sypro Orange (Biorad, 170-3120) or Comassie Blue R-250 (Amresco, 0472-10G) and the band intensities were measured using imageJ 1.38X software.Citation31 The apparent purities were 80% or greater.
Electrophoretic mobility shift assays and DNA footprints.
The 5′ 32P end labeled DNA substrates were generated () and purified as previously described.Citation6 The binding reactions in were performed in 13 uL reactions: 10 uL of a solution containing 7.5 mM TRIS-HCl (pH 7.5), 50 mM NaCl, 2.75 mM MgCl2, 0.5 mM CaCl2, 0.8 mM ditriothritol, 9 ng of target DNA, 50 ng of poly dI-dC (Sigma Aldrich, P4929-5UN); and 3 uL of protein diluted to an appropriate concentration in protein storage buffer (see above). The binding reactions were incubated at 25°C for 20 min and then loaded on a 1X TBE (89 mM Tris base, 89 mM boric acid, 2 mM EDTA) native 5% polyacrylamide gel. The gels were run at 230 V for 30 min. Gels were dried and visualized on a phosphorimager screen.
The binding reactions for DNase I footprints were similar but lacked poly dI-dC and were performed under conditions that gave approximately 40–60% bound species (see also refs. Citation6 and Citation32). 0.012 units of DNase I were used. Binding reactions treated with DNase were fractionated on native polyacrylamide to isolate the bound, free, and reference fractions. The bound and reference fractions were analyzed on a denaturing 6% polyacrylamide gel. The reference DNA fraction was from reactions that did not contain transposon protein. Missing nucleoside footprints were as previously described except for the use of the binding conditions noted above for DNase I footprints.Citation18,Citation30
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Abbreviations
| LTR | = | long terminal repeat |
| UTR | = | untranslated region |
| TPRT | = | target primed reverse transcription |
| TE | = | transposable element |
| RLE | = | restriction-like endonuclease |
| PDR | = | polymerase chain reaction |
| EMSA | = | electrophoretic mobility shift assay |
| ZF | = | zinc finger |
| Bm | = | Bombyx mori |
| Av | = | Adineta vaga |
| Lp | = | Limulus polyphemus |
| Hm | = | Hydra magnipapillata |
| bp | = | base pair |
| nt | = | nucleotide |
| APE | = | apurinic-apyrimidinic endonuclease |
Figures and Tables
Figure 1 Restriction-like endonuclease bearing non-LTR retrotransposon structure and insertion mechanism. (A) A generalized Open Reading Frame (ORF) structure diagram of each of the major recognized RLE-bearing non-LTR retrotransposons clades is depicted along with the target site(s) and the name of a representative element(s) is given.Citation12,Citation21,Citation22,Citation24,Citation27,Citation28,Citation33,Citation34 In the structure diagrams the ORF is depicted by a rectangle. The lines flanking ORF rectangle are the 5′ and 3′ untranslated regions, respectively. The two major structural variants of R2 are given. The element names in bold text and gray filled structures are the subject of the paper. Abbreviations: R9 element from Adineta vaga (R9Av), R8 from Hydra magnipapillata (R8Hm), R2 from Limulus polyphemus (R2Lp), R2 from Bombyx mori (R2Bm), R4 element from Ascaris lumbricoides (R4Al), NeSL from Caenorhabditis elegans (NeSL-1Ce), CRE2 from Crithidia fasciculata (CRE2Cf), Genie-1 from Giardia lamblia (Genie-1Gl), Zinc Finger (ZF), Myb motif (Myb), reverse transcriptase (RT), cysteine-histidine rich motif (cchc), and restriction-like endonuclease (RLE). The lines flanking the ORFs are the 5′ and 3′ untranslated regions, respectively. Elements and conserved domains are not drawn to scale. (B) Model of R2 insertion as shown for the R2Bm element.Citation26 The R2Bm RNA contains higher order RNA structures that function as protein binding motifs. Two subunits of protein are bound to single RNA, forming a pseudo-dimer of protein linked through the RNA. The R2Bm protein bound to the 3′ UTR RNA adopts a protein conformation that binds upstream of the insertion site (insertion site = arrow) through an unidentified protein R2 protein domain. Protein bound to the 5′ RNA adopts a protein conformation that binds downstream of the insertion site through the N-terminal ZF and Myb motifs. Insertion is proposed to be catalyzed by the two protein subunits in four steps. Step 1: the RLE from the upstream subunit is responsible for first-strand cleavage. Step 2: the RT of the upstream subunit catalyzes first-strand TPRT using the cleaved DNA as a primer. Step 3: the downstream subunit cleaves the second DNA strand. Step 4: the downstream subunit provides the polymerase to perform second-strand synthesis using the cleaved DNA as a primer. Step 4 has not yet been shown to occur in vitro.
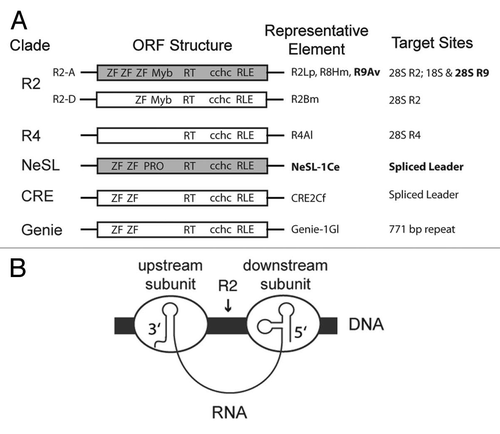
Figure 2 R2-A group structure and target sites. A ribosomal array unit is depicted with the 18S, 5S, and 28S ribosomal genes indicated by individual black boxes separated by intervening sequences (lines). The relative positions of R9, R8, and R2 insertion sites are marked by arrows. The domain structure of R9Av (gray) is depicted. Abbreviations and symbols are as in . R9Av is flanked by a 126 bp target site duplication (TSD) (black rectangles). The portions of R9Av cloned, expressed, and tested for DNA binding activity are indicated. The R9Av subclones are named based on which ZF and Myb domains are included in the clone (see ZF numbering above the R9Av structure).
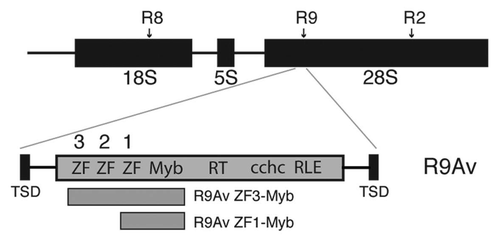
Figure 3 Electrophoretic mobility shift assay (EMSA) of the R9Av ZF3-Myb polypeptide bound to potential target DNAs. (A) Diagram of the target site DNAs used in the EMSA reactions shown in panels B–E. Target 1 consisted of the segment of the 28S that becomes duplicated upon R9 insertion (126 bp TSD region) along with 112 bp of upstream flanking sequence (net 238 bp). Target 2 was the 126 bp TSD region along with 101 bp of downstream flanking sequence (net 227bp). Target 3 was the 112 bp of upstream flanking sequence. Target 4 was the 101 bp of downstream flanking sequence. Target 1 was used for the footprint assays in . Below the targets is a ruler. The numbers correspond to base pair positions relative to the presumptive R9 bottom strand cleavage site (i.e., the presumptive site of TPRT), with negative numbers corresponding to target sequences upstream of the TPRT site and positive numbers downstream. Top strand cleavage site is expected to occur at bp 126, thus generating the TSD upon insertion.Citation28 (B) R9Av ZF3-Myb polypeptide EMSA on Target 1. All lanes represent 13 ul binding reactions containing 9 ng (9.4 nM) target DNA end-labeled with 32P in the presence of 50 ng of cold poly dIdC DNA as a competitor. Lane 1 is the DNA reference lane (no protein). Lane 2 contains 240 nM of R9Av ZF3-Myb polypeptide. Lane 3 contains 80 nM of R9Av ZF3-Myb polypeptide. Lane 4 contains 27 nM of R9Av ZF3-Myb polypeptide. Lane 5 contains 9 nM of R9Av ZF3-Myb polypeptide. The triangle above Lanes 2–5 indicates the R9Av ZF3-Myb polypeptide titration series (240 nM to 9 nM). (C) R9Av ZF3-Myb polypeptide EMSA on Target 2 (9.8 nM). Lanes and symbols are as in panel B. (D) R9Av ZF3-Myb polypeptide EMSA on Target 3. All lanes are as in panel B except that 20 nM of end-labeled target DNA was used. (E) R9Av ZF3-Myb polypeptide EMSA on Target 4. All lanes are as in panel B except that 22 nM of end-labeled target DNA was used.
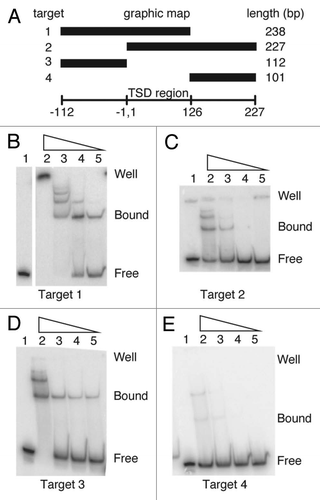
Figure 4 DNA footprints of the R9Av ZF3-Myb and polypeptides. (A) DNase I footprint on Target 1. The 238 bp Target 1 DNA was 5′ end labeled on either the top (left panel) or bottom strand (right panel). Lanes 1, adenine-plus-guanosine ladders (L). Lanes 2, DNase I pattern of naked DNA (R). Lanes 3, two polypeptides (i.e., dimer) bound DNA (Bdim). Lanes 4, single polypeptide (i.e., monomer) bound DNA (Bmon) The numbers to the left of the footprint correspond to base pair positions relative to the presumptive R9 bottom strand cleavage site (i.e., the presumptive site of TPRT) as in . Regions of DNA that are protected from DNase I degradation by the presence of the R9Av ZF3-Myb polypeptide are marked with thick black lines. Short thin black lines mark polypeptide binding induced DNase I hypersensitive sites. (B) Missing nucleoside footprint of R9Av ZF3-Myb polypeptide bound to 5′-end-labeled hydroxy-radical-treated 238 bp Target 1 DNA. Missing nucleoside footprinting is a binding interference based assay. Hydroxyradical treatment of target DNA generates abasic sites and cleaves the DNA backbone at the abasic site. Protein is then added to the treated DNA in a binding reaction. Abasic sites that interfere with protein binding are under-represented in the bound fraction and over represented in the free fraction. Binding reactions are fractionated into the component bound and free fractions by EMSA prior to being analyzed on denaturing polyacrylamide gels. Lanes 1, adenine-plus-guanosine ladders. Lanes 2, missing nucleoside reference pattern. Lanes 3, bound DNA fraction. Lanes 4, Free DNA fraction. The numbers to the left are as in . Nucleosides that, when missing, interfere with binding are marked with dashed lines. (C) Missing nucleoside footprint of R9Av ZF1-Myb polypeptide bound to 5′-end-labeled hydroxy-radical-treated 238 bp Target 1 DNA. Lanes are as in panel B. (D) Summary of the R9Av footprints. The presumptive bottom strand cleavage/TPRT site is indicated by the arrowhead. Base pair positions are numbered as in (i.e., the relative to the site of TPRT). Regions of DNA that are protected from DNase I degradation by the presence of the R9Av ZF3-Myb polypeptide are marked with thick black lines. Short thin black lines mark polypeptide binding induced DNase I hypersensitive sites. Nucleosides that, when missing, interfere with R9Av ZF3-Myb binding and R9Av ZF1-Myb binding, respectively, are marked with dashed lines. Jagged lines indicate that only the relevant portion of Target 1 is being shown.
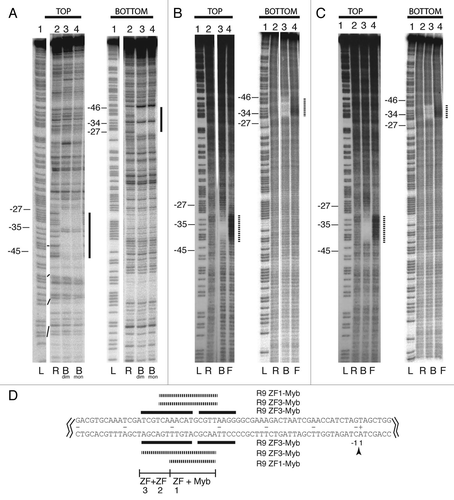
Figure 5 Target recognition by NeSL-1, a non-R2 clade element. (A) Structure of NeSL-1. Symbols and abbreviations are as in . Additional abbreviation: protease domain (pro). The region of NeSL-1 that was cloned and analyzed for DNA binding activity is indicated. (B) EMSA . EMSA using a 125 bp DNA fragment encompassing the spliced leader exon along with flanking sequences. Lane 1: reference DNA lane containing no protein. Lane 2: protein plus DNA lane. (C) DNase I footprint. Abbreviations and symbols are as in . Base pair numbering scheme is relative to the site of TPRT, with negative numbers corresponding to target sequences upstream of the TPRT site and positive numbers downstream. (D) Summary of footprint on target sequence. Symbols are as in . Jagged lines indicate that only the relevant section of the 125 bp target sequence that was used in the footprint analysis is shown. The leader sequence is in bold text. The intron sequence is in normal text. The presumptive cleavage sites are marked with arrowheads. The expected site of TPRT is at the bottom strand cleavage site. The base pair numbering is centered around this cleavage site. Sequences protected from DNase I degradation are marked with horizontal lines.
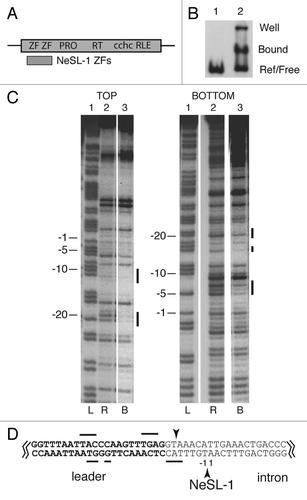
Figure 6 Summary of RLE-bearing non-LTR retrotransposon DNA binding modes and implications for the integration model. Abbreviations and symbols are as in . Myb and ZF indicates that a subunit of the listed non-LTR retrotransposon protein binds to target DNA at the indicated position via the Myb and/or ZF motifs. The order of the listing of Myb and ZF indicates the determined binding order along the DNA. A single question mark indicates that the position, existence, and role of the protein subunit has been determined, but the protein motif used to secure the protein to the DNA has not been determined. This subunit binds 3′ RNA, cleaves the bottom strand DNA, and performs TPRT. Two question marks indicate that the existence and position of the subunit on the DNA is hypothetical; however, it would be expected to bind to DNA using the aforementioned undetermined DNA binding domain used in R2Bm. The subunit with two question marks is further speculated to bind 3′ RNA, cleave the bottom strand, and perform TPRT (as does its cognate in R2Bm). The subunit binding via the variable N-terminal Myb and/or ZFs is known (R2Bm) or speculated (R2Lp, R9Av, and NeSL-1) to bind 5′ RNA, cleave the top DNA strand, and (hypothetically) perform second strand synthesis—processes known to be highly variable between lineages. Alternatively, a high degree of plasticity may exist in how the ordered series nucleic acid binding (RNA and DNA), DNA cleavage, and polymerization functions are performed by the two subunits.
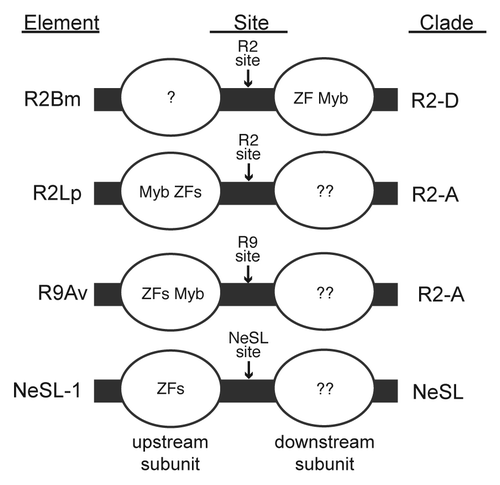
Table 1 Primers used for cloning and for generating target DNAs
Acknowledgments
The authors wish to thank Dr. Irina Arkhipova and Dr. Andre Pires da Silva for generously providing A. vaga and C. elegans genomic DNA, respectively. The authors thank Dr. Kimberly Bowles, Dr. Brad Reveal, Athena Jagdish, and Matthew Nelson for critical reading of the manuscript and for useful discussions. Finally the authors wish to thank and Aiswarya Pat for help with cloning and Micki Christensen for copy-editing. Funding was generously provided by NSF.
References
- Malik HS, Burke WD, Eickbush TH. The age and evolution of non-LTR retrotransposable elements. Mol Biol Evol 1999; 16:793 - 805; PMID: 10368957
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, et al. Initial sequencing and analysis of the human genome. Nature 2001; 409:860 - 921; PMID: 11237011; http://dx.doi.org/10.1038/35057062
- Han JS, Boeke JD. LINE-1 retrotransposons: modulators of quantity and quality of mammalian gene expression?. Bioessays 2005; 27:775 - 784; PMID: 16015595; http://dx.doi.org/10.1002/bies.20257
- Ye J, Eickbush TH. Chromatin structure and transcription of the R1- and R2-inserted rRNA genes of Drosophila melanogaster. Mol Cell Biol 2006; 26:8781 - 8790; PMID: 17000772; http://dx.doi.org/10.1128/MCB.01409-06
- Beck CR, Collier P, Macfarlane C, Malig M, Kidd JM, Eichler EE, et al. LINE-1 retrotransposition activity in human genomes. Cell 2010; 141:1159 - 1170; PMID: 20602998; http://dx.doi.org/10.1016/j.cell.2010.05.021
- Christensen SM, Eickbush TH. R2 target-primed reverse transcription: ordered cleavage and polymerization steps by protein subunits asymmetrically bound to the target DNA. Mol Cell Biol 2005; 25:6617 - 6628; PMID: 16024797; http://dx.doi.org/10.1128/MCB.25.15.6617-6628.2005
- Luan DD, Korman MH, Jakubczak JL, Eickbush TH. Reverse transcription of R2Bm RNA is primed by a nick at the chromosomal target site: a mechanism for non-LTR retrotransposition. Cell 1993; 72:595 - 605; PMID: 7679954; http://dx.doi.org/10.1016/0092-8674(93)90078-5
- Eickbush TH. Craig NL, Craigie R, Gellert M, Lambowitz AM. R2 and Related Site-Specific Non-Long Terminal Repeat Retrotransposons. Mobile DNA II 2002; Washington, DC ASM Press 813 - 835
- Moran JV, Gilbert N. Craig NL, Craigie R, Gellert M, Lambowitz AM. Mammalian LINE-1 Retrotransposons and Related Elements. Mobile DNA II 2002; Washington, DC ASM Press 836 - 869
- Eickbush TH, Jamburuthugoda VK. The diversity of retrotransposons and the properties of their reverse transcriptases. Virus Res 2008; 134:221 - 234; PMID: 18261821; http://dx.doi.org/10.1016/j.virusres.2007.12.010
- Yang J, Malik HS, Eickbush TH. Identification of the endonuclease domain encoded by R2 and other site-specific, non-long terminal repeat retrotransposable elements. Proc Natl Acad Sci USA 1999; 96:7847 - 7852; PMID: 10393910; http://dx.doi.org/10.1073/pnas.96.14.7847
- Burke WD, Malik HS, Jones JP, Eickbush TH. The domain structure and retrotransposition mechanism of R2 elements are conserved throughout arthropods. Mol Biol Evol 1999; 16:502 - 511; PMID: 10331276
- Moran JV, Holmes SE, Naas TP, DeBerardinis RJ, Boeke JD, Kazazian HHJ. High frequency retrotransposition in cultured mammalian cells. Cell 1996; 87:917 - 927; PMID: 8945518; http://dx.doi.org/10.1016/S0092-8674(00)81998-4
- Feng Q, Moran JV, Kazazian HHJ, Boeke JD. Human L1 retrotransposon encodes a conserved endonuclease required for retrotransposition. Cell 1996; 87:905 - 916; PMID: 8945517; http://dx.doi.org/10.1016/S0092-8674(00)81997-2
- Christensen S, Pont-Kingdon G, Carroll D. Comparative studies of the endonucleases from two related Xenopus laevis retrotransposons, Tx1L and Tx2L: target site specificity and evolutionary implications. Genetica 2000; 110:245 - 256; PMID: 11766845; http://dx.doi.org/10.1023/A:1012704812424
- Mandal PK, Bagchi A, Bhattacharya A, Bhattacharya S. An Entamoeba histolytica LINE/SINE pair inserts at common target sites cleaved by the restriction enzyme-like LINE-encoded endonuclease. Eukaryot Cell 2004; 3:170 - 179; PMID: 14871947; http://dx.doi.org/10.1128/EC.3.1.170-179.2004
- Thompson BK, Christensen SM. Independently derived targeting of 28S rDNA by A- and D-clade R2 retrotransposons: plasticity of integration mechanism. Mobile Genet Elements 2011; 1:29 - 37
- Christensen SM, Bibillo A, Eickbush TH. Role of the Bombyx mori R2 element N-terminal domain in the target-primed reverse transcription (TPRT) reaction. Nucleic Acids Res 2005; 33:6461 - 6468; PMID: 16284201; http://dx.doi.org/10.1093/nar/gki957
- Christensen S, Eickbush TH. Footprint of the retrotransposon R2Bm protein on its target site before and after cleavage. J Mol Biol 2004; 336:1035 - 1045; PMID: 15037067; http://dx.doi.org/10.1016/j.jmb.2003.12.077
- Volff JN, Korting C, Froschauer A, Sweeney K, Schartl M. Non-LTR retrotransposons encoding a restriction enzyme-like endonuclease in vertebrates. J Mol Evol 2001; 52:351 - 360; PMID: 11343131
- Malik HS, Eickbush TH. NeSL-1, an ancient lineage of site-specific non-LTR retrotransposons from Caenorhabditis elegans. Genetics 2000; 154:193 - 203; PMID: 10628980
- Burke WD, Malik HS, Rich SM, Eickbush TH. Ancient lineages of non-LTR retrotransposons in the primitive eukaryote, Giardia lamblia. Mol Biol Evol 2002; 19:619 - 630; PMID: 11961096
- Eickbush TH, Malik HS. Craig NL, Craigie R, Gellert M, Lambowitz AM. Origins and Evolution of Retrotransposons. Mobile DNA II 2002; Washington, DC ASM Press 1111 - 1146
- Kojima KK, Fujiwara H. Cross-genome screening of novel sequence-specific non-LTR retrotransposons: various multicopy RNA genes and microsatellites are selected as targets. Mol Biol Evol 2004; 21:207 - 217; PMID: 12949131; http://dx.doi.org/10.1093/molbev/msg235
- Kojima KK, Fujiwara H. Long-term inheritance of the 28S rDNA-specific retrotransposon R2. Mol Biol Evol 2005; 22:2157 - 2165; PMID: 16014872; http://dx.doi.org/10.1093/molbev/msi210
- Christensen SM, Ye J, Eickbush TH. RNA from the 5′ end of the R2 retrotransposon controls R2 protein binding to and cleavage of its DNA target site. Proc Natl Acad Sci USA 2006; 103:17602 - 17607; PMID: 17105809; http://dx.doi.org/10.1073/pnas.0605476103
- Kojima KK, Kuma K, Toh H, Fujiwara H. Identification of rDNA-specific non-LTR retrotransposons in Cnidaria. Mol Biol Evol 2006; 23:1984 - 1993; PMID: 16870681; http://dx.doi.org/10.1093/molbev/msl067
- Gladyshev EA, Arkhipova IR. Rotifer rDNA-specific R9 retrotransposable elements generate an exceptionally long target site duplication upon insertion. Gene 2009; 448:145 - 150; PMID: 19744548; http://dx.doi.org/10.1016/j.gene.2009.08.016
- Stage DE, Eickbush TH. Origin of nascent lineages and the mechanisms used to prime second-strand DNA synthesis in the R1 and R2 retrotransposons of Drosophila. Genome Biol 2009; 10:R49; PMID: 19416522; http://dx.doi.org/10.1186/gb-2009-10-5-r49
- Hayes JJ, Tullius TD. The missing nucleoside experiment: a new technique to study recognition of DNA by protein. Biochemistry 1989; 28:9521 - 9527; PMID: 2611245; http://dx.doi.org/10.1021/bi00450a041
- Abramoff MD, Magalhaes PJ. Image Processing with ImageJ. Biophotonics International 2004; 11:36 - 42
- Brenowitz B, Senear DF, Kingston RE. Ausubel FM. DNase I Footprint Analysis of Protein-DNA Binding. Current Protocols in Molecular Biology 2003; Hoboken, NJ John Wiley and Sons, Inc 12.4 - 12.5
- Burke WD, Muller F, Eickbush TH. R4, a non-LTR retrotransposon specific to the large subunit rRNA genes of nematodes. Nucleic Acids Res 1995; 23:4628 - 4634; PMID: 8524653; http://dx.doi.org/10.1093/nar/23.22.4628
- Teng SC, Wang SX, Gabriel A. A new non-LTR retrotransposon provides evidence for multiple distinct site-specific elements in Crithidia fasciculata miniexon arrays. Nucleic Acids Res 1995; 23:2929 - 2936; PMID: 7659515; http://dx.doi.org/10.1093/nar/23.15.2929