Abstract
MicroRNAs coordinate networks of mRNAs, but predicting specific sites of interactions is complicated by the very few bases of complementarity needed for regulation. Although efforts to characterize the specific requirements for microRNA (miR) regulation have made some advances, no general model of target recognition has been widely accepted. In this work, we describe an entirely novel approach to miR target identification. The genomic events responsible for the creation of individual miR loci have now been described with many miRs now known to have been initially formed from transposable element (TE) sequences. In light of this, we propose that limiting miR target searches to transcripts containing a miR’s progenitor TE can facilitate accurate target identification. In this report we outline the methodology behind OrbId (Origin-based identification of microRNA targets). In stark contrast to the principal miR target algorithms (which rely heavily on target site conservation across species and are therefore most effective at predicting targets for older miRs), we find OrbId is particularly efficacious at predicting the mRNA targets of miRs formed more recently in evolutionary time. After defining the TE origins of > 200 human miRs, OrbId successfully generated likely target sets for 191 predominately primate-specific human miR loci. While only a handful of the loci examined were well enough conserved to have been previously evaluated by existing algorithms, we find ~80% of the targets for the oldest miR (miR-28) in our analysis contained within the principal Diana and TargetScan prediction sets. More importantly, four of the 15 OrbId miR-28 putative targets have been previously verified experimentally. In light of OrbId proving best-suited for predicting targets for more recently formed miRs, we suggest OrbId makes a logical complement to existing, conservation based, miR target algorithms.
Introduction
During the latter half of the 20th century one of the greatest achievements in genetic research was the meticulous cataloging of epistatic relationships between genetic loci. While new relationships brought new insights, they also created massive networks of seemingly endlessly interacting genetic pathways. In 1993, however, Lee et al. described an entirely new short noncoding RNA that, despite its size, would ultimately be recognized as an important player in deciphering complex genetic interactions.Citation1 These small microRNAs (miRs) are only ~20 nts in length () and are capable of coordinating the expressions of networks of mRNAs (mRNAs) through complementary basepairing.Citation1 Strikingly, over 1,900 unique human miRs have been clonedCitation2 since the first were discovered in 2001.Citation3-Citation5 As such, it is of little surprise that miR research has seen a recent explosion of interest, especially considering that a single miR has the potential to control expression of dozens of genes and miR misregulations are commonly associated with oncogenesis (recently reviewed in ref. Citation6).
Figure 1. MiR biology and origins. (A) MiR generation. MiRs can occur inter- or intragenically and be transcribed by either RNA Polymerase II or III.Citation24 Following transcription, the “pre-miR” hairpin (middle) is excised from the initial transcript (or pri-miR) (top) by Drosha. Once in the cytoplasm, the hairpin or stem loop is cleaved and denatured by Dicer to excise the ~20 nt mature miR (bottom). (B) MiR seeds. A seed match between a miR (top) and target mRNA (bottom) is illustrated. The nucleotides in a miR generally referred to as a “seed” (nts 2 through 8) and a “seed match” in a mRNA are depicted in red. Basepairing is indicated by vertical lines. (C) Cartoon depicting the molecular origin of many miR loci. MiRs were initially formed by the neighboring insertions of related TEs. A pri-miR is depicted just above the genome with an arrow indicating readthrough Pol-III transcription from a (+) strand Alu SINE into a neighboring (-) strand Alu. As illustrated, transcriptional readthrough would generate a RNA stem loop whose stems (loaded into the RISC machinery if processed) would correspond to the terminal nucleotides of the neighboring Alus. Figure adapted from.Citation23
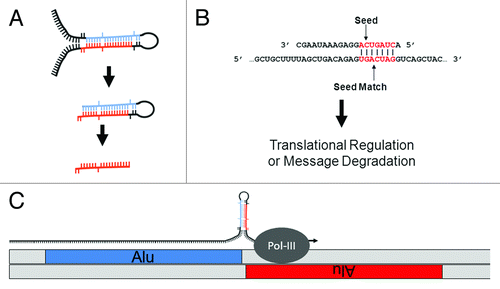
Whereas novel miR discovery has been forthcoming, progress in deciphering miR regulations has proven exceptionally challenging. This is largely due to miRs requiring very little sequence complementarity to the mRNAs they coordinate. In contrast to siRNAs which depend upon almost perfect complementarity to direct message degradation, miR target recognition and consequent repression can be mediated through as few as 7 bps of complementarity. Generally thought to most frequently occur in the 5′ miR sequence, these 7 participating nts are typically referred to as the miR “seed” and the complement in a mRNA as the “seed match”Citation7-Citation9 (). The recurrent observation of complementarity between seed and seed match in a few initially characterized miR-target interactions lead to the majority of miR target recognition algorithms basing target searches on perfect seed matches. Following this, most algorithms differ primarily by the significance they attribute to seed match conservation between species, the presence of multiple seed matches in a given mRNA target, and the extent of complementarity between the proposed target and remainder of the miR (recently reviewed refs. Citation10-Citation14). While algorithms have been developed that do not require target site conservation across species (focusing instead on thermodynamic stability and target site secondary structure (e.g., PITACitation15 and rna22Citation16), the principal, most widely accepted target prediction algorithms (DIANA-microT,Citation17 miRanda,Citation18 PicTar,Citation19,Citation20 and TargetScanCitation21) each incorporate target site conservation into their prediction methodologies. Although efforts to characterize the specific requirements for miR target recognition continue to advance, to date the principal target algorithms typically suggest several hundred putative mRNA targets for each individual miR. As such is the case, no model of miR target prediction has been widely accepted.
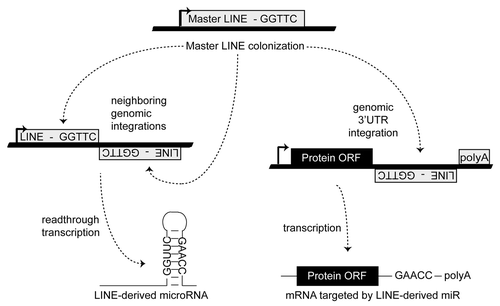
Similar in rationale to the principal miR target prediction algorithms (although not requiring target site conservation across species), we have developed an entirely novel approach to miR target identification. First suggested by Smalheiser and Torvik,Citation22 the molecular events responsible for the genomic formation of many miR loci from transposable element (TE) sequences have now been describedCitation22-Citation28(). Having recently performed a series of detailed genomic analyses describing the TE origins of ~2,400 distinct miRs,Citation23 we hypothesized that a miR and its mRNA target sites might actually be formed in parallel by the ongoing colonization of a common ancestral transposable element (). In light of this, we propose that limiting miR target searches to mRNAs containing the TE initially giving rise to a miR can significantly hone accurate target identification. In this work we outline the methodology behind, and initial findings for, a novel miR target prediction strategy: OrbId (Origin-based Identification of microRNA targets). In all, we have successfully generated target sets for 191 unique miRs after applying OrbId to a set of 208 distinct human miRs of defined TE origin.Citation23 While the majority of OrbId putative targets were for recently formed miR loci, we did generate targets for the evolutionarily older miR-28 family and find our results largely in agreement with both traditional target prediction strategiesCitation17,Citation21 and existing experimental evidence.Citation29 Thus, the mRNA targets of a given miR can largely be predicted based on shared transposable element origins.
Figure 2. Establishing a miR regulatory network. MiR regulatory networks are formed when an advantageous regulation arises from a series of random TE insertions into expressed genomic loci, and the formation of a TE juxtaposition by the positive and negative strand insertions of related TEs. Thick lines indicate genomic DNA and thin lines denote RNA. Figure adapted from.Citation23
Results
Targets predicted for 92% of human miRs with defined TE origins
OrbId operates under the premise that a miR and its mRNA target sites were formed in parallel by the colonization of a common progenitor transposable element (). Utilizing this premise, we have successfully generated putative target sets for 191 of 208 human miRs with defined TE origins.Citation23 In stark contrast to the principal miR target algorithms currently utilized (which typically predict several hundred putative mRNA targets for individual miRsCitation17-Citation19,Citation21), we find OrbId predicts significantly fewer mRNA targets per miR (average 7.9, median 3) (). In all, 59 produced a single mRNA target, 120 distinct miRs were suggested to have between 2 and 25 target mRNAs, and 12 were predicted to target > 25 mRNAs (max = 94, putative targets for miR-574) (Table S1). In order to ensure strict adherence to the OrbId operating methodology, sequence alignments of unique mRNA target sites, miRs, and progenitor TEs were independently verified (, Table S2).
Table 1. OrbId summary. The full EnsemblCitation36 set of 178,375 unique human mRNA transcripts including 5′UTR, 3′UTR, and ORF annotations were compiled in and retrieved using the Biomart mining utilityCitation37. “Human miRs analyzed” correspond to the full set of human miR mature sequences identified by Borchert et al. as originating from TEsCitation23 and were obtained from the miR Registry miRBase.Citation2
Figure 3. MiR-28 predicted target three way alignments. Alignments between OrbId predicted miR-28 target mRNAs (middle), a consensus L2B LINE (L2Plat1o) (top), and miR-28 (bottom). (*), base identity in the three aligning sequences. (^), base identity (indicating base pairing) between the miR and mRNA target only. (:), GU basepairing between miR and mRNA target. 3′ UTR or 5′ UTR targeting is indicated. Uracils are shown as thymines and UTRs have been reverse complemented for illustrative purposes.

Target sites are generally not preferentially located in 3′ UTRs
MiRs have now been conclusively shown to regulate target mRNAs through interactions with 5′ untranslated regions (UTR) and open reading frame (ORF) sequences similar to 3′ UTR interactions.Citation30-Citation35 As such, it is somewhat surprising that most target prediction algorithmsCitation16-Citation18,Citation20 predominately screen mRNA 3′ UTRs for miR regulatory sites. In this analysis, we assessed all publically available human mRNA sequence regardless of functional annotation.Citation36,Citation37 Strikingly, not only did we find strong evidence supporting 5′ UTR and ORF regulations, we did not observe a general bias for 3′ UTR target sites (). In all, of 1529 unique predicted regulations, 970, 410 and 149 are located within 3′ UTR, 5′ UTR and ORF sequences respectively. When the average lengths of human 3′ UTR (386 nt), 5′ UTR (117 nt) and ORF (647 nt) sequences are taken in consideration, we find no significant bias for targeting to occur in either UTR preferentially. However, we did find target sites were approximately 12 times as likely to occur in noncoding UTR sequences than in ORF coding regions. Importantly, while we observed no general bias for miR targeting of 3′ UTR, 5′ UTR or ORF sequences, individual miR families showed significant targeting preferences. Of note, the targets of the principal mariner transposon derived miR family (miR-548) were located almost exclusively (> 99%) within 3′ UTR sequences, the targets of the principal LINE derived miR family (miR-28) were similarly biased to occur within 3′ UTR sequences (> 96%), but, in sharp contrast, the targets of a novel Alu SINE derived miR family (Alu-miR) were located predominately (> 81%) within 5′ UTR sequences (, Figure S1). Additionally, while less than 10% of putative targets were predicted to occur in ORFs (despite ORFs accounting for > 56% of the total transcript sequence examined(), we identify two miRs, miR-544 and miR-301a-5p which are predicted to preferentially (> 90%) target ORF sequences (, Figure S1).
Table 2. OrbId prediction set for select TE-derived human miRs. “miR Name” refers to miRBaseCitation2 annotation while “Ensembl Gene ID” and “Gene Name” were obtained using the Biomart mining utilityCitation37. “Diana, TS” refers to whether a predicted target is contained within publically accessible Diana (D) and TargetScan (TS) predictionsCitation17,Citation21. “Region” refers to the location of a predicted target site within a given mRNA. MiR-28–5p corresponds to the participating member of the miR-28 family. MiR-1254–1 is a member of the Alu-miR family. MiR-603 is a member of the miR-548 family
LINE L2B (miR-28) family.
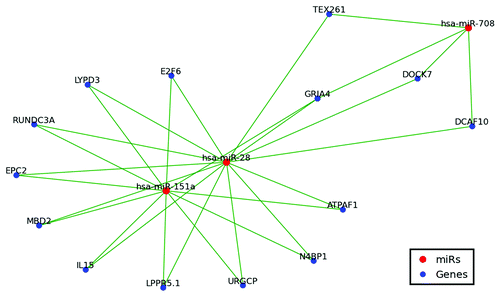
First identified in 2003Citation22 as arising from L2B LINE elements, miR-28 and miR-151 have long been recognized as being related, and their numerous representative sequences across mammalia are collectively referred to as the miR-28 family.Citation2 Supporting this relationship, and despite there only being an ~10% likelihood that a given miR in this analysis would target the same mRNA as any other miR, we find ~76% of miR-28 and miR-151 proposed targets (11 of 15 and 11 of 14 respectively) common to both miRs (, Table S1). Our analyses also indicate the likelihood of a third, until now overlooked, member of the miR-28 family, miR-708. While initially formed from the same LINE element that gave rise to miR-28 and miR-151Citation23 and baring significant pre-miR homology to both miR-28 and miR-151Citation2 (Figure S2), we find ~31% of miR-708 targets also constitute miR-28 family targets (, Table S1). Additionally, as the miR-28 family was the oldest in our analysis, miR-28 was one of the few miRs with publically available Diana and TargetScan predictions. Encouragingly we find ~80% of our miR-28 target predictions contained within the principal Diana and TargetScan prediction setsCitation16,Citation20 (). Furthermore, over 25% of our putative miR-28 targets (4 of 15) have already been experimentally verified and shown to indeed regulate the mRNAs predicted by OrbId (ref. Citation29 data not shown).
Figure 4. MiR-28, miR-151 and miR-708 target network. Only shared targets are depicted including 14 of 15 miR-28–5p targets, 11 of 14 miR-151a-5p targets, and 4 of 13 miR-708 targets. Green lines indicate miR regulation.
MiRs formed from Alu repeats
In contrast to the miR-548 and miR-28 families, the targets of a novel Alu SINE derived miR family (miR-566) were located predominately (> 80%) within 5′ UTR sequences. While not as closely related as the miR-28 family, these Alu-derived miRs share several target relationships. While they may not constitute a traditional miR family based on common molecular origin, they could be considered to be a family in the sense of common targeting. In all, miRs -566, -1254, -1268, -1273, -1285, -1968, -1972, and -1973 appear to establish a significant network of target regulations (Figure S3). Intriguingly, our findings are largely in agreement with previous reports suggesting that 3′UTR embedded Alu repeats frequently house novel, primate-specific miR target sites.Citation38-Citation40
Discussion
The genomic events responsible for the initial formation of numerous miR loci have recently been described.Citation23 The majority of these loci appear to have initially arisen from transposable element (TE) sequences. In addition to forming miR loci, we now hypothesize that TE mobilizations also generate miR regulatory networks by simultaneously integrating into existing mRNA expression cassettes (). Thus, the principle objective of this work was to utilize common TE ancestry to facilitate accurate prediction of miR-mRNA target interactions. To accomplish this, we have developed a novel methodology titled OrbId (Origin-based Identification of microRNA targets) (). OrbId contrasts sharply with current miR target algorithmsCitation16-Citation18,Citation20 as these methodologies rely heavily on target site conservation across species and have therefore been primarily effective at predicting targets for well conserved miRs. OrbId is better suited for predicting the mRNA targets of evolutionarily younger miRs for which target site conservation searches are impractical. For example, the 70 human miR loci known to have been formed from primate-specific Alu repeats,Citation23,Citation24 rodent-specific miRs formed from rodent specific B1 SINEs,Citation23 or the marsupial-specific miRs formed from marsupial-specific transposable elements.Citation25
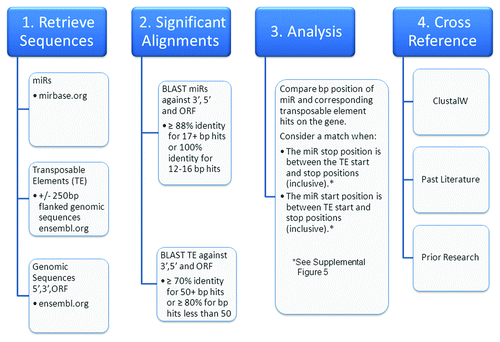
Figure 5. OrbId methodology flowchart. A high level overview of the steps taken to determine miR and transposable element concurrent alignments within the human transcriptome.
OrbId may also prove valuable in identifying taxon-specific targets of more conserved miRs. Requiring target site conservation across species has been effective at predicting many of the targets for conserved miRs. By design, however, traditional conservation-based miR target algorithms miss any targets arising from TE mobilizations following the initial establishment of a miR regulatory network. For example, if ongoing TE colonizations occur following speciation events, separate species might well acquire distinct, novel targets for existing miRs. Although beyond the scope of this analysis, more comprehensive species wide implementations of OrbId will be needed to fully evaluate the prevalence of such events.
Future analyses will unquestionably broaden the range of OrbId utility as the existing repertoire of defined miR-TE relationships continues to expand through the ongoing characterizations of additional miR loci and novel TE sequences. Importantly, de Koning et al. recently suggested that over two-thirds of the human genome were actually formed from repetitive elements.Citation44 While highly intriguing, the extent of the repetitive composition of the human genome remains a significant point of debate and attempts to fully clarify this issue remain ongoing. Should the work of de Koning et al. prove largely accurate, the incorporation of this information into current OrBId methodology would clearly result in marked increases in the definable number of putative miR::target relationships. Additionally, while this would likely predominately facilitate putative target identification for evolutionarily older miRs, it would also almost certainly require increased stringency to avoid concurrent increases in false positives. As a result of electing to limit our OrBId analysis to identifying the targets of miRs whose TE origins have been clearly definedCitation23 using RepBase annotations,Citation42,Citation43 this analysis was confined to the evaluation of ~16% of currently annotated human miR loci (resulting in target predictions for 191 unique human miRs). While our OrbId analysis primarily dealt with miRs predominately unexamined by the principal miR target prediction algorithms, in striking contrast to the hundreds of putative mRNAs generally predicted by the principal algorithms,Citation16-Citation18,Citation20 OrbId averaged ~8 putative mRNA targets per unique miR. While the average number of mRNAs a typical miR regulates remains poorly defined, we suggest our predictions most likely only constitute a subset of actual miR regulations (largely due to the high degree of complementarity we required for putative target interaction). However, since OrbId target sets are derived through a rationale based on molecular origin, we suggest that the OrbId putative target lists reported here likely contain a markedly higher proportion of actual endogenous miR targets than the hundreds of predicted mRNA targets obtained through less stringent algorithms. Additionally and in terms of laboratory and clinical efforts, we suggest that a manageable number of likely endogenous relationships based on a molecular rationale is in many ways advantageous to more encompassing sets of hundreds of putative targets.
Importantly, > 95% of the miRs included in our analysis have not been examined by the principal target prediction algorithms (most likely due to either their repetitive nature or their being primate specific and not conserved across species). We do find, however, that the OrBId target predictions for the few miRs in our analysis that have previously been examined are largely in agreement with more established algorithms. For example, we find ~80% of our putative miR-28 targets are contained within the principal Diana and TargetScan predictions (). Excitingly, four of our 13 putative miR-28 3′ UTR targets have actually previously been verified experimentally.Citation29 Additionally, three of these experimentally verified miR-28 targets, N4BP1, E2F6, and TEX261 are expressed alongside miR-28 in blood cell lineages and have each been speculated to contribute to myeloproliferative neoplasms.Citation29 While experimental corroborations such as these are encouraging, the majority of our novel OrBId miR target predictions will clearly ultimately require direct experimental validation. It is tempting to speculate, however, that experimental verification of many of our miR interactions might well be forthcoming as this work represents the first time putative target sets have been reported for the majority of the 191 distinct miRs examined in this analysis thereby constituting the first real examination of potential target interactions for ~10% of all currently characterized human miRs.
In conclusion, we report here a new approach for miR target prediction that relies on TE origins. In all probability a universal description of miR target interaction has not yet been characterized because there is no universal description of miR target interaction. Complicating factors such as GU base-pairing, nucleotide editing, target secondary structure and RNA-interacting protein effectsCitation41 make strict thermodynamic modeling largely incapable of honing in on actual mRNA targets. Likely a closer estimation of true mRNA regulations, OrbId predicts far fewer mRNA targets per miR than existing algorithms through employing a molecular, origin-based rationale. Importantly, incorporating logical molecular cues such as target site conservation has previously been successfully exploited to circumvent the limitations of mathematical modeling alone.Citation16-Citation18,Citation20 Similarly based on genetic rationale, this work introduces a novel consideration that helps to circumvent many of the difficulties in accurate target identification.Citation41 We suggest that since TEs are present in multiple copies across the genome,Citation36 and miRs target sequences through complementary basepairing, requiring a miR target site to occur in the same TE from which a miR was initially formed represents a logical addition to miR target prediction. In contrast to the principal miR target algorithms currently utilizedCitation16-Citation18,Citation20 (which rely heavily on target site conservation across species and have therefore been primarily effective at predicting targets for well conserved miRs), OrbId has been designed to predict the mRNA targets of evolutionarily younger miRs and therefore makes a strategically logical complement to existing miR target algorithms.
Materials and Methods
Retrieving miR, transposable element mRNA and genomic sequences
In 2011, Borchert et al.Citation23 established a connection between miRs and transposable elements (TE) providing evidence for the role of repetitive elements in miR origin. Unique TEs associated with the origins of > 200 human miRs were retrieved from the data set created from the work of Borchert et al. and used as the basis for this analysis. Single FASTA files containing the full set of human miR mature sequences were downloaded from the miR Registry housed at Sanger (http://www.mirbase.org).Citation2 Flanked genomic sequences were obtained for human miRs corresponding to genomes currently available in Ensembl (+/− 250 base pair flanks).Citation36 Unique miR accession numbers from the miR Registry were attached to the corresponding flanked genomic sequence then utilized as the origin-based TE sequence. Next, the full set of Ensembl human 5′UTR, 3′UTR, and ORF sequences were compiled in and retrieved using the Biomart mining utility.Citation37 Of 178,375 unique human transcripts, 68,892,718 nts corresponded to 3′ UTR sequence, 20,940,347 nts corresponded to 5′ UTR sequence and 115,422,049 nts corresponded to ORF sequence making the average 3′ UTR, 5′ UTR and ORF lengths examined in this study 386, 117 and 647 nts respectively.
Correlating miR target sites with progenitor TEs
It is important to note that all alignment analyses were identically run in parallel by three independent research teams and cross examined for verification. Significant alignments between the miR and TE sequences with the human 5′UTR, 3′UTR, and ORF sequences were obtained via BLAST (BLASTN 2.2.15 with -FF, -W7 flags). Beyond requiring a common molecular origin for each member of a putative miR::mRNA interaction, the majority of false positive relationships were largely avoided through requiring long, nearly perfect complementarities. Strongly agreeing with similarly stringent statistical searches for miR targets,Citation45 this strategy resulted in the identification of numerous long runs of perfect complementarity between putative miRs and targets and found no significant bias for that complementarity to occur near miR 5′ ends or in mRNA 3′UTRs. For the miR sequences, significant alignments were strictly defined as ≥ 88% identity for ≥ 17 bp hits or 100% identity for 12–16 bps. For TE sequences, significant alignments were strictly defined as ≥ 70% identity for 50+ bp hits or ≥ 80% for bp hits less than 50. Using the proceeding search algorithm we determined alignment matches along the human 5′UTR, 3′UTR, and ORF sequences between each miR and its corresponding TE. Our algorithm looked at each miR::mRNA alignment and searched for overlapping TE alignments in the same region of that transcript. If such TE alignments were found, the transcript was recorded as a target for that miR. We defined a miR as hitting the same region as its corresponding TE if either of two following criterion were satisfied: (1) The miR ending alignment position was between the TE beginning and ending alignment positions (inclusive), or (2) The miR beginning alignment position was between the TE beginning and ending alignment positions (inclusive). If at least part of the miR alignment is within the TE alignment region on a gene, then this method counted the transcript as a miR target (Figure S5). Additionally, as control, we randomly generated 10 scrambled sets of matched, size appropriate miR repeat pairs to search for targets using OrBId. Importantly, we identified no putative targets for scrambled controls in the human transcriptome.
| Abbreviations: | ||
| bp | = | basepair |
| LINE | = | long interspersed repeated element |
| LTR | = | long terminal repeat |
| miR | = | microRNA |
| mRNA | = | messenger RNAs |
| nt | = | nucleotide |
| ORF | = | open reading frame |
| Pol III | = | RNA polymerase III |
| pre-miR | = | miR hairpin |
| pri-miR | = | initial miR transcript |
| RISC | = | RNA induced silencing complex |
| RNAi | = | RNA interference |
| SINE | = | short interspersed repeated elements |
| siRNA | = | small interfering RNA |
| TE | = | transposable element |
| tRNA | = | transfer RNA |
| UTR | = | untranslated region |
Additional material
Download Zip (1.8 MB)Acknowledgments
This work was funded by the School of Biological Sciences, the College of Arts and Sciences at Illinois State University, USDA-NIFA-AFRI2011–67021–30114 to DAR and National Institutes of Health (1R15CA137608) to EDL.
Declaration of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplementary Material
Supplemental material may be downloaded here: www.landesbioscience.com/journals/mge/article/21617
Notes
† These authors contributed equally to this work
References
- Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993; 75:843 - 54; http://dx.doi.org/10.1016/0092-8674(93)90529-Y; PMID: 8252621
- Kozomara A, Griffiths-Jones S. miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res 2011; 39:Database issue D152 - 7; http://dx.doi.org/10.1093/nar/gkq1027; PMID: 21037258
- Lagos-Quintana M, Rauhut R, Lendeckel W, Tuschl T. Identification of novel genes coding for small expressed RNAs. Science 2001; 294:853 - 8; http://dx.doi.org/10.1126/science.1064921; PMID: 11679670
- Lee RC, Ambros V. An extensive class of small RNAs in Caenorhabditis elegans. Science 2001; 294:862 - 4; http://dx.doi.org/10.1126/science.1065329; PMID: 11679672
- Lau NC, Lim LP, Weinstein EG, Bartel DP. An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science 2001; 294:858 - 62; http://dx.doi.org/10.1126/science.1065062; PMID: 11679671
- Farazi TA, Spitzer JI, Morozov P, Tuschl T. miRNAs in human cancer. J Pathol 2011; 223:102 - 15; http://dx.doi.org/10.1002/path.2806; PMID: 21125669
- Hutvágner G, Zamore PD. A microRNA in a multiple-turnover RNAi enzyme complex. Science 2002; 297:2056 - 60; http://dx.doi.org/10.1126/science.1073827; PMID: 12154197
- Lai EC. Micro RNAs are complementary to 3′ UTR sequence motifs that mediate negative post-transcriptional regulation. Nat Genet 2002; 30:363 - 4; http://dx.doi.org/10.1038/ng865; PMID: 11896390
- Zeng Y, Wagner EJ, Cullen BR. Both natural and designed micro RNAs can inhibit the expression of cognate mRNAs when expressed in human cells. Mol Cell 2002; 9:1327 - 33; http://dx.doi.org/10.1016/S1097-2765(02)00541-5; PMID: 12086629
- Witkos TM, Koscianska E, Krzyzosiak WJ. Practical Aspects of microRNA Target Prediction. Curr Mol Med 2011; 11:93 - 109; http://dx.doi.org/10.2174/156652411794859250; PMID: 21342132
- Min H, Yoon S. Got target? Computational methods for microRNA target prediction and their extension. Exp Mol Med 2010; 42:233 - 44; http://dx.doi.org/10.3858/emm.2010.42.4.032; PMID: 20177143
- Saito T, Saetrom P. MicroRNAs–targeting and target prediction. New Biotechnol 2010; 27:243 - 9; http://dx.doi.org/10.1016/j.nbt.2010.02.016
- Thomas M, Lieberman J, Lal A. Desperately seeking microRNA targets. Nat Struct Mol Biol 2010; 17:1169 - 74; http://dx.doi.org/10.1038/nsmb.1921; PMID: 20924405
- Yue D, Liu H, Huang Y. Survey of Computational Algorithms for MicroRNA Target Prediction. Curr Genomics 2009; 10:478 - 92; http://dx.doi.org/10.2174/138920209789208219; PMID: 20436875
- Kertesz M, Iovino N, Unnerstall U, Gaul U, Segal E. The role of site accessibility in microRNA target recognition. Nat Genet 2007; 39:1278 - 84; http://dx.doi.org/10.1038/ng2135; PMID: 17893677
- Miranda KC, Huynh T, Tay Y, Ang YS, Tam WL, Thomson AM, et al. A pattern-based method for the identification of MicroRNA binding sites and their corresponding heteroduplexes. Cell 2006; 126:1203 - 17; http://dx.doi.org/10.1016/j.cell.2006.07.031; PMID: 16990141
- Kiriakidou M, Nelson PT, Kouranov A, Fitziev P, Bouyioukos C, Mourelatos Z, et al. A combined computational-experimental approach predicts human microRNA targets. Genes Dev 2004; 18:1165 - 78; http://dx.doi.org/10.1101/gad.1184704; PMID: 15131085
- John B, Enright AJ, Aravin A, Tuschl T, Sander C, Marks DS. Human MicroRNA targets. PLoS Biol 2004; 2:e363; http://dx.doi.org/10.1371/journal.pbio.0020363; PMID: 15502875
- Lall S, Grun D, Krek A, Chen K, Wang YL, Dewey CN, et al. A genome-wide map of conserved microRNA targets in C. elegans. Curr Biol 2006; 16:460 - 71
- Krek A, Grün D, Poy MN, Wolf R, Rosenberg L, Epstein EJ, et al. Combinatorial microRNA target predictions. Nat Genet 2005; 37:495 - 500; http://dx.doi.org/10.1038/ng1536; PMID: 15806104
- Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 2005; 120:15 - 20; http://dx.doi.org/10.1016/j.cell.2004.12.035; PMID: 15652477
- Smalheiser NR, Torvik VI. Mammalian microRNAs derived from genomic repeats. Trends Genet 2005; 21:322 - 6; http://dx.doi.org/10.1016/j.tig.2005.04.008; PMID: 15922829
- Borchert GM, Holton NW, Williams JD, Hernan WL, Bishop IP, Dembosky JA, et al. Comprehensive analysis of microRNA genomic loci identifies pervasive repetitive-element origins. Mob Genet Elements 2011; 1:8 - 17; http://dx.doi.org/10.4161/mge.1.1.15766; PMID: 22016841
- Borchert GM, Lanier W, Davidson BL. RNA polymerase III transcribes human microRNAs. Nat Struct Mol Biol 2006; 13:1097 - 101; http://dx.doi.org/10.1038/nsmb1167; PMID: 17099701
- Devor EJ, Peek AS, Lanier W, Samollow PB. Marsupial-specific microRNAs evolved from marsupial-specific transposable elements. Gene 2009; 448:187 - 91; http://dx.doi.org/10.1016/j.gene.2009.06.019; PMID: 19577616
- Piriyapongsa J, Jordan IK. A family of human microRNA genes from miniature inverted-repeat transposable elements. PLoS One 2007; 2:e203; http://dx.doi.org/10.1371/journal.pone.0000203; PMID: 17301878
- Yan Y, Zhang Y, Yang K, Sun Z, Fu Y, Chen X, et al. Small RNAs from MITE-derived stem-loop precursors regulate abscisic acid signaling and abiotic stress responses in rice. Plant J 2011; 65:820 - 8
- Yao C, Zhao B, Li W, Li Y, Qin W, Huang B, et al. Cloning of novel repeat-associated small RNAs derived from hairpin precursors in Oryza sativa. Acta Biochim Biophys Sin (Shanghai) 2007; 39:829 - 34; http://dx.doi.org/10.1111/j.1745-7270.2007.00346.x; PMID: 17989873
- Girardot M, Pecquet C, Boukour S, Knoops L, Ferrant A, Vainchenker W, et al. miR-28 is a thrombopoietin receptor targeting microRNA detected in a fraction of myeloproliferative neoplasm patient platelets. Blood 2010; 116:437 - 45; http://dx.doi.org/10.1182/blood-2008-06-165985; PMID: 20445018
- Lee I, Ajay SS, Yook JI, Kim HS, Hong SH, Kim NH, et al. New class of microRNA targets containing simultaneous 5′-UTR and 3′-UTR interaction sites. Genome Res 2009; 19:1175 - 83; http://dx.doi.org/10.1101/gr.089367.108; PMID: 19336450
- Lytle JR, Yario TA, Steitz JA. Target mRNAs are repressed as efficiently by microRNA-binding sites in the 5′ UTR as in the 3′ UTR. Proc Natl Acad Sci U S A 2007; 104:9667 - 72; http://dx.doi.org/10.1073/pnas.0703820104; PMID: 17535905
- Moretti F, Thermann R, Hentze MW. Mechanism of translational regulation by miR-2 from sites in the 5′ untranslated region or the open reading frame. RNA 2010; 16:2493 - 502; http://dx.doi.org/10.1261/rna.2384610; PMID: 20966199
- Ørom UA, Nielsen FC, Lund AH. MicroRNA-10a binds the 5’UTR of ribosomal protein mRNAs and enhances their translation. Mol Cell 2008; 30:460 - 71; http://dx.doi.org/10.1016/j.molcel.2008.05.001; PMID: 18498749
- Schnall-Levin M, Zhao Y, Perrimon N, Berger B. Conserved microRNA targeting in Drosophila is as widespread in coding regions as in 3’UTRs. Proc Natl Acad Sci U S A 2010; 107:15751 - 6; http://dx.doi.org/10.1073/pnas.1006172107; PMID: 20729470
- Zhou X, Duan X, Qian J, Li F. Abundant conserved microRNA target sites in the 5′-untranslated region and coding sequence. Genetica 2009; 137:159 - 64; http://dx.doi.org/10.1007/s10709-009-9378-7; PMID: 19578934
- Birney E, Andrews D, Caccamo M, Chen Y, Clarke L, Coates G, et al. Ensembl 2006. Nucleic Acids Res 2006; 34:Database issue D556 - 61; http://dx.doi.org/10.1093/nar/gkj133; PMID: 16381931
- Durinck S, Moreau Y, Kasprzyk A, Davis S, De Moor B, Brazma A, et al. BioMart and Bioconductor: a powerful link between biological databases and microarray data analysis. Bioinformatics 2005; 21:3439 - 40; http://dx.doi.org/10.1093/bioinformatics/bti525; PMID: 16082012
- Lehnert S, Van Loo P, Thilakarathne PJ, Marynen P, Verbeke G, Schuit FC. Evidence for co-evolution between human microRNAs and Alu-repeats. PLoS One 2009; 4:e4456; http://dx.doi.org/10.1371/journal.pone.0004456; PMID: 19209240
- Smalheiser NR, Torvik VI. Alu elements within human mRNAs are probable microRNA targets. Trends Genet 2006; 22:532 - 6; http://dx.doi.org/10.1016/j.tig.2006.08.007; PMID: 16914224
- Zhang R, Wang YQ, Su B. Molecular evolution of a primate-specific microRNA family. Mol Biol Evol 2008; 25:1493 - 502; http://dx.doi.org/10.1093/molbev/msn094; PMID: 18417486
- Smalheiser NR, Torvik VI. Complications in mammalian microRNA target prediction. Methods Mol Biol 2006; 342:115 - 27; PMID: 16957371
- Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O, Walichiewicz J. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet Genome Res 2005; 110:462 - 7; http://dx.doi.org/10.1159/000084979; PMID: 16093699
- Kohany O, Gentles AJ, Hankus L, Jurka J. Annotation, submission and screening of repetitive elements in Repbase: RepbaseSubmitter and Censor. BMC Bioinformatics 2006; 7:474; http://dx.doi.org/10.1186/1471-2105-7-474; PMID: 17064419
- de Koning AP, Gu W, Castoe TA, Batzer MA, Pollock DD. Repetitive elements may comprise over two-thirds of the human genome. PLoS Genet 2011; 7:e1002384; http://dx.doi.org/10.1371/journal.pgen.1002384; PMID: 22144907
- Smalheiser NR, Torvik VI. A population-based statistical approach identifies parameters characteristic of human microRNA-mRNA interactions. BMC Bioinformatics 2004; 5:139; http://dx.doi.org/10.1186/1471-2105-5-139; PMID: 15453917
- Griffiths-Jones S, Bateman A, Marshall M, Khanna A, Eddy SR. Rfam: an RNA family database. Nucleic Acids Res 2003; 31:439 - 41; http://dx.doi.org/10.1093/nar/gkg006; PMID: 12520045
- Griffiths-Jones S, Moxon S, Marshall M, Khanna A, Eddy SR, Bateman A. Rfam: annotating non-coding RNAs in complete genomes. Nucleic Acids Res 2005; 33:Database issue D121 - 4; http://dx.doi.org/10.1093/nar/gki081; PMID: 15608160