
Abstract
MicroRNAs (miRNAs) constitute a recently discovered class of noncoding RNAs that play key roles in the regulation of gene expression. Despite being only ~20 nucleotides in length, these highly versatile molecules have been shown to play pivotal roles in development, basic cellular metabolism, apoptosis, and disease. While over 24,000 miRNAs have been characterized since they were first isolated in mammals in 2001, the functions of the majority of these miRNAs remain largely undescribed. That said, many now suggest that characterization of the relationships between miRNAs and transposable elements (TEs) can help elucidate miRNA functionality. Strikingly, over 20 publications have now reported the initial formation of thousands of miRNA loci from TE sequences. In this review we chronicle the findings of these reports, discuss the evolution of the field along with future directions, and examine how this information can be used to ascertain insights into miRNA transcriptional regulation and how it can be exploited to facilitate miRNA target prediction.
Introduction
Functional roles for microRNAs have now been described in virtually every basic biological process including (in part): control of the cell cycle, the regulation of apoptosis, insulin production, lipid metabolism, hypoxia response, immune regulation and viral defense.Citation1 Furthermore, patterns of miRNA expression are highly regulated both spatially and temporally during embryonic development, which suggests that these molecules play key roles in cell fate determination and the differentiation and maintenance of tissue identity.Citation2 Also of note, differential expressions of miRNAs have been found to be characteristically associated with a number of pathologies (e.g., various types of cancer, cardiovascular disease and neurological disorders) with some of these altered miRNA expressions actually playing causal roles in particular malignancies.Citation3 As a result, miRNAs are progressively becoming the focus of considerable research as the potential for their use as diagnostic and prognostic biomarkers, therapeutic targets, and as regulators of basic cellular metabolism continue to advance.
As we propose that determining the genomic events initially giving rise to miRNAs can provide novel insight into their individual functions and regulations, this review will focus specifically on the relationship between miRNAs and TEs and summarize the mounting body of evidence suggesting that the majority of miRNAs were initially formed from TE sequences. Also of note, although beyond the scope of this review, in addition to the relationship between TEs and miRNAs, numerous other functional relationships between various TEs and noncoding RNAs have now been documented. This suggests that integral sequence-based relationships between TEs and noncoding RNAs (ncRNAs) are perhaps more prevalent than initially appreciated (e.g., Alu-mediated turnover of long non-coding RNAs (lncRNAs), long-terminal repeat (LTR) TEs providing regulatory sequences for long intergenic non-coding RNAs (lincRNAs), and the formation of other types of short noncoding RNAs (endogenous siRNAs and piRNAs) from TE sequences (for a general review see Hadjiargyrou 2013Citation4).
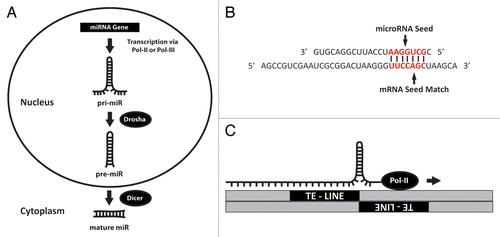
Before examining how a miRNA locus can be initially formed from TE sequences, we must first detail the basic steps involved with miRNA expression. MiRNA biogenesis typically begins with expression of an initial miRNA transcript that is several thousand nucleotides (nt) in length.Citation5 Next, these long RNA molecules are processed in the nucleus by Drosha to generate a ~70 nt stem loop known as a pre-miRNA that is exported to the cytoplasm once excised. After arriving in the cytoplasm, pre-miRNAs enter the RNA interference pathway where DICER cleaves and denatures these stem loops producing the final, mature single stranded miRNAs, now ~20 nt in sizeCitation6 (). Once ready, these functionally mature miRNAs typically engage in the regulation of gene expression by binding to complementary base pairs in the 3′UTRs of target mRNAs, typically resulting in gene silencing through either repressing translation or triggering mRNA degradation.Citation1 Interestingly while the mechanism of miRNA biogenesis has been fairly well described, the most integral facet of miRNA functionality, the specific mRNAs they target, continues to be elusive. Numerous groups have attempted to tackle this issue by devising various strategies for alignment-based search algorithms to identify targets, but these efforts have met with only limited success. To date, no generally accepted strategy for miRNA target prediction has been broadly embraced by the miRNA research community, primarily due to the inefficiency of current methods largely arising from the ability of miRNAs to bind target mRNAs with only a few nucleotides of sequence complementarityCitation8 ().
Figure 1. MiRNA biogenesis and mechanism of origination. (A) Synthesis of microRNAs begins in the nucleus when miRNA genes are transcribed via Pol-II or Pol-III into a precursor (pri-miRNA) molecule that is several hundred nucleotides in length. Subsequent processing of this transcript by Drosha results in a stem loop ~70 nts in length known as a “pre-miRNA”. This RNA hairpin is then exported into the cytoplasm where it is trimmed by Dicer into a functional, mature ~22 nt miRNA. (B) MiRNA mediated gene regulation typically requires base pairing between a specific region within the miRNA (generally referred to as a “seed” comprising nucleotides 2 through 8) and a complimentary “seed match” region in the mRNA. Base pairing in this figure is indicated by bold vertical lines. The relevant regions of the miRNA and mRNA are shown in red. (C) As reported in this review, it is now known that the molecular origins of many miR loci are a result of TE insertions into adjacent positions within the genome. The cartoon depicts a pri-miR transcript being generated from transcription across such an area of converging TEs. The arrow indicates the direction of Pol-II transcription as it reads through a leading strand LINE element into a neighboring negative strand containing the same TE. As shown it is evident how such activity would result in the formation of a RNA hairpin that could then be processed via the mechanism illustrated in (A). Figure adapted from reference Citation7.
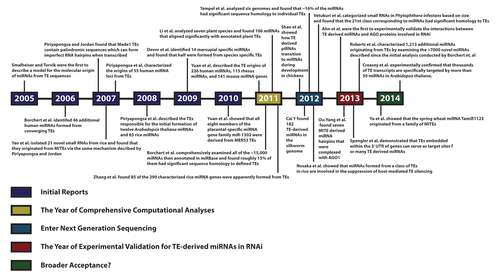
Importantly, several groups now suggest that a fundamental insight into how miRNAs were originally formed (first provided in 2005 when Smalheiser and TorvikCitation9 hypothesized that miRNA hairpins were formed as a result of the insertion of two similar transposable elements into the same genomic locus) can be exploited to help elucidate miRNA functionality. In their initial report, Smalheiser and Torvik showed that transcription across a juxtaposition of converging TEs followed by RNAi processing initially led to the formation of several functional miRNAs (). While this relationship between miRNAs and TEs was largely underappreciated when it was first proposed almost a decade ago, the general model for initial miRNA genomic locus formation from TEs has since been corroborated by numerous independent reports and is now becoming generally accepted as the mechanism responsible for the formation of thousands of distinct miRNAs in plants, animals and fungi. With that in mind this review will consist of a summary chronicle of the body of reports now independently supporting the initial formation of microRNAs from transposable element sequences (outlined in ) followed by a discussion on the evolution of the field, current and future directions, and an in depth examination of the utility of this information in elucidating microRNA function.
Figure 2. Timeline illustrating published reports of microRNAs originating from TEs. Beginning in 2005 with the first description of the mechanism by which adjacent TE insertions could result in miRNA formation, 23 papers spanning almost a decade are listed in chronological order including the most recent publications as of this writing. Colors corresponding to the categories of research progress as used in this review are shown for clarity and reference.
MiRNAs Found to Have Been Formed from TE Sequences: A chronology
2005–2010: Initial reports
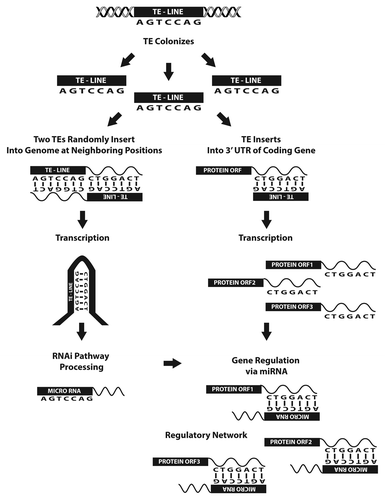
In 2005 Smalheiser and TorvikCitation9 were the first to describe a model for the molecular origin of miRNAs from TE sequences. Their initial examination of human, mouse, and rat miRNA loci identified 11 miRNA hairpins readily shown to have been initially formed from various repetitive sequences (LINES, SINEs, LTRs and simple repeats) via the model depicted in . In addition, the authors went on to show that these miRNAs were also complementary to sequences in a large number of mRNAs that contained related TE sequences in their 3′ UTRs, leading them to hypothesize that miRNA targets might also have arisen from TE sequences. Furthermore, in a related report the following year, this same group demonstrated that highly conserved Alu elements within the 3′ UTRs of many human mRNAs bare complementarity to 30 distinct human miRNAs,Citation10 providing further evidence for the initial development of miRNA mediated regulatory networks based on TE-derived targets (as illustrated in ).
Figure 3. Cartoon depicting the cellular events responsible for the formation of many miRNAs as well as the network of genes they regulate. As described in , random TE insertions into the genome at neighboring positions can lead to the formation of miRNAs. During the extensive period of time it would take for this event to occur the same TE also likely inserted into noncoding regions of protein coding transcripts elsewhere in the genome. As illustrated, this series of events can result in the formation of a network of genes capable of regulation by the TE-derived miRNA. Figure adapted from reference Citation7.
A more comprehensive study in 2006 by Borchert et al.,Citation11 utilizing the same basic methodology as Smalheiser and Torvik,Citation9 was able to identify 46 additional human miRNAs formed from converging TEs by including an examination of the sequences immediately flanking miRNA hairpins. In addition to expanding the repertoire of miRNA formations from converging TEs, this group also identified 43 additional miRNAs apparently processed from hairpins found in the 3′ tails of transcribed Alu repeats clustered on human chromosome 19, suggesting the existence of second, unresolved mechanism for microRNA formation from TE sequences.
In 2007, Piriyapongsa and JordanCitation12 comprehensively investigated the relationship between the seven members of the hsa-miR-548 family and the Made1 MITE (miniature inverted repeat transposable element) sequence believed to be responsible for their initial formation. Although the mechanism behind the formation of the hsa-miR-548 loci was not directly addressed in the report by Borchert et al., Piriyapongsa and Jordan recognized that the origin of these miRNAs did not agree with Smalheiser’s model.Citation9 Instead, they found that Made1 TEs contain palindromic sequences that form imperfect RNA hairpins when transcribed and suggested that the initial creation of these miRNAs occurred through a second, distinct mechanism of miRNA production from TEs similar to those Borchert et al. identified on human chromosome 19.Citation11
In a second report in 2007,Citation13 through utilizing an alternative computational strategy, Piriyapongsa et al. characterized the origins of 55 human miRNA loci from TEs, independently corroborating the findings of SmalheiserCitation9 and BorchertCitation11 as well as initially describing the origins of nine previously undescribed human miRNA loci from TEs. Moreover, as the genomes of distinct taxa are largely populated by unique TE compositionsCitation14,Citation15 and the authors identified several miRNAs formed from TEs unique to particular taxa, their findings led them to suggest that TEs are an unappreciated source of taxon-specific microRNAs. In addition, the group also went on to predict 85 novel TE-derived miRNA genes based on the conservation of hairpin forming potential within various transposable elements. Strikingly, they found 15 of these perfectly aligned with experimentally cloned miRNAs, indicating the utility of including TE sequences in searches for putative miRNAs.
Also in 2007, providing the first evidence that similar mechanisms for miRNA locus formation are responsible for the creation of miRNAs in plants, Yao et al.Citation16 isolated 21 novel small RNAs from rice. Interestingly, the hairpin precursors corresponding to these small RNAs were found to have originated from MITEs apparently via the same mechanism Piriyapongsa and JordanCitation12 described as being responsible for the formation of the miR-548 family from MITE sequences in humans. Although conjecturing that the majority of these small RNAs represented an intermediate between siRNAs and miRNAs, several of these small RNAs have now been annotated as belonging to the MITE-derived osa-miR-2121 family.Citation17
In 2008, Piriyapongsa et al.Citation18 next undertook a comprehensive examination of the genomic loci corresponding to the then characterized Oryza sativa and Arabadopsis thaliana miRNAs describing the TEs responsible for the initial formation of 12 of the Arabidopsis miRNAs (6.5%) and 83 of the rice miRNAs (35.9%). Of note, in contrast to animal miRNAs whose sequences are generally found to diverge from their progenitor elements over time and consequently not be perfectly maintained, in Arabidopsis, ten of the 12 TE-derived miRNAs and 38 of the 83 TE-derived miRNAs in rice were found to be 100% identical to consensus TEs. In addition, in this work Piriyapongsa et al. also identified several examples of individual plant TEs encoding both siRNAs and miRNAs, leading them to hypothesize (much like Yao et al.Citation16) that many miRNAs may have originally functioned as siRNAs, and that this siRNA-to-miRNA transition might illustrate how siRNAs initially charged with silencing TEs could later be exploited for additional levels of gene regulation.
In 2009, Devor et al.Citation19 performed a comprehensive characterization of the miRNAs they experimentally identified in Monodelphis domestica, including an examination of the correlation between these molecules and TEs. In addition to the 174 miRNAs they found in M. domestica that were conserved across mammals, the group also identified 14 miRNAs that were unique to marsupials, including three exclusive to M. domestica. Strikingly, when mapped against repeat data sets they found that seven (half of the lineage-specific microRNAs) aligned with TEs (six with L1/L2 LINEs and one with a Mariner DNA transposon), and that these repeat elements were themselves marsupial specific. Further analysis revealed that two of three M. domestica specific miRNAs originated from within a large genomic cluster of 39 miRNAs and that this entire region was flanked on both ends by a marsupial specific L1 element, leading the team to believe that this juxtaposition was responsible for the formation of the entire cluster of miRNAs via the mechanism first proposed by Smalheiser et al.Citation9 Importantly, by demonstrating that species-specific miRNAs arose from species-specific TEs, the authors’ report further strengthened the argument originally suggested by Piriyapongsa et al.,Citation12 in that taxon-specific TEs frequently give rise to taxon-specific miRNAs uniquely attuned to the organism, as well as further supporting the broader idea that TEs are routinely involved in the emergence of new miRNAs.
In 2010 Yuan et al.Citation20 investigated the origin and evolution of the placental-specific miRNA gene family miR-1302 and showed that all eight members of this family were derived from MER53 TEs. The group also identified 36 potential paralogs of this miRNA in the human genome and another 58 orthologs conserved across placental mammals and suggested that all of these were similarly formed from MER53 TEs.
2011: The year of comprehensive computational analyses
In 2011, Yuan et al.Citation21 published an analysis of how mammalian miRNA families originally formed from TEs uniquely expanded within three genomes (human, rhesus, and mouse). Using a novel strategy, the authors looked at the coverage density of TEs within these genomes and determined if individual miRNA genes originated from particular TEs based on sequence homology. By employing this computational methodology the group successfully described the TE origins of 226 human miRNAs, 115 rhesus miRNAs, and 141 mouse miRNA genes from various LINEs, SINEs, LTRs and DNA transposons.
Next in 2011, Borchert et al.Citation22 performed the first ever comprehensive analysis of the genomic events responsible for miRNA formation (examining all of the ~15,000 miRNAs annotated in miRBaseCitation17,Citation23 at the time) by employing a computational methodology developed to align miRNA sequences to the principle data sets for TEs and noncoding RNAs (ncRNAs). In all, the authors found that roughly 15% of analyzed miRNAs had significant sequence homology to defined TEs. The authors proposed that the majority (~89%) of these miRNAs originated via the model depicted in , with the remaining ~11% instead corresponding to characteristic hairpin-forming sequences within individual TEs (e.g., MITE sequences previously suggested to produce miRNAsCitation12). Of the 2,392 miRNAs Borchert et al. found to have TE origins, DNA transposons were most frequently responsible for miRNA generation (891). The rest originated from LTR retrotransposons (414), non-LTR retrotransposon (814), LINEs (312), SINEs (353), satellites (137) and others (136). Interestingly, sequences contained within the “other” category had significant sequence identity to known noncoding RNA sequences such as snoRNAs and tRNAs, each of which have been speculated to have contributed to the formation of novel mobile genetic elements.Citation24,Citation25 Based on their findings the authors further advanced the hypothetical proposition that miRNA based regulatory systems first arose and subsequently persisted over time as a result of the obvious advantage conveyed by the ability to regulate host genes containing portions of the TE from which a miRNA was formed (). Since these gene networks have been found in organisms as primitive as protozoa, the authors went on to speculate that miRNA based regulation of multiple genes may have been the catalyst in the evolution of more sophisticated developmental systems.
Also in 2011, Li et al.Citation26 found that a substantial number of previously described, experimentally isolated plant miRNAs were homologous to TEs as well as to TEs contained within mRNA transcripts. In all, in an examination of seven plant species the authors found 106 miRNAs aligning significantly with annotated plant TEs, and similar to the report by Zhang et al.,Citation27 the authors found that ~80% of these TE-derived miRNAs were apparently initially formed from MITE TE sequences.
Lastly in 2011, Zhang et al.Citation27 examined the origin and evolution of miRNA loci in flowering plants. After conducting genome wide analyses of Oryza sativa and Arabidopsis thaliana they described four potential molecular mechanisms responsible for the formation of miRNAs—two of which involve TEs. In particular, in agreement with the earlier work of Yao et al.,Citation16 the authors found that many of the miRNA genes in rice had notable sequence overlap with MITEs. In all, they found 85 of the 290 characterized rice miRNA genes were apparently formed from TEs, with over half of these corresponding to MITEs. Also, in agreement with previous work by SmalheiserCitation9 and Borchert,Citation11 preliminary target predictions for these miRNAs suggested that many of the TE-derived miRNAs (45 of 85) characterized in this work had target sites bearing complete homology with the same TE, giving rise to individual miRNAs. This lead the authors to propose that the transposition of this TE into other genes could yield other target sites for the miRNA to interact with and subsequently allow for the creation of regulatory systems that could enhance the evolutionary capacity of the organism (as illustrated in ).
2012: Enter next generation sequencing
In 2012 Shao et al.Citation28 employed next generation sequencing (NGS) to categorize small RNAs found in chickens and demonstrated how TE derived piRNAs transition to form miRNAs during embryonic development. They examined the expression levels of miRNAs throughout development and found that most were dynamically regulated throughout formation with many of them targeting signal transduction pathways related to reproduction and embryogenesis. It was also shown that the TEs giving rise to the initial piRNAs were the most abundant ones in the genome.
Also in 2012, Cai et al.Citation29 found numerous TE derived small RNAs (182 miRNAs) in the Bombyx mori (silkworm) genome. In the first study of its kind in the silkworm, the authors systematically discovered TE-associated small RNAs transcribed from the B. mori genome through employing a deep RNA-sequencing strategy yielding 182, 788 and 4,990 TE-associated small RNAs corresponding to miRNAs, siRNAs and piRNAs, respectively.
Next in 2012, Tempel et al.Citation30 undertook an extensive study to map all miRNA precursor sequences in miRBaseCitation17,Citation23 to several genomes in order to determine if any had overlapping sequences with TEs. They used an automated method called ncRNA classifier in order to catalog the interaction between the TEs and pre-ncRNAs. By analyzing six genomes (frog, human, mouse, nematode, rat and sea squirt) the group found that ~16% of the miRNAs (strikingly similar to the ~15% reported by Borchert et al.Citation22 in 2011) had significant sequence homology to individual MITEs, DNA transposons, LTR/ERV, CR1/RTE, L1s, SINEs, and other non-LTRs.
Also in 2012, Nosaka et al.Citation31 showed that miRNAs formed from a class of TEs in rice are involved in the suppression of host-mediated TE silencing. In plants TEs are typically suppressed epigenetically via small RNA directed DNA methylation, but in this instance the authors showed that miR-820 family members originating from CACTA DNA transposons were actually targeting and repressing one of the methyltransferase genes, OsDRM2, responsible for epigenetic suppression. This purported ability of the TE to utilize miRNAs as a countermeasure to host silencing and subsequently allow further TE insertion into the genome elucidates a novel function of TE derived miRNAs and provides insight into the possible evolutionary forces driving the relationship between TEs and miRNAs.
Lastly in 2012, Vetukuri et al.Citation32 examined data obtained from small RNA mediated RNAi processes in the fungus Phytophthora infestans, the oomycete pathogen responsible for late blight in Solanacea. They categorized the small RNAs based on size (21nt, 25/26nt, and 32nt) and found that the majority were homologous to LTR retrotransposons within the fungal genome. Notably, the 21 nt class of small RNAs corresponding to miRNAs showed the most homology to the transposable elements. Interestingly, the group also identified six putative miRNAs with characteristics of both plant and metazoan miRNAs.
2013: The year of experimental validation for TE-derived miRNAs in RNAi
In 2013 Ahn et al.Citation33 examined miRNAs specifically formed from palindromic MER (Medium Reiteration frequency) TEs in the genomes of primates, rodents, and rabbits. After identifying three specific miRNAs derived from MER96, they next experimentally validated the interactions between these MER-derived miRNAs with the catalytic AGO1, AGO2, and AGO3 proteins involved in the RNAi gene silencing pathway. Importantly, this work constituted the first ever definitive demonstration that miRNAs derived from TEs can be processed via the same RNAi mechanism as other non-TE derived miRNAs.
Also in 2013, driven by the contested authenticity of many of the annotated miRNAs in rice due to sequence homology with TEs, Ou-Yang et al.Citation34 examined the association between miRNAs originating from TEs and the RISC (RNA-induced silencing complex) proteins responsible for miRNA function. To this end, the group characterized seven miRNAs substantially corresponding to MITEs TE sequences that were complexed with AGO1 in immunoprecipitation assays, further substantiating that TE-derived miRNAs are in fact involved with RISC regulations in the same way as other silencing ncRNAs.
Next in 2013, Spengler et al.Citation35 experimentally validated that miRNAs and miRNA target sites derived from common human TEs such as LINE2, MIR, and Alus are functional. Importantly, the authors experimentally demonstrated that TEs embedded within the 3′ UTR of genes can serve as the source of target sites for many miRNAs. Specifically, 3′ UTR embedded Alus were shown to be the origin of target sites for miR-24 and miR-122, and an Alu-derived microRNA, miR-1285–1, was shown to regulate genes containing target sites with homologous Alu elements.
Finally in 2013, building on the previous comprehensive analysis by Borchert et al.,Citation22 Roberts et al.Citation7 characterized 1,213 additional miRNA origins from TE elements by examining the > 7,000 novel miRNAs described since the earlier analysis, bringing the total number of miRNA loci origins defined by this group to 3,605. In all, these studies have comprehensively defined the origins of roughly 15% of the miRNAs currently annotated in miRBase as being formed from TE sequences.
2014: Broader acceptance?
As recently as early 2014 Yu et al.Citation36 showed that the spring wheat miRNA TamiR-1123 originated from a family of MITEs. A gene involved in the vernalization of spring wheat, Vrn-A1a, contains a MITE in its promoter that is able to be transcribed into a stable RNA hairpin. This MITE also contains sequences that are homologous to TamiR-1223, and both the MITE derived RNA hairpin and TamiR-1223 were detected when a gel containing small RNAs was probed with labeled TamiR-1223. Based on this evidence, the authors concluded that TamiR-1223 originated from the MITE, and that Vrn-A1a is potentially regulated by this TE-derived microRNA.
Most recently, in an extremely exciting report in a 2014 Letter to Nature, Creasey et al.Citation37 used parallel analysis of RNA ends (PARE) sequencing to experimentally demonstrate that thousands of transposon transcripts are specifically targeted by more than 50 miRNAs for cleavage and processing by RNA-dependent RNA polymerase 6 (RDR6) in Arabidopsis thaliana.
Discussion
Taken together the reports chronicled here clearly indicate that the relationship between miRNAs and TEs is much more significant than what was originally believed when the model was first proposed in 2005. Specifically, there is now abundant evidence indicating that transposable elements are directly involved in the initial formation of an appreciable number of miRNA loci, a quantity likely much higher than currently defined due to the progressive degeneration of TEs and the limited availability of complete miRNA and TE data sets. After fully reviewing the body of these works, we suggest that a thorough understanding of the connections between specific TEs and microRNAs can be directly employed to: (Utility I) ascertain insights into miRNA transcriptional regulation, and also to (Utility II) facilitate accurate miRNA target prediction.
Utility I
Knowing the TEs giving rise to a miRNA can provide a deeper understanding into the elements controlling their expression. This was first demonstrated in 2006Citation11 when 43 human miRNAs located immediately downstream of Alu transposable elements were shown to be transcribed by RNA polymerase III (Pol-III) as the 3′ ends of distinct Alu transcripts. Amidst analyzing the set of all known human miRNA loci for TE relationships, this study described a large cluster of primate-specific miRNAs located on chromosome 19 (C19MC)Citation38 in which individual miRNAs were consistently flanked upstream (~100 bp) by intact Alu repeats. Although the accepted paradigm prior to this report was that miRNAs were exclusively transcribed by RNA polymerase II (Pol-II), this group elected to examine whether these miRNAs were actually being transcribed by Pol-III as the 3′ tails of Alu repeats as Alu elements were well documented as being transcribed by Pol-III. These researchers found that the Alus located upstream of the miRNAs in the C19MC contained the sequences necessary for Pol-III expression.Citation39,Citation40 Significantly, in each of their experimental assays (cell free transcription, expression constructs, and chromatin immunoprecipitation), the authors found that Pol-III was associated with the C19MC miRNA promoters and responsible for the expression of these miRNAs directly contradicting the assumption that miRNAs were exclusively transcribed via Pol-II. Importantly, through identifying the particular TEs responsible for the formation of the miRNA genomic loci in the C19MC, these authors obtained direct insights into the mechanisms behind the transcriptional regulation of these miRNAs, clearly illustrating the value of determining the TEs responsible for forming individual miRNA loci in ascertaining their transcriptional regulation.
Utility II
Beyond the transcriptional insights provided by knowing the genomic origins of individual miRNAs, perhaps the greatest potential utility of this information is in facilitating accurate miRNA target prediction. Several groups have now suggested that a subset of miRNAs may preferentially target TE sequences located in mRNA 3′UTRs.Citation10,Citation12,Citation35,Citation41 Based on this, Filstein et al.Citation41 recently developed a genuinely novel approach to identifying miRNA targets through speculating that a miRNA and its related mRNA target site might actually be created concurrently by the continuing mobilization of a common ancestral TE (). In their initial report this group suggested that the accuracy of target identification algorithms could be significantly improved by limiting searches to mRNAs that contain the TEs identified as being responsible for the formation of individual miRNAs. After developing a computational methodology based on this, OrBId (Origin Based Identification), the authors generated putative mRNA target sets for 191 human miRNAs with defined TE origins. While the authors found their methodology was best suited for predicting targets of taxon-specific miRNA loci formed more recently in evolutionary time, they also found this strategy was capable of successfully predicting targets for the evolutionarily older, mammalian-conserved miR-28 family—targets found to be largely in agreement with both conventional target prediction algorithmsCitation42,Citation43 and existing experimental evidence.Citation44 While further validation and a more comprehensive comparison of OrBId target sets to those generated by existing methods will be required to further substantiate this innovative target prediction strategy, this work clearly advances the idea that the mRNA targets of miRNAs with defined TE origins can successfully be predicted based on a common TE origin shared by a miRNA and target site. Importantly, this point was recently significantly corroborated in a report by Spengler et al.Citation35 (discussed in the preceding chronology) which definitively experimentally demonstrated that common human TEs such as Alu elements contained within the 3′UTRs of active genes function endogenously as miRNA target sites.
In terms of future directions of the field as a whole, since all the conclusions drawn in this review are limited by the currently available data, further advances in our understanding of the relationships between microRNAs and TEs will depend upon and advance along with novel, ongoing miRNA and TE discovery and annotation. Furthermore, we suggest that only a fraction of characterizable TE::miRNA relationships have been defined to date. First, not all microRNAs and TEs have been identified. RepBase, the most commonly used repetitive element database, and its microRNA equivalent database—miRBase—are constantly being updated and therefore new elements for examination are provided with every update to these resources.Citation15,Citation40,Citation45
Novel, ongoing miRNA
Additionally, in terms of miRNA discovery, currently annotated miRNAs are biased toward evolutionarily older and non-repetitive sequences, as it was typical of many of the initial miRNA cloning efforts to discard all sequences that were homologous to transposable elements.Citation38,Citation46 Furthermore, early miRNA cloning studies also commonly discarding pools of “tRNA degradation products”Citation38,Citation46 (e.g., short RNAs corresponding to tRNAs and snoRNAs) that were ~20 nt in size. Today, however, next generation sequencing (NGS) of RNA populations immunoprecipitating with miRNA protein complexes will likely help redefine many of these previously omitted short RNAs as functional miRNAs. Clearly, recent reports dealing with TE-derived miRNA endogenous functions such as the one by Spengler et al.Citation35 discussed above, as well as recent studies identifying tRNA and snoRNACitation26,Citation43 sequence fragments complexed with RNAi machinery, suggest that these previously overlooked short noncoding RNAs are actually being processed by the RISC machinery and participate in miRNA-like regulations. As many SINEs were initially formed from tRNA sequences,Citation26,Citation43 and snoRNAs have also been shown to propagate through genomes and increase in number by retrotransposition,Citation26,Citation43 it will be interesting to see if these tRNA- and snoRNA-derived miRNA like elements were formed through similar mechanisms and behave similarly to other TE-derived miRNAs or if they instead constitute a distinct set of small RNAs with unique properties.
TE discovery and annotation
In terms of TE discovery, identification of the elements responsible for miRNA origins (as well as their mRNA targets that arose from TE insertion into expressed sequences) becomes increasingly less accurate the farther back in evolutionary time an insertion event occurred. Given that miRNAs have been well characterized as regulators in both situations of nutrient deprivation and abiotic stress,Citation4,Citation11,Citation35,Citation38,Citation41 although harder to describe, we speculate that the cryptic beginnings of some of the more archaic miRNA loci might also be explained via TE origins. Within this model, selective pressure to preserve only the sequences required for stem loop structure, target recognition, and transcriptional regulation could account for the degradation of other noncritical components, and thus result in increased difficulty when identifying TE-derived miRNAs and their associated TE containing mRNA targets. More simply, genomes are highly plastic, and constituent sequences that provide no significant advantage are eventually degenerated (e.g., if only a 30 bp portion of a 7 kb LINE insertion conveyed a meaningful benefit, the 30 bp segment would be maintained while the remainder of the 7 kb would ultimately degrade over time). Fortunately new algorithms are being developed that focus on identifying TEs that have degenerated beyond the ability of Repeat MaskerCitation15 to identify them. One such program, Greedier,Citation47 has recently been used to facilitate the discovery of TEs across eukaryotes by taking into account the fragmentation of repeats. This method is proving particularly useful at identifying degenerate TEs within genomes that would escape identification by RepeatMasker alone. Programs such as this, or others employing alternate strategies for characterizing degenerate TE sequences, may well be utilized in future analyses to successfully characterize more ancient miRNA::TE relationships.
Beyond the relationship between miRNAs and TEs continuing to advance through novel miRNA and TE discovery, the acceptance of this relationship by the broader miRNA community is also of importance. Strikingly, it should be noted that miRNAs are not the only TE-derived small RNAs known to participate in RNAi. In fact, although not directly discussed in this review, it is widely accepted that both of the two other principle classes of short RNAs engaged in RNAi (endogenous siRNAs and piRNAs) are also processed from and correspond to transposable elements (recently reviewed by HadjiargyrouCitation4). In contrast, despite several groups having now independently published reports (as summarized here) clearly describing the genomic origins of thousands of individual microRNAs from an array of transposable element sequences, it is still common practice to discard small RNA sequence reads that readily align to TEs when attempting to experimentally identify novel miRNAs.Citation17,Citation23 We suggest this represents a significant error in the conventional microRNA discovery pipeline that should be corrected by those continuing to eliminate sequences corresponding to TEs from consideration as miRNAs.
Furthermore, beyond the functional implications of characterizing miRNA::TE relationships, these connections may well also play underappreciated roles in speciation. As an example, Filstein et al.Citation41 found the genomic origins of more recently established, taxon-specific miRNA-loci to be readily definable, characterizing 113 human miRNA genomic loci as having been formed from primate-specific Alu TEs. As they found Alu insertions into expressed regions of the human genome were responsible for the formation of numerous human miRNA loci,Citation48 the authors speculated that the continued expansion of Alus within the genome does not constitute a failure of the RNAi pathway to inhibit Alu transposition, but instead represents a beneficial genetic partnership whereby the insertion of Alu elements into noncoding sections of transcripts has culminated in minor alterations of gene expression that have ultimately led to a heightened rate of adaptation for the human genome that could potentially be in part responsible for our unique complexities in comparison to other primates.
In conclusion, while the events leading to the initial formation of many miRNA loci may never be fully characterized, the cumulative efforts of the reports summarized in this review unequivocally demonstrate that a notable percentage of miRNAs were initially formed from TE sequences, with many more likely to be identified as new miRNAs and TEs are elucidated. As these additional relationships continue to be defined, it will be interesting to see how much of an impact the development and evolution of programs like OrBId will have on our ability to identify endogenous miRNA targets, as well as how much experimental demonstrations that TE-derived microRNAs actually do participate in endogenous regulations (like those recently published in 2013 and 2014 by Ahn et al.,Citation33 Ou-Yang et al.,Citation34 Spengler et al.Citation35 and Creasey et al.Citation37) will lead the broader miRNA community to further embrace the relevance of these microRNAs and their relationship to transposable elements.
| Abbreviations: | ||
| bp | = | base pair |
| C19MC | = | chromosome 19 microRNA cluster |
| kb | = | kilobase pairs |
| lincRNA | = | long intergenic non-coding RNA |
| LINE | = | long interspersed repeated element |
| lncRNA | = | long non-coding RNA |
| LTR | = | long terminal repeat |
| MER | = | medium reiteration |
| miR | = | microRNA |
| MIR | = | mammalian wide interspersed repeat |
| miRNA | = | microRNA |
| mRNA | = | messenger RNA |
| MITE | = | miniature inverted repeat transposable elements |
| ncRNA | = | non-coding RNA |
| NGS | = | next generation sequencing |
| nt | = | nucleotide |
| OrBId | = | origin based identification of microRNA targets algorithm |
| PARE | = | parallel analysis of RNA ends |
| piRNA | = | piwi-interacting RNA |
| Pol-II | = | RNA polymerase II |
| Pol-III | = | RNA polymerase III |
| RDR6 | = | RNA-dependent RNA polymerase 6 |
| RISC | = | RNA-induced silencing complex |
| RNAi | = | RNA interference |
| SINE | = | short interspersed repeated elements |
| siRNA | = | small interfering RNA |
| snoRNA | = | small nucleolar RNA |
| TE | = | transposable element |
| tRNA | = | transfer RNA |
| UTR | = | untranslated region |
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This research was funded in part by the Department of Biology, the College of Arts and Sciences at the University of South Alabama and also supported by NSF CAREER grant 1350064 awarded by Division of Molecular and Cellular Biosciences, with co-funding provided by the NSF EPSCoR program.
References
- Kim VN. MicroRNA biogenesis: coordinated cropping and dicing. Nat Rev Mol Cell Biol 2005; 6:376 - 85; http://dx.doi.org/10.1038/nrm1644; PMID: 15852042
- He L, Hannon GJ. MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet 2004; 5:522 - 31; http://dx.doi.org/10.1038/nrg1379; PMID: 15211354
- Huang Y, Wang JP, Yu XL, Wang ZV, Xu TS, Cheng XC. [Non-coding RNAs and diseases]. Mol Biol (Mosk) 2013; 47:531 - 43; http://dx.doi.org/10.7868/S0026898413040174; PMID: 24466743
- Hadjiargyrou M, Delihas N. The Intertwining of Transposable Elements and Non-Coding RNAs. Int J Mol Sci 2013; 14:13307 - 28; http://dx.doi.org/10.3390/ijms140713307; PMID: 23803660
- Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993; 75:843 - 54; http://dx.doi.org/10.1016/0092-8674(93)90529-Y; PMID: 8252621
- Hutvágner G, Zamore PD. A microRNA in a multiple-turnover RNAi enzyme complex. Science 2002; 297:2056 - 60; http://dx.doi.org/10.1126/science.1073827; PMID: 12154197
- Roberts JT, Cooper EA, Favreau CJ, Howell JS, Lane LG, Mills JE, Newman DC, Perry TJ, Russell ME, Wallace BM, et al. Continuing analysis of microRNA origins: Formation from transposable element insertions and noncoding RNA mutations. Mob Genet Elements 2013; 3:e27755; http://dx.doi.org/10.4161/mge.27755; PMID: 24475369
- Smalheiser NR, Torvik VI. Complications in mammalian microRNA target prediction. Methods Mol Biol 2006; 342:115 - 27; PMID: 16957371
- Smalheiser NR, Torvik VI. Mammalian microRNAs derived from genomic repeats. Trends Genet 2005; 21:322 - 6; http://dx.doi.org/10.1016/j.tig.2005.04.008; PMID: 15922829
- Smalheiser NR, Torvik VI. Alu elements within human mRNAs are probable microRNA targets. Trends Genet 2006; 22:532 - 6; http://dx.doi.org/10.1016/j.tig.2006.08.007; PMID: 16914224
- Borchert GM, Lanier W, Davidson BL. RNA polymerase III transcribes human microRNAs. Nat Struct Mol Biol 2006; 13:1097 - 101; http://dx.doi.org/10.1038/nsmb1167; PMID: 17099701
- Piriyapongsa J, Jordan IK. A family of human microRNA genes from miniature inverted-repeat transposable elements. PLoS One 2007; 2:e203; http://dx.doi.org/10.1371/journal.pone.0000203; PMID: 17301878
- Piriyapongsa J, Mariño-Ramírez L, Jordan IK. Origin and evolution of human microRNAs from transposable elements. Genetics 2007; 176:1323 - 37; http://dx.doi.org/10.1534/genetics.107.072553; PMID: 17435244
- Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O, Walichiewicz J. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet Genome Res 2005; 110:462 - 7; http://dx.doi.org/10.1159/000084979; PMID: 16093699
- Kohany O, Gentles AJ, Hankus L, Jurka J. Annotation, submission and screening of repetitive elements in Repbase: RepbaseSubmitter and Censor. BMC Bioinformatics 2006; 7:474; http://dx.doi.org/10.1186/1471-2105-7-474; PMID: 17064419
- Yao C, Zhao B, Li W, Li Y, Qin W, Huang B, Jin Y. Cloning of novel repeat-associated small RNAs derived from hairpin precursors in Oryza sativa. Acta Biochim Biophys Sin (Shanghai) 2007; 39:829 - 34; http://dx.doi.org/10.1111/j.1745-7270.2007.00346.x; PMID: 17989873
- Kozomara A, Griffiths-Jones S. miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res 2011; 39:D152 - 7; http://dx.doi.org/10.1093/nar/gkq1027; PMID: 21037258
- Piriyapongsa J, Jordan IK. Dual coding of siRNAs and miRNAs by plant transposable elements. RNA 2008; 14:814 - 21; http://dx.doi.org/10.1261/rna.916708; PMID: 18367716
- Devor EJ, Peek AS, Lanier W, Samollow PB. Marsupial-specific microRNAs evolved from marsupial-specific transposable elements. Gene 2009; 448:187 - 91; http://dx.doi.org/10.1016/j.gene.2009.06.019; PMID: 19577616
- Yuan Z, Sun X, Jiang D, Ding Y, Lu Z, Gong L, Liu H, Xie J. Origin and evolution of a placental-specific microRNA family in the human genome. BMC Evol Biol 2010; 10:346; http://dx.doi.org/10.1186/1471-2148-10-346; PMID: 21067568
- Yuan Z, Sun X, Liu H, Xie J. MicroRNA genes derived from repetitive elements and expanded by segmental duplication events in mammalian genomes. PLoS One 2011; 6:e17666; http://dx.doi.org/10.1371/journal.pone.0017666; PMID: 21436881
- Borchert GM, Holton NW, Williams JD, Hernan WL, Bishop IP, Dembosky JA, Elste JE, Gregoire NS, Kim JA, Koehler WW, et al. Comprehensive analysis of microRNA genomic loci identifies pervasive repetitive-element origins. Mob Genet Elements 2011; 1:8 - 17; http://dx.doi.org/10.4161/mge.1.1.15766; PMID: 22016841
- Wang X, Liu XS. Systematic Curation of miRBase Annotation Using Integrated Small RNA High-Throughput Sequencing Data for C. elegans and Drosophila. Front Genet 2011; 2:25; http://dx.doi.org/10.3389/fgene.2011.00025; PMID: 22303321
- Weber MJ. Mammalian small nucleolar RNAs are mobile genetic elements. PLoS Genet 2006; 2:e205; http://dx.doi.org/10.1371/journal.pgen.0020205; PMID: 17154719
- Li Z, Ender C, Meister G, Moore PS, Chang Y, John B. Extensive terminal and asymmetric processing of small RNAs from rRNAs, snoRNAs, snRNAs, and tRNAs. Nucleic Acids Res 2012; 40:6787 - 99; http://dx.doi.org/10.1093/nar/gks307; PMID: 22492706
- Li Y, Li C, Xia J, Jin Y. Domestication of transposable elements into MicroRNA genes in plants. PLoS One 2011; 6:e19212; http://dx.doi.org/10.1371/journal.pone.0019212; PMID: 21559273
- Zhang Y, Jiang WK, Gao LZ. Evolution of microRNA genes in Oryza sativa and Arabidopsis thaliana: an update of the inverted duplication model. PLoS One 2011; 6:e28073; http://dx.doi.org/10.1371/journal.pone.0028073; PMID: 22194805
- Shao P, Liao JY, Guan DG, Yang JH, Zheng LL, Jing Q, Zhou H, Qu LH. Drastic expression change of transposon-derived piRNA-like RNAs and microRNAs in early stages of chicken embryos implies a role in gastrulation. RNA Biol 2012; 9:212 - 27; http://dx.doi.org/10.4161/rna.18489; PMID: 22418847
- Cai Y, Zhou Q, Yu C, Wang X, Hu S, Yu J, Yu X. Transposable-element associated small RNAs in Bombyx mori genome. PLoS One 2012; 7:e36599; http://dx.doi.org/10.1371/journal.pone.0036599; PMID: 22662121
- Tempel S, Pollet N, Tahi F. ncRNAclassifier: a tool for detection and classification of transposable element sequences in RNA hairpins. BMC Bioinformatics 2012; 13:246; http://dx.doi.org/10.1186/1471-2105-13-246; PMID: 23009561
- Nosaka M, Itoh J, Nagato Y, Ono A, Ishiwata A, Sato Y. Role of transposon-derived small RNAs in the interplay between genomes and parasitic DNA in rice. PLoS Genet 2012; 8:e1002953; http://dx.doi.org/10.1371/journal.pgen.1002953; PMID: 23028360
- Vetukuri RR, Åsman AK, Tellgren-Roth C, Jahan SN, Reimegård J, Fogelqvist J, Savenkov E, Söderbom F, Avrova AO, Whisson SC, et al. Evidence for small RNAs homologous to effector-encoding genes and transposable elements in the oomycete Phytophthora infestans. PLoS One 2012; 7:e51399; http://dx.doi.org/10.1371/journal.pone.0051399; PMID: 23272103
- Ahn K, Gim JA, Ha HS, Han K, Kim HS. The novel MER transposon-derived miRNAs in human genome. Gene 2013; 512:422 - 8; http://dx.doi.org/10.1016/j.gene.2012.08.028; PMID: 22926102
- Ou-Yang F, Luo QJ, Zhang Y, Richardson CR, Jiang Y, Rock CD. Transposable element-associated microRNA hairpins produce 21-nt sRNAs integrated into typical microRNA pathways in rice. Funct Integr Genomics 2013; 13:207 - 16; http://dx.doi.org/10.1007/s10142-013-0313-8; PMID: 23420033
- Spengler RM, Oakley CK, Davidson BL. Functional microRNAs and target sites are created by lineage-specific transposition. Hum Mol Genet 2014; 23:1783 - 93; http://dx.doi.org/10.1093/hmg/ddt569; PMID: 24234653
- Yu M, Carver BF, Yan L. TamiR1123 originated from a family of miniature inverted-repeat transposable elements (MITE) including one inserted in the Vrn-A1a promoter in wheat. Plant Sci 2014; 215-216:117 - 23; http://dx.doi.org/10.1016/j.plantsci.2013.11.007; PMID: 24388522
- Creasey KM, Zhai J, Borges F, Van Ex F, Regulski M, Meyers BC, Martienssen RA. miRNAs trigger widespread epigenetically activated siRNAs from transposons in Arabidopsis. Nature 2014; 508:411 - 5; http://dx.doi.org/10.1038/nature13069; PMID: 24670663
- Bentwich I, Avniel A, Karov Y, Aharonov R, Gilad S, Barad O, Barzilai A, Einat P, Einav U, Meiri E, et al. Identification of hundreds of conserved and nonconserved human microRNAs. Nat Genet 2005; 37:766 - 70; http://dx.doi.org/10.1038/ng1590; PMID: 15965474
- Mighell AJ, Markham AF, Robinson PA. Alu sequences. FEBS Lett 1997; 417:1 - 5; http://dx.doi.org/10.1016/S0014-5793(97)01259-3; PMID: 9395063
- Jurka J. Evolutionary impact of human Alu repetitive elements. Curr Opin Genet Dev 2004; 14:603 - 8; http://dx.doi.org/10.1016/j.gde.2004.08.008; PMID: 15531153
- Filshtein TJ, Mackenzie CO, Dale MD, Dela-Cruz PS, Ernst DM, Frankenberger EA, He C, Heath KL, Jones AS, Jones DK, et al. OrbId: Origin-based identification of microRNA targets. Mob Genet Elements 2012; 2:184 - 92; http://dx.doi.org/10.4161/mge.21617; PMID: 23087843
- Kiriakidou M, Nelson PT, Kouranov A, Fitziev P, Bouyioukos C, Mourelatos Z, Hatzigeorgiou A. A combined computational-experimental approach predicts human microRNA targets. Genes Dev 2004; 18:1165 - 78; http://dx.doi.org/10.1101/gad.1184704; PMID: 15131085
- Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 2005; 120:15 - 20; http://dx.doi.org/10.1016/j.cell.2004.12.035; PMID: 15652477
- Girardot M, Pecquet C, Boukour S, Knoops L, Ferrant A, Vainchenker W, Giraudier S, Constantinescu SN. miR-28 is a thrombopoietin receptor targeting microRNA detected in a fraction of myeloproliferative neoplasm patient platelets. Blood 2010; 116:437 - 45; http://dx.doi.org/10.1182/blood-2008-06-165985; PMID: 20445018
- Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O, Walichiewicz J. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet Genome Res 2005; 110:462 - 7; http://dx.doi.org/10.1159/000084979; PMID: 16093699
- Berezikov E, van Tetering G, Verheul M, van de Belt J, van Laake L, Vos J, Verloop R, van de Wetering M, Guryev V, Takada S, et al. Many novel mammalian microRNA candidates identified by extensive cloning and RAKE analysis. Genome Res 2006; 16:1289 - 98; http://dx.doi.org/10.1101/gr.5159906; PMID: 16954537
- Li X, Kahveci T, Settles AM. A novel genome-scale repeat finder geared towards transposons. Bioinformatics 2008; 24:468 - 76; http://dx.doi.org/10.1093/bioinformatics/btm613; PMID: 18089620
- Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, Smith HO, Yandell M, Evans CA, Holt RA, et al. The sequence of the human genome. Science 2001; 291:1304 - 51; http://dx.doi.org/10.1126/science.1058040; PMID: 11181995