Abstract
DNA methylation is one of the best studied epigenetic modifications observed in prokaryotes as well as eukaryotes. It affects nearby gene expression. Most DNA methylation reactions in prokaryotes are catalyzed by a DNA methyltransferase, the modification enzyme of a restriction-modification (RM) system. Its target recognition domain (TRD) recognizes a specific DNA sequence for methylation. In this commentary, we review recent evidence for movement of TRDs between non-orthologous genes and movement within a gene. These movements are likely mediated by DNA recombination machinery, and are expected to alter the methylation status of a genome. Such alterations potentially lead to changes in global gene expression pattern and various phenotypes. The targets of natural selection in adaptive evolution might be these diverse methylomes rather than diverse genome sequences, the target according to the current paradigm in biology. This “epigenetics-driven adaptive evolution” hypothesis can explain several observations in the evolution of prokaryotes and eukaryotes.
Roles of Epigenetic DNA Methylation and Its Diversity
Among the possible epigenetic modifications of genes, DNA methylation has been well studied both in eukaryotes and prokaryotes. Recent innovations in genome sequencing technology have led to detection of methylated bases even in large genomes and have revealed relationships between DNA methylation and gene expression regulation among others.Citation1 The various roles of DNA methylation have been well studied in model prokaryotes. For example, methylation status of target sites is used to switch between on/off states for nearby gene expression. Differential recognition between fully-methylated sites and hemi-methylated sites are utilized for strand discrimination during mismatch repair in Escherichia coli.Citation2 Methylation status around the genome replication origin leads to regulation of replication in Caulobacter crescentus.Citation2
DNA methylation in prokaryotes is often performed by a DNA methyltransferase, which forms a restriction-modification (RM) system.Citation3 RM systems show methyltransferase (modification) activity and restriction enzyme activity, where the restriction enzyme cuts unmethylated DNA. Both activities show high specificity with respect to DNA sequence recognition. Most of them recognize specific four- to eight-bp long target sequence, unlike eukaryotic DNA methyltransferases that recognize two to three nucleotides.Citation1
Because DNA methylation affects gene expression, its changes may lead to changes in cell physiology and contribute to adaptive evolution.
Such changes can involve destruction and reconstruction of constituent genes by various mutations, especially frameshift mutations. Length variation of single nucleotide repeats within an open reading frame may lead to phase variation of modification genes,Citation4 bringing about global changes in the transcriptome.
Changes in expression of RM genes may lead to diversity in the level of DNA methylation at target sequences for a particular genome. A modification enzyme or special regulatory protein may work as a transcriptional regulator of a RM system. For example, reverse promoters and antisense RNA within coding regions affect gene expression.Citation5 The promoter region of RM systems can include its own target sequence of the systems, and the methylation of promoter region leads to repression of expression.Citation6,Citation7
Changes in the repertoire of RM systems within a genome may result from horizontal gene transfer. By genome context comparison analysis, many RM systems are found to be linked with genes of mobile genetic elements such as those for transposases and integrases or are found to be in a transposon-like structure without transposases.Citation8 Diversity in RM system repertoire has been observed through intraspecific genome comparisons, for example, in Helicobacter pylori and Neisseria meningitidis.Citation9,Citation10 These observations suggest the gain and loss of RM genes by horizontal transfer.
The target sequence of an RM system is primarily determined by the region within a gene called the target recognition domain (TRD).Citation11 TRD sequence variation among the same ortholog group within a species is observed in the Type III mod gene (methyltransferase gene) and likely corresponds to variation in the target sequence.Citation12 Diversity of TRD sequences of Type I S (specificity) genes, composed of two TRD regions, is also well known.Citation13-Citation15 A genome inversion between two Type I S genes in a head-to-head manner causes swapping of TRD sequences between the genes.Citation16,Citation17 The previously discovered allelic diversity of TRD regions were, however, only observed between the same orthologous group or between the same domain sites.
In this commentary, we discuss two novel TRD movement mechanisms that may lead to changes in the DNA methylation target sequence.Citation18,Citation19 These mechanisms may increase variety of target DNA sequences and may also be responsible for diversity in global gene expression patterns.
Change in Sequence Specificity of DNA Methylation by Movement of Target Recognition Domains
By comparison of RM systems in various strains of Helicobacter pylori, we found two novel mechanisms for movement of the associated TRDs: movement between non-orthologous Type III mod genes and movement between different domain sites of Type I S genes. Both mechanisms utilize DNA recombination at sequences flanking TRD regions.
TRD movement between non-orthologous Type III mod genes
H. pylori has five orthologs of Type III mod genes at five different loci, four of which have large allelic diversity in the TRD region.Citation12,Citation19,Citation20 We found that TRDs with the same or nearly same amino acid sequence are shared by different orthologs (). For example, TRD homology group A is observed in loci 1 and 3; TRD C in locus 1 and 3 and TRD D in loci 1, 2 and 4. Sequences outside of the TRD are not well conserved between the orthologs, so how can this TRD sharing occur?
Figure 1. Movement of target DNA recognition domains between non-orthologous genes of Type III mod genes. (A) Gene organization in mod genes. TRD, target recognition domain. Roman numerals, amino-acid sequence motifs conserved among m6A DNA methyltransferases. (B) A likely process of the movement of target recognition domains: DNA recombination at conserved DNA sequences flanking the target recognition domain that encode the conserved amino-acid motifs. (C) Repertoire of orthologs of mod genes in global strains of Helicobacter pylori. Members of the same homology group of target recognition domains are in the same color. Small vertical bars in green and small vertical bars in orange: start codon and stop codon generated by frameshift mutations. Modified from Furuta et al.Citation19
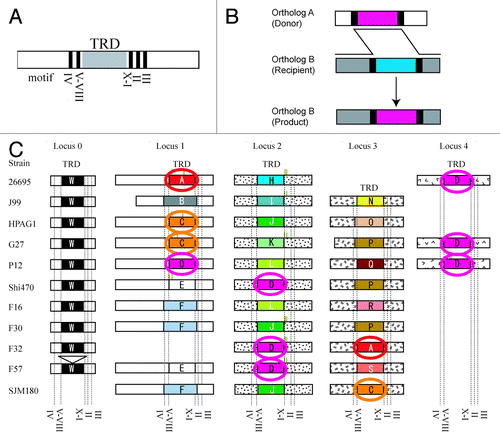
Sequence comparison revealed that the TRD region in all of the orthologs is flanked by amino-acid sequence motifs conserved among DNA methyltransferasesCitation21 (). The DNA sequences encoding these motifs have weak sequence similarity and seem to be subjects for DNA recombination during the movement of TRD region between non-orthologous genes ().
We also found similar TRD sequences in different ortholog genes for other Helicobacter species. This TRD mobility mechanism may occur not only within a species but also between species.Citation19 Notably, some of these TRD homology groups are found in various classes such as Bacilli, Clostridia, Fusobacteria in addition to most of Proteobacteria.Citation19
TRD movement between two domain sites within a Type I S gene and within a Type IIG S gene
A Type I RM system consists of three genes: restriction, modification and specificity. The specificity gene is known to determine the recognition sequence of the whole Type I RM system and is essential for both restriction and modification enzyme activities.Citation3 The specificity gene has two TRD sites, TRD1 and TRD2, each of which recognizes one half of a bipartite recognition sequence (). H. pylori has four orthologs (six loci in total) of Type I S genes. In three of them (five loci in total), TRD1 and TRD2 are flanked by the same pair of short sequences (). For these three orthologs, we found that a TRD sequence can move between TRD1 and TRD2 sites within a gene (). For example, TRD homology groups labeled a, b, c, d and f are observed at both the TRD1 site and the TRD2 site of the gene on locus 1. The amino acid sequences of the same TRD homology groups are identical or almost identical to each other. This movement likely occurred by DNA recombination at the flanking sequences (). This novel process was designated as Domain Movement (DoMo).Citation18
Figure 2. Domain movement (DoMo) between two domain sites within a gene for the specificity subunit of Type I RM systems. (A) Organization of the specificity (S) gene. TRD1 and TRD2 recognize a 5′ half site and 3′ half site, respectively. Copy number of tandem repeats in the middle striped region defines the distance between the two half sites. (B) A likely process of replacement of TRD sequences by DNA recombination between the flanking sequences, x and y. (C) A likely process of domain movement by recombination between the flanking sequences, x and y. (D) Repertoire of S genes in two loci of global strains of H. pylori. The number in the central white box indicates copy number of the tandem repeat sequences. A white circle indicates a start codon, whereas a black circle indicates a stop codon. Modified from Furuta et al.Citation18
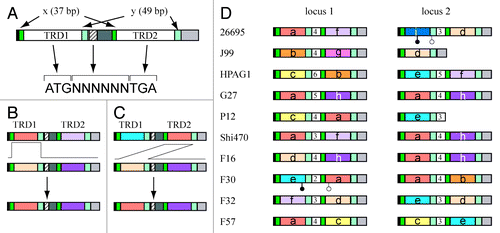
TRD movement also took place between genes at different loci presumably taking advantage of the similarity of the flanking sequences. For example, TRD homology groups labeled a, b, c, d, e, f and h are shared by locus 1 and locus 2 ().
Domain Movement was also found for a S gene of Type IIG restriction-modification system in H. pylori.Citation18
Differences from Other Gene Diversification Mechanisms
Various mechanisms for changing gene sequences are known. For example, exon shuffling and alternative splicing observed in eukaryotes result in changes to domain combinations.Citation22,Citation23 Recruitment of domain sequences from pseudogenes at a different locus by gene conversion is a well-known mechanism for antigenic variation of cell surface component genes in several bacteria.Citation24-Citation26 The uniqueness of the mechanisms we have discovered is in movement between different intragenic sites, either inter-locus or intra-locus. Our mechanisms occur without exon-intron structure or presumably other mobile genetic elements. Domain Movement found for TRDs in Type I S genes occurs between two domains within a gene in contrast to gene conversion involving two genes. Therefore Domain Movement represents a novel mechanism for gene/protein alteration.
Adaptive Evolution through Alteration in DNA Methylation Specificity?
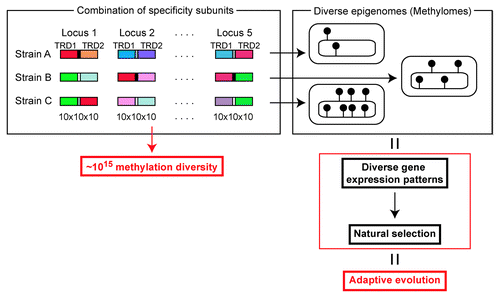
What is the biological significance of these gene mechanisms for TRD movement? They very likely lead to alteration and diversification of DNA sequences targeted for methylation. Mechanisms we found may lead to drastic variations of target recognition sequences per lineage. Especially in the case of Domain Movement for Type I S genes, each locus has about 10 types of TRD sequences that can be present at two sites. If we suppose that repeat length between the two domain sites has 10 variations, each locus has 10 × 10 × 10 = 103 diversity in recognition sequence. H. pylori carries Type I S genes in up to five different loci. Therefore the overall methylation sequence diversity totals to 1015 (). One genome sequence can take any one of these 1015 methylome states.
Figure 3. Epigenetics-driven adaptive evolution, a hypothesis. Movements of target DNA recognition domains generate a wide diversity in sequence specificity in a DNA methyltransferase at one locus. Combination of DNA methyltransferases of multiple loci results in huge overall diversity in DNA sequences to be methylated. If one locus can show 1000 DNA sequence specificities, five such loci would generate 1015 specificities in DNA methylation. One genome sequence may take one of a huge number of epigenome states differing in DNA methylation pattern. Each of these epigenomes (methylomes) may define a specific pattern of global gene expression and a specific set of phenotypic traits. The diverse epigenomes may be the target of natural selection in adaptive evolution. See text for evidence and further detail.
Recent innovation in genome sequencing has made it possible to reveal genome methylation at the single nucleotide resolution.Citation27 This technology may be used for determination of methylome status in vivo and for comparison of diverse methylomes in various strains. The level of expression of genes involved in DNA methylation is another potentially important factor affecting methylome diversity. Although transcription of all S loci in an H. pylori strain was confirmed by transcriptome analysis,Citation28 methylation activity of them must be confirmed by the methylome analysis.
DNA methylation affects nearby transcription. Therefore diversification of methylation specificity should lead to diversification of a cell’s transcriptome. Each epigenome (methylome) may correspond to a specific transcriptome and a specific set of phenotypes. These diverse epigenomes may provide targets of natural selection in adaptation. The paradigm for adaptive evolution in current biology is “selection from preformed genome diversity.” We propose that the epigenome diversity is at least as important as the genome diversity and can provide targets of selection.
Furthermore, alteration of expression of restriction-modification systems discussed above suggests that epigenome variation might be inducible by environmental and internal factors. Adaptive evolution is now often explained by selection from genome sequence variants, but several phenomena are difficult to explain by this paradigm.Citation29 For example, it is difficult to explain by current paradigm about the crossing of a valley on the fitness landscape during acquisition of complex adaptive traits. This “epigenetics-driven adaptive evolution” hypothesis may overcome these difficulties in the current paradigm in evolution and explain recent findings in the epigenetics of various forms of life.Citation1,Citation2
Acknowledgments
We are grateful to David Dryden for comments on the manuscript and Jacob Albritton for editing grammar, style and contents. This work was supported by the Grants-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (21370001 to I.K., 24790412 to Y.F.) and from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) (24113506 to I.K., 24119503 to Y.F.); the Global COE Project of Genome Information Big Bang from MEXT to I.K.; Grant in Promotion of Basic Research Activities for Innovative Biosciences from Bio-oriented Technology Research Advance Institution to I.K. and the Takeda Science Foundation to Y.F.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Suzuki MM, Bird A. DNA methylation landscapes: provocative insights from epigenomics. Nat Rev Genet 2008; 9:465 - 76; http://dx.doi.org/10.1038/nrg2341; PMID: 18463664
- Wion D, Casadesús J. N6-methyl-adenine: an epigenetic signal for DNA-protein interactions. Nat Rev Microbiol 2006; 4:183 - 92; http://dx.doi.org/10.1038/nrmicro1350; PMID: 16489347
- Roberts RJ, Belfort M, Bestor T, Bhagwat AS, Bickle TA, Bitinaite J, et al. A nomenclature for restriction enzymes, DNA methyltransferases, homing endonucleases and their genes. Nucleic Acids Res 2003; 31:1805 - 12; http://dx.doi.org/10.1093/nar/gkg274; PMID: 12654995
- Fox KL, Srikhanta YN, Jennings MP. Phase variable type III restriction-modification systems of host-adapted bacterial pathogens. Mol Microbiol 2007; 65:1375 - 9; http://dx.doi.org/10.1111/j.1365-2958.2007.05873.x; PMID: 17714447
- Mruk I, Liu Y, Ge L, Kobayashi I. Antisense RNA associated with biological regulation of a restriction-modification system. Nucleic Acids Res 2011; 39:5622 - 32; http://dx.doi.org/10.1093/nar/gkr166; PMID: 21459843
- Beletskaya IV, Zakharova MV, Shlyapnikov MG, Semenova LM, Solonin AS. DNA methylation at the CfrBI site is involved in expression control in the CfrBI restriction-modification system. Nucleic Acids Res 2000; 28:3817 - 22; http://dx.doi.org/10.1093/nar/28.19.3817; PMID: 11000275
- Christensen LL, Josephsen J. The methyltransferase from the LlaDII restriction-modification system influences the level of expression of its own gene. J Bacteriol 2004; 186:287 - 95; http://dx.doi.org/10.1128/JB.186.2.287-295.2004; PMID: 14702296
- Furuta Y, Abe K, Kobayashi I. Genome comparison and context analysis reveals putative mobile forms of restriction-modification systems and related rearrangements. Nucleic Acids Res 2010; 38:2428 - 43; http://dx.doi.org/10.1093/nar/gkp1226; PMID: 20071371
- Budroni S, Siena E, Dunning Hotopp JC, Seib KL, Serruto D, Nofroni C, et al. Neisseria meningitidis is structured in clades associated with restriction modification systems that modulate homologous recombination. Proc Natl Acad Sci U S A 2011; 108:4494 - 9; http://dx.doi.org/10.1073/pnas.1019751108; PMID: 21368196
- Vale FF, Mégraud F, Vítor JM. Geographic distribution of methyltransferases of Helicobacter pylori: evidence of human host population isolation and migration. BMC Microbiol 2009; 9:193; http://dx.doi.org/10.1186/1471-2180-9-193; PMID: 19737407
- Klimasauskas S, Nelson JL, Roberts RJ. The sequence specificity domain of cytosine-C5 methylases. Nucleic Acids Res 1991; 19:6183 - 90; http://dx.doi.org/10.1093/nar/19.22.6183; PMID: 1659688
- Srikhanta YN, Fox KL, Jennings MP. The phasevarion: phase variation of type III DNA methyltransferases controls coordinated switching in multiple genes. Nat Rev Microbiol 2010; 8:196 - 206; http://dx.doi.org/10.1038/nrmicro2283; PMID: 20140025
- Andres S, Skoglund A, Nilsson C, Krabbe M, Björkholm B, Engstrand L. Type I restriction-modification loci reveal high allelic diversity in clinical Helicobacter pylori isolates. Helicobacter 2010; 15:114 - 25; http://dx.doi.org/10.1111/j.1523-5378.2010.00745.x; PMID: 20402814
- Tsuru T, Kawai M, Mizutani-Ui Y, Uchiyama I, Kobayashi I. Evolution of paralogous genes: Reconstruction of genome rearrangements through comparison of multiple genomes within Staphylococcus aureus.. Mol Biol Evol 2006; 23:1269 - 85; http://dx.doi.org/10.1093/molbev/msk013; PMID: 16601000
- Waldron DE, Lindsay JA. Sau1: a novel lineage-specific type I restriction-modification system that blocks horizontal gene transfer into Staphylococcus aureus and between S. aureus isolates of different lineages. J Bacteriol 2006; 188:5578 - 85; http://dx.doi.org/10.1128/JB.00418-06; PMID: 16855248
- Cerdeño-Tárraga AM, Patrick S, Crossman LC, Blakely G, Abratt V, Lennard N, et al. Extensive DNA inversions in the B. fragilis genome control variable gene expression. Science 2005; 307:1463 - 5; http://dx.doi.org/10.1126/science.1107008; PMID: 15746427
- Dybvig K, Sitaraman R, French CT. A family of phase-variable restriction enzymes with differing specificities generated by high-frequency gene rearrangements. Proc Natl Acad Sci U S A 1998; 95:13923 - 8; http://dx.doi.org/10.1073/pnas.95.23.13923; PMID: 9811902
- Furuta Y, Kawai M, Uchiyama I, Kobayashi I. Domain movement within a gene: a novel evolutionary mechanism for protein diversification. PLoS One 2011; 6:e18819; http://dx.doi.org/10.1371/journal.pone.0018819; PMID: 21533192
- Furuta Y, Kobayashi I. Movement of DNA sequence recognition domains between non-orthologous proteins. Nucleic Acids Res 2012; 40:9218 - 32; http://dx.doi.org/10.1093/nar/gks681; PMID: 22821560
- Srikhanta YN, Gorrell RJ, Steen JA, Gawthorne JA, Kwok T, Grimmond SM, et al. Phasevarion mediated epigenetic gene regulation in Helicobacter pylori.. PLoS One 2011; 6:e27569; http://dx.doi.org/10.1371/journal.pone.0027569; PMID: 22162751
- Malone T, Blumenthal RM, Cheng X. Structure-guided analysis reveals nine sequence motifs conserved among DNA amino-methyltransferases, and suggests a catalytic mechanism for these enzymes. J Mol Biol 1995; 253:618 - 32; http://dx.doi.org/10.1006/jmbi.1995.0577; PMID: 7473738
- Patthy L. Genome evolution and the evolution of exon-shuffling--a review. Gene 1999; 238:103 - 14; http://dx.doi.org/10.1016/S0378-1119(99)00228-0; PMID: 10570989
- Keren H, Lev-Maor G, Ast G. Alternative splicing and evolution: diversification, exon definition and function. Nat Rev Genet 2010; 11:345 - 55; http://dx.doi.org/10.1038/nrg2776; PMID: 20376054
- Barbour AG. Antigenic variation of a relapsing fever Borrelia species. Annu Rev Microbiol 1990; 44:155 - 71; http://dx.doi.org/10.1146/annurev.mi.44.100190.001103; PMID: 2252381
- Brayton KA, Palmer GH, Lundgren A, Yi J, Barbet AF. Antigenic variation of Anaplasma marginale msp2 occurs by combinatorial gene conversion. Mol Microbiol 2002; 43:1151 - 9; http://dx.doi.org/10.1046/j.1365-2958.2002.02792.x; PMID: 11918803
- Haas R, Meyer TF. The repertoire of silent pilus genes in Neisseria gonorrhoeae: evidence for gene conversion. Cell 1986; 44:107 - 15; http://dx.doi.org/10.1016/0092-8674(86)90489-7; PMID: 2866848
- Clark TA, Murray IA, Morgan RD, Kislyuk AO, Spittle KE, Boitano M, et al. Characterization of DNA methyltransferase specificities using single-molecule, real-time DNA sequencing. Nucleic Acids Res 2012; 40:e29; http://dx.doi.org/10.1093/nar/gkr1146; PMID: 22156058
- Sharma CM, Hoffmann S, Darfeuille F, Reignier J, Findeiss S, Sittka A, et al. The primary transcriptome of the major human pathogen Helicobacter pylori. Nature 2010; 464:250 - 5; http://dx.doi.org/10.1038/nature08756; PMID: 20164839
- Whitlock MC, Phillips PC, Moore FBG, Tonsor SJ. Multiple fitness peaks and epistasis. Annu Rev Ecol Syst 1995; 26:601 - 29; http://dx.doi.org/10.1146/annurev.es.26.110195.003125