
Abstract
Objectives: Bacteroides fragilis, a Gram-negative anaerobic bacterium, is alternately a gut commensal or virulent pathogen and is an important reservoir for horizontal gene transfer (HGT) of bacterial resistance and virulence genes in the human gastrointestinal tract. We identified a unique conjugative transposon (CTn) in a multidrug resistant clinical isolate of B. fragilis (BF-HMW615); we named this element CTnHyb because it included a hybrid mosaic of foreign elements. This study reports the characterization of CTnHyb and discusses the potential impact on horizontal spread of resistance genes.
Results: CTnHyb contains several efflux pump genes and several genes that confer or may confer antibiotic resistance to tetracycline, kanamycin, metronidazole and spectinomycin (truncated gene). CTnHyb also contains a mosaic of mobile elements from Gram-positive organisms. CTnHyb is easily transferred from BF-HMW615 (the original isolate) to BF638R (lab strain) and integrated into the BF638R chromosome. The “foreign” (from Gram-positive bacteria) nucleotide sequences within CTnHyb were > 99% preserved indicating that the gene acquisition from the Gram-positive bacteria was very recent.
Conclusion: CTnHyb is a novel CTn residing in a multidrug resistant strain of B. fragilis. The global nature and wide phylogenetic reach of HGT means that any gene in any bacterium can potentially be mobilized. Understanding the mechanisms that drive the formation and transfer of these elements and, potentially, ways to limit the transfer are necessary to prevent a devastating spread of resistance elements.
Introduction
The fluidity of the human gut microbiome has been recognized for decades but the recent data explosion from the analysis and sequencing of the gut microbiota is clarifying the vast extent of this gene transfer. Not only are genes regularly transferred among permanent residents of the gut, but organisms such as Staphylococcus and Streptococcus that do not colonize the intestine can participate in this genetic “swap meet”Citation1 (when they pass through the gut).
Of the resident gut population, ~99% of the microbiota belongs to two divisions (superkingdoms) of Bacteria–the Bacteroidetes (48%) (including Bacteroides species) and the Firmicutes (51%).Citation2 Bacteroides species, the dominant bacterial genus in the human gut are known to harbor many conjugative and mobilizable elements.Citation3
Conjugative transposons, also known as integrative conjugative elements (ICEs), are a subset of mobile elements that also include plasmids and transposons.Citation3 Like transposons, CTns can integrate into diverse sites in the host chromosome. The CTns do not exclude each other as do plasmids, so a strain can accumulate more than one CTn. Furthermore, there is some evidence that the presence of more than one copy of the CTn in the strain results in a stimulation of transposition (transactivation).Citation4 Theoretically, this implies that as CTns with antibiotic resistance genes accumulate in the environment, the transfer of these genes to other bacteria will also increase and may result in upward spiraling of antibiotic resistance.Citation5
We recently investigated a multidrug resistant clinical isolate of Bacteroides fragilis (BF-HMW615) isolated from a pediatric appendiceal specimen.Citation6 BF-HMW615 is resistant to multiple antibiotics, including metronidazole. Our analysis identified a nimJ gene which conferred increased MICs to metronidazole when introduced into a susceptible strainCitation7 and nimJ is carried on a novel conjugative transposon. We are now reporting the identification of this novel conjugative transposon, CTnHyb (for “hybrid”), which contains genes from Gram-positive bacteria. CTnHyb is transferable to B. fragilis 638R and thus is confirmed as a mobile element. The CTnHyb has (besides from Bacteroides spp) exact nucleotide homologs from at least three phylogenetically distinct Gram-positive organisms. The extent of the “hybrid” nature of this CTn, to our knowledge, has not been reported before in Bacteroides.
Results
Identification of CTnHyb in BF-HMW615 by comparative genome analysis
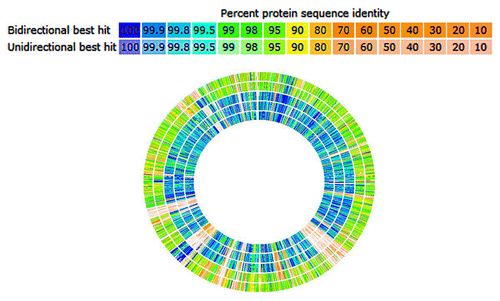
Comparative RAST-based genomic analysisCitation8,Citation9 indicated a few continuous regions of BF-HMW615 chromosome (> 50,000 bp) that contained genes with no homologs in B. fragilis ATCC 9343, BF638R, BF Y46H or the multidrug resistant clinical isolates BF-HMW610 and BF-HMW616. The blue lines indicate that BF-HMW615 has homologs and red lines indicate absence of homologs (). One of these segments (indicated by red arrows, ) included the tetQ gene as well as multiple Bacteroides tra genes (implicated in conjugative transposition). Thus, we considered this a potential CTn. Initial BLAST analysis of this non-homologous segment in BF-HMW 615 indicated that some of the genes have been horizontally transferred from other species.
Figure 1. Genomic sequence comparison using the RAST Server (http://rast.nmpdr.org/). HMW 615 is compared with HMW 610, HMW 616, BF 9343, BF638R, and BF YCH46, respectively from perimeter of circle inward; HMW 615 sequence is inferred (not shown). The sequence of HMW 615 has been taken from the published supercontigs and rearranged according to the Mauve prediction. Hits on the comparison organisms are displayed graphically. Percent protein sequence identity is indicated (legend).
Transfer of CTnHyb to B. fragilis 638R
We mobilized CTnHyb from BF-HMW615 to BF638R by mating. The new 638R-CTnHyb mutant was selected using tetracycline and rifampicin (BF638R is rifampicin resistant, tetracycline sensitive; BF-HMW615 is rifampicin sensitive, tetracycline resistant). To rule out the small chance of the tetR/rifR transconjugant of BF-HMW615 origin, we confirmed the BF638R origin by PCR amplification and partial sequencing of two BF638R genes (BF638R_2089 and BF638R_4382) which are not present in BF-HMW615. There was no evident tetracycline-mediated increase in frequency of CTnHyb.
Determination of the CTnHyb ends and the point of insertion into the BF638R chromosome
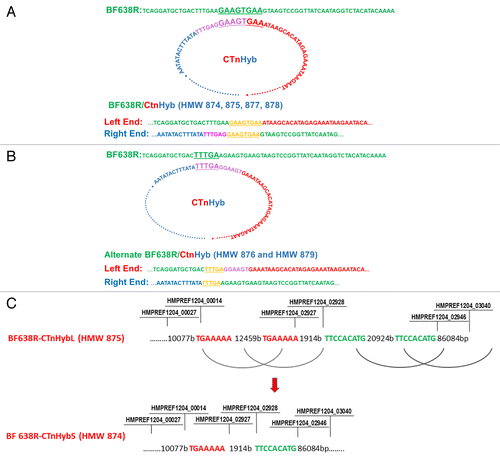
We selected six colonies (HMW 874, HMW 875, HMW 876, HMW 877, HMW 878, and HMW 879) from independent mating experiments (of BF638R and BF-HMW615) to determine the insertion point of CTnHyb. The insertion points were determined by the SRP technique as described in Materials and Methods. The isolates HMW 874, HMW 875, HMW 877, and HMW 878 had identical ends and insertion points, whereas HMW 876 and HMW 879 had slightly altered ends and insertion point.
The genome of BF-HMW615 has been sequenced but the published sequence is in supercontigs: the sizes range from 1.1 (largest supercontig) to 1.14 (smallest supercontig). Our analysis indicated that the transferred CTnHyb fragment was partially contained in two supercontigs annotated as 1.1 and 1.3. Our PCR data connecting the supercontigs 1.1 and 1.3 was supported by MAUVE alignment of BF-HMW615 supercontigs with BF638R (). Also, an unannotated transposase was identified, through PCR, in the junction sequence of 1.1 and 1.3.
Figure 2. MAUVE-based alignment of BF 638R and BF-HMW615 and schematic indication of CTnHyb insertion into BF 638R. The MAUVE Contig Mover module was used to order (and orient) the BF-HMW615 supercontigs relative to the BF 638 reference genome. Colored block is a region of the genome that is homologous to a similarly colored part of another genome without extensive genomic rearrangement. Regions outside blocks lack detectable homology among the input genomes. Only a small part of the alignment is shown to visualize the insertion; nimJ and tetQ are indicated for orientation.

The predicted crossovers that would result in the sequences in 638R/CTnHyb isolates is depicted in , respectively. The left end of CTnHyb is dnaK2 (HMPREF1204_0027 in reverse complement supercontig 1.1 of BF-HMW615) and the right end is between HMPREF1204_03040 and HMPREF1204_03041 (in contig 1.3 in BF-HMW615). The insertion is in dnaK2 (BF638R_1256, between 1507197 and 1507222 bp).
Figure 3. (A) Predicted events leading to integration of CTnHyb into the BF638R chromosome to result in BF638R/CTnHyb. The circular form of CTnHyb recombines with BF638R at the underlined sequence (GAAAGTGAA). (B) Alternate integration of CTnHyb into the BF638R chromosome. Predicted events leading to integration of CTnHyb into the BF638R chromosome to result in BF638R/CTnHyb found in HMW 876 and HMW 879 . The circular form of CTnHyb recombines with BF638R at the underlined sequence (TTTGA). (C) Predicted model of the deletions in CTnHybL leading to CTnHybS. The locus tag labels serve as approximate reference points. The bases are referred to as “bases” or “b”. The regions of homology that are predicted to recombine are color coded. The nimJ gene is among the genes deleted in the first deletion and metronidazole has a lower MIC for BF638R/CTnHybS than for BF638R/CTnHybL, as expected ().
Determination of size of CTnHyb
The two transconjugants, HMW 874 and HMW 875, were used to determine the length of CtnHyb. Surprisingly, two different size inserts had been incorporated into the BF638R chromosome. HMW 875 had a larger insert (thus named, CTnHybL) of 131,471 bp and HMW 874 had a smaller insert (thus named, CTnHybS) of 98,099 bp. Sequence analysis indicated that both inserts had the same ends but there were two internal segments missing in the CTnHybS (98099 bp), leading to the transconjugant with the smaller insertion as depicted in . Primers were designed across distances of 13327 bp (if the first deletion had not occurred) and 21512 bp (if the second deletion had not occurred), respectively, to detect whether the deletions were present in various isolates. PCR and subsequent sequencing across these regions indicated that the deletions were present not only in HMW874, but were also present in chromosomal DNA of BF-HMW 615 and in HMW875. The amplicons spanning the deletions (861 and 579 bp, respectively) were sequenced to confirm that they represented the deletion region. Thus, it appears that these deletions can occur in BF-HMW 615 as well as in BF638R/CTnHybL indicating the volatile nature of CTnHyb.
Determination of the circular form of CtnHyb
The primers (2783 and 2784) were used in a PCR reaction on BF-HMW615 and the BF638R/CTnHyb transconjugants to determine if we could detect a circular intermediate, which is a critical intermediary step in CTn transfer. The resultant amplicons indicated that there was indeed a junction between the two ends of CTnHyb indicating the presence of circular intermediate in BF-HMW 615. Interestingly, the circular intermediates were also detected in 4 independent isolates of BF638R/CTnHyb which indicated that CTnHyb can continue to make conjugative circle intermediates even after transfer to BF638R.
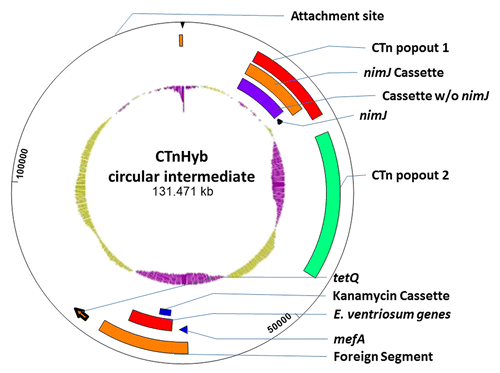
To visualize this circular intermediate, we constructed a GenBank (gb) file for CTnHyb from: 1) the sequence included within supercontig 1.1, 2) an unannotated transpose downstream of the predicted HMPREF1204_0001, 3) a short sequence obtained by PCR of the supercontig 1.1 and 1.3 junctions, and 4) the sequence contained within supercontig 1.3. The 4 gb files were concatenated using the SeqNinja program (DNASTAR, Inc., Madison, WI). The concatenated file was annotated and viewed using SeqBuilder (DNASTAR, Inc., Madison, WI) (). The nimJ and tetQ genes are indicated, as are the “foreign element” (from Gram-positive bacteria) extending from HMPREF1204_2965 to HMPREF1204_2980 and the predicted crossover point. The inner most circle is the GC% (green is higher than average, purple is lower than average).
Figure 4. CTnHyb: Circularized form and predicted nested ICEs. Circular intermediates were detected in HMW 615, and in 4 isolates of BF638R/CTnHybL using CTnHyb/BF 638R Junction primers at either end of CTnHyb (Primers 2783 and 2784). The continuous sequence was generated using 4 GenBank files consisting of 1) the known Broad sequence within supercontig 1.1, an unannotated transpose downstream of the predicted HMPREF1204_0001, a short nucleotide segment and the sequence within supercontig 1.3 (we obtained the 2nd and 3rd sequence from manual PCR and sequencing). The GenBank files were concatenated using the SeqNinja program (DNA Star) and the generated sequence was annotated and visualized with the SeqBuilder program (DNA Star). The inner most circle is the GC% (green is higher than average BF GC % and purple is lower than average BF GC %). The predicted attachment site, the nimJ, mefA and tetQ genes and the predicted nested ICEs are indicated on the figure. 1) CTnHyb “Popout 1”: (missing in CTnHybS) 12466 bases long, extending from HMPREF1204_00014 to HMPREF1204_00001 of contig 1.1 and the unannotated transposase and HMPREF1204_02924 to HMPREF1204_02926 of contig 1.3); 2) NimJ cassette, 9955 bases, present in 3 copies in BF HMW 615; 3) Cassette (minus nimJ) is found in other BF strains; 3) nimJ gene; 4) CTnHyb “Popout 2 (missing in CTnHybS): 20,933 bases long; extending from HMPREF1204_02928 to HMPREF1204_02946; 5) The “foreign segment” extends from HMPREF1204_2965 to HMPREF1204_2980; 6) mefA (HMPREF 1204_02965, 1232 bp) is a 95% match to mefA genes on ICEs from Staphylococcus, Streptococcus, and EnterococcusCitation10, and a close mefA homolog was also found in CTnGermCitation11; 7) Eubacterium ventriosum cassette (7178 bp not counting the kanamycin cassette within); 8) kanamycin cassette (1517 bp); HMPREF1204_2969-HMPREF1204_2971 is 100% match to nucleotide regions on ICEs from Streptococcus. However, the transposase (HMPREF 1204_02971) in the “kanamycin cassette” is not homologous to the Staphylococcus or Enterococcus transposases, but is hom0logous to a transposase from Blautia hansenii—a novel genus of Gram-positive, anaerobic, non-sporulating coccobacillus-shaped bacteria that includes several former coccobacillary shaped species of Clostridia and Ruminococcus.Citation12 HMPREF 1204_02971 is truncated at a 10 bp palindrome (ACTTCCGCCG, bp 195–204 and 218–227 within HMPREF 1204_2970) which is characteristic of transposase insertions. Bacterial repetitive extragenic palindromic sequences are known DNA targets for Insertion Sequence elements.Citation13 This palindrome, however, is contained within the coding region of HMPREF1204_02971.
CTnHyb contains 144 genes
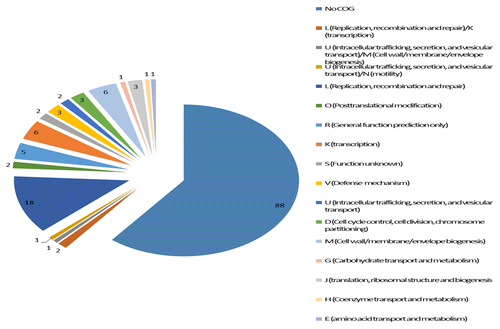
BLAST analysis of the CTnHyb indicated that it contained CTn specific tra genes (traE, traG, traJ, traK, traM and traN, excisionases, transposases and other DNA-associated proteins, a tetracycline resistance gene (tetQ), 3 putative pump system genes coding for efflux pumps (MefA, ABC, and RND type transporters), genes coding for hemagglutinin and thioredoxin (both may be important in virulence), and genes coding for metronidazole, kanamycin and tetracycline resistance. The genes contained within CTnHyb are shown in Table S2. RNA-Seq results for all of the BF-HMW 615 genes will be published as part of a larger study comparing the total transcriptome of BF clinical isolates (Husain F, Veeranagouda Y, Wexler HM, unpublished data) but the RNA-Seq counts for the CTnHyb genes are presented here, as well. The distribution of genes in CTnHyb according to COG (Cluster of Orthologous Gene class) is shown in .
Figure 5. Distribution of genes in CTnHyb according to COG (Cluster of Orthologous Gene class). COG ID numbers and classification were taken from the Integrated Microbial Genomes and Metagenomes (IMG) at the Department of Energy Joint Genome Institute. (http://img.jgi.doe.gov/cgi-bin/w/main.cgi?section=FindGenes&page=geneSearch). Eighty-eight genes in CTnHyb do not belong to any COG. Similarly, RAST analysis of the entire BF-HMW 615 genome sequence indicates that 69% of the putative genes are not assigned to any subsystem (a subsystem is a method of categorization that can be thought of as roughly equivalent to a COG). Of the remaining 57 genes in CTnHyb(L), almost one third of the genes (n = 18) are in COG L (replication, recombination and repair) which includes such genes as DNA primases, excionases, integrases and transferases. Another 11 genes are annotated as “viral proteins” and are not assigned to a COG class, even though several are also functionally annotated as excisionase or integrase proteins.
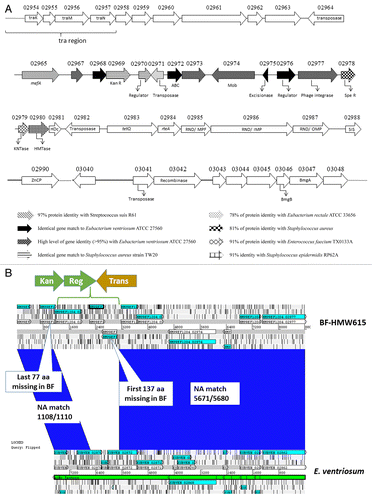
CtnHyb includes a “foreign segment” containing genes homologous to a variety of Gram-positive bacteria (; ). HMPREF 1204_02969 (coding for aminoglycoside 3′-phosphotransferase conferring kanamycin resistance) is 100% identical to Staphylococcus epidermidis RP62A aphA (SEA0010). HMPREF 1204_02965 encodes a MefA type efflux pump and is 100% homologous to mefA genes in many Streptococcus species. Downstream of these genes is a 6790 bp segment (HMPREF1204_2967 through HMPREF1204_02977) that is homologous to a nucleotide segment in Eubacterium ventriosum 27560 (a Gram-positive gut anaerobe) from EUBVEN_02875 thru EUBVEN_02862 except for a short missing stretch (). The “missing piece” is replaced by a 3617 bp sequence (HMP1204_02969, HMP1204_02970 and HMP1204_02971, GenBank Accession AGXR01000023.1 46489–48006) that is highly conserved in both genome sequences and ICEs in Staphylococcus, Streptococcus and Enterococcus. In S. pneumonia, for example, it is present on mobile elements carrying multidrug resistance determinantsCitation14 and in Enterococcus faecalis RE25 it is present on a 50-kb conjugative multidrug resistance plasmid (pRE25)Citation15 (). We don’t know whether the insertion of the 3617 bp segment (containing HMPREF1204_02969, 02970 and 02971) happened before or after the Eubacterium segment was transferred into BF-HMW 615. Also, whether the acquisition of the E. ventriosum genes is a result of a BF CTnHyb-like element having moved into Eubacterium and then back into BF is not clear at this point.
Figure 6. (A) Schematic representation of the “foreign” segment in CTnHyb. White arrows correspond to conserved genes in Bacteroides sp strains. See the figure for explanation of other arrows. APH(3′): aminoglycoside 3′-phosphotransferase; ABC: ABC transporter ; Mob: mobilization protein; Spe R: spectinomycin adenylyltransferase; KNTase: nucleotidyltransferase; HMTase: methlytransferase; HDc: Metal dependent phosphohydrolases with conserved “HD” motif; SIS: Sugar Isomerase; ZnCP: Zinc peptidase; BmgA and BmgB: mobilization proteins. (B) ACT alignment of area of homology between Eubacterium ventriosum and CTnHyb (BF-HMW 615). A segment (7671 bp) of the E. ventriosum genome is nearly completely conserved except for a three gene insert that replaces part of EUBVEN_02872, EUBVEN_02873 and part of EUBVEN_02874. In the conserved portions, only 11 nucleotides differed.
CTnHybL contains a novel metronidazole resistance gene
CTnHyb contains a cassette with a recently reported metronidazole resistance (“nimJ”) gene, found in two metronidazole resistant clinical BF isolates (BF-HMW 615 and BF-HMW 616).Citation7 Downstream of nimJ is an unannotated transposase of the IS4 class (in opposite orientation) which spans the junction of supercontigs 1.1 and 1.3. It is in reverse orientation to nimJ and contains the consensus promoter sequence TAnnTTTG that is found on insertion sequences containing the cfiA (imipenemase) gene.Citation16 nimJ (HMPREF1204_00002) is present within CTnHybL but is not present in CTnHybS (see ). As expected, BF638R/CTnHybL had increased MICs for metronidazole (1.0–1.5 µg/ml) while BF638R/CTnHybS had MICs in the same range as BF638R (0.38–0.5 µg/ml). Additionally, MICs of erythromycin for BF638R/CTnHybL and BF638R/CTnHybS were increased from those of BF638R ().
Table 1. Minimal inhibitory concentrations of strains and constructs
CTnHyb contains multiple transposase genes of the IS4 category
In addition to the transposase gene adjacent to nimJ, CTnHyb contains two additional transposase genes of the IS4 class that are divergently transcribed from their adjacent genes. HMPREF1204_2983 is adjacent to HMPREF1204_2984 (tetQ) and contains the consensus promoter sequence, and HMPREF1204_2964 is another IS4 family transposase adjacent to HMPREF1204_2965 (mefA).
Expression of HMPREF1204_02969 (conferring kanR) and HMPREF1204_02965 (mefA) in E. coli
HMPREF1204_2969 (encoding aminoglycoside 3′-phosphotransferase for kanamycin resistance) and HMPREF1204_02965 (mefA) were cloned into pSportI and introduced into E. coli: HMPREF1204_2969 into E. coli AG100 to determine changes in kanamycin MICs and HMPREF1204_02965 into both E. coli AG100 and E. coli Kam43 (a pump deficient mutant, lacking AcrAB, AcrEF and TolCCitation17) to determine erythromycin MIC changes due to mefA expression. E. coli/pSportI::HMPREF1204_2969 grew in the presence of 40 µg/ml kanamycin, while E. coli/pSportI (without the kanamycin gene) did not grow, indicating that HMPREF1204_2969 was fully functional in E. coli. HMPREF1204_02965 (mefA) did not confer erythromycin resistance in either AG100 or KAM43. The mating of BF-HMW615 and E. coli AG100 did result in DNA transfer that yielded E. coli that were kanamycin resistant, but the transconjugants were not stable and did not yield further generations on purification.
Discussion
The importance of the gut flora in human health and disease is at the forefront of scientific and public awareness and major efforts are underway to characterize and sequence the gut microbiota as part of the massive Human Microbiome Project (HMP) at the Broad Institute/NIH. The gut bacteria, in terms of total cell and gene numbers in the humanCitation18, “represents a virtual inner organ”Citation19. BF is a human gut commensal that only accounts for 2% of the total Bacteroides but it is the agent of > 70% of Bacteroides infections.Citation5 As a commensal, it hydrolyzes complex polysaccharides and produces volatile fatty acids used by the host as source of energyCitation5 and is important in immune development.Citation20,Citation21 However, it is very virulent when it escapes the gut and has been associated with nearly all types of infections. Additionally, it may be a reservoir of resistance genes that can get passed, by horizontal gene transfer (HGT), to other organisms resident in or passing through the gut.
Horizontal gene transfer among gut microbiota is particularly intense; gut microbes, therefore, may be a major reservoir for antibiotic resistance genes.Citation22,Citation23 Indeed, the taxonomically different representatives of gut microbiota may share the pool of closely related antimicrobial resistance genes. HGT is also a crucial event in the development of virulence traits.Citation24
One of the major elements responsible for HGT are conjugative transposons (CTns). CTns are similar to transposons (that integrate into the host semi-randomly) but also carry the genes necessary for conjugal transfer to other cells. The first complete sequence of the transfer region of a Bacteroides conjugative transposon was described in 2001.Citation25 The many known conjugative transposons and other mobile elements that are present in strains of Bacteroides were reviewed recentlyCitation3 and include CTnDOT (the most widely studied Bacteroides CTn),Citation26 CTnGerm,Citation11 CTn341,Citation27 CTnBSTCitation28 and CTn12256.Citation29 CTnGerm and CTnBst were reported to carry a variety of genes with high similarity to genes from aerobic bacteriaCitation11,Citation28 and CTn12256 was described as a chimeric transposon composed of two independently active mobile elements.Citation29 Sequence comparisons between CTnHyb and other Bacteroides CTns, including CTnDOT, CTnGerm or CTnBST did not reveal any significant homology.
CTnHyb is unique and noteworthy among the many conjugative transposons and other mobile elements present in strains of Bacteroides. First, several genes coding for antibiotic resistance proteins are contained within CTnHyb. The tetQ and nimJ genes conferred tetracycline and metronidazole resistance, respectively, in BF638R and HMPREF1204_2969 conferred kanamycin resistance in E.coli. The encoded efflux pumps including the RND and mefA pumps may also contribute to drug resistance. Also, there is a truncated gene for spectinomycin resistance. Second, CTnHyb has a long sequence stretch of homologous sequences from a Gram-positive species (i.e., Eubacterium) that includes a mosaic of resistance genes from other aerobic Gram-positive species. The degree of nucleotide conservation (6779/6790 nucleic acids) within the area homologous to Eubacterium ventriosum indicates a very recent transfer to BF, otherwise some degree of adaptation to codon usage bias patterns of BF would be expected.Citation30 The manner in which the various “foreign” elements are arranged within CTnHyb suggests an “Ellis Island” effect where incoming CTns are preferentially drawn to genomic regions that are composed of other mobilizable elements; ICEs are nested within each other () (note: Ellis Island was the gateway for millions of immigrants to the United States from 1892 to 1954). This type of modular transfer within conjugative elements has been described in other bacteria and in one CTn in Bacteroides and may be an important mechanism for the accumulation of resistance and virulence genes in the gut and the subsequent development of resistance or pathogenicity islands.Citation31
Target site selection is an important characteristic for each transposon and determines dissemination and stability.Citation32 We detected a circular intermediate of CTnHyb in BF HMW 615 and in HMW615/BF638R transconjugants and predicted two potential crossover points that would yield sequences consistent with the sequences found in the resultant BF638R/CTnHyb transconjugants. In CTnDOT, the most extensively studied Bacteroides CTn, a tyrosine recombinase called IntDOT catalyzes integration into, and excision out of, the bacterial host chromosome.Citation33 The core (GTANNTTT), are inverted repeat sequences that flank target sites in the chromosome and in CTnDOT, where strand exchange takes place catalyzed by IntDOT. The target sites, attB, on the host chromosome consist of a pair of inverted repeat core sites (B and B’). The complementary sites on CTnDOT, attDOT sites, have the core sites D and D’. CTnBST, another Bacteroides CTn, appears to integrate more site specifically than CTnDOT, with a 6-amino-acid signature that is associated with the catalytic regions of members of the tyrosine recombinase family.Citation32 Although the core site sequence is present (~5037 times in BF638R), it is not at the crossover position of CTnHyb.
Transfer of several of the widely studied Bacteroides CTns, including CTnDOT, is mediated by low levels of tetracycline and it is believed that the wide use of antibiotics therapeutically and in animal feed (with subsequent contamination of both meat and manure-fertilized crops) influence the introduction of these mobile-element bearing-organisms into the human gut. It is possible that elimination of the inducers (i.e., antibiotic) might be achieved by radically reducing their use. In the case of CTnHyb, however, transfer (from the multidrug resistant clinical isolate to the susceptible lab strain) could be easily achieved without any need for induction by tetracycline so change in tetracycline use may not affect transfer frequency. The global nature and wide phylogenetic pool of the horizontal transfer described in recent years means that any gene in any bacterium can potentially be mobilized and resistance phenotypes can be established in a diverse range of organisms worldwide. Understanding the mechanisms that drive this transfer and ways to limit the transfer are necessary to quell the spread of resistance elements.Citation26,Citation34-Citation36
Materials and Methods
Strains and culture conditions
Strains used in this study are listed in . All strains were grown as describedCitation40 using Brain Heart Infusion media supplemented with 15 μg /ml hemin (BHIS) for Bacteroides isolates (Anaerobe Systems, Morgan Hill, CA) and Luria Bertani (LB) agar or broth (Sigma) for Escherichia coli. The multidrug resistant clinical isolates BF-HMW610, BF-HMW615 and BF-HMW616 have been previously described.Citation6,Citation41,Citation42 E. coli AG100 was used as the host to test for the kanamycin (kan) and spectinomycin resistance phenotypes. E. coli Kam43 (a pump deficient mutant, lacking AcrAB, AcrEF and TolCCitation17) was a kind gift from Dr Tomofuso Tsuchiya (Okayama University, Japan). Ampicillin (50 μg/ml), erythromycin (10 μg/ml), and kanamycin (40 μg/ml) were used for selection as indicated.
Table 2. Strains and plasmids used in this study
Molecular methods
DNA extraction, restriction digestions, gel electrophoresis and analysis were done as previously described.Citation40 The size and sequence of the transferred CTn was determined by PCR and sequencing of the CTn at regular intervals. Based on BF-HMW 615 sequence, primers were designed to yield 150 to 200 bases products targeting DNA approximately every ~20 KB on either side of the tetQ gene (HMPREF1204_02983). Using these primers, the PCR amplification was done with genomic DNA from BF-HMW 615, BF638R, and the selected BF-HMW 615/BF638R transconjugants as templates. The exact boundaries of CTnHyb insertion into BF638R, the insertion points in BF638R, the segments deleted in CTnHybS, and the sequence of the junction of contig 1.1, the unannotated transposase, and contig 1.3 were determined by semi-random priming (SRP)-PCR.Citation43 The primers used for sequencing the BF638R/CTnHyb junctions and the gaps found within the CTnHyb in one of the isolates are listed in Table S1.
Genome sequencing
BF-HMW615 (along with two other multidrug resistant isolates BF-HMW610 and BF-HMW616) were submitted to the Broad Institute and sequenced as part of the Human Microbiome Project, Bacteroides group Sequencing Project, Broad Institute of Harvard and MIT (http://www.broadinstitute.org/). The Broad sequencing project utilized 454 Whole Genome Shotgun methodology and Newbler (454 Life Sciences) assembly. This sequencing project was supported by the National Institute of Allergy and Infectious Disease/National Institutes of Health-funded Genome Sequencing Center for Infectious Diseases at the Broad Institute. BF-HMW610, BF-HMW615 and BF-HMW616 have been given the Broad designations HMPREF1203, HMPREF1204, and HMPREF1205, respectively. For the sake of consistency, the BF-HMW 615 genes are referred to by their designation HMPREF1204_, etc.; these are the designations used in GenBank FASTA files of the genome sequences and associated annotations were downloaded from the Broad Institute.
Genomic analysis
The RAST (Rapid Annotation using Subsystem Technology) Annotation ServerCitation8 was used for comparative genome analysis. All sequences submitted to RAST were downloaded from either the Broad Institute (for the clinical isolates) or NCBI (for the reference strains) and submitted to the RAST server.Citation8,Citation9 Genomes were compared by the sequence comparison feature of the SEED server.Citation9 Comparisons of known CTns and the BF-HMW 615 genome sequence were analyzed as described earlier.Citation7 The Double ACT server (http://www.hpa-bioinfotools.org.uk) was used to generate the comparison file with the BLASTN function and a cutoff of 1000–2000. Results were viewed using the ACT viewer.Citation44 MAUVE alignment software, which is specifically designed for multiple genome alignment in the presence of large-scale evolutionary events such as rearrangement and inversion,Citation45 was used to help predict the arrangement and orientation of supercontigs in the BF-HMW615 genome. The MAUVE Contig Mover module was used to order and orient the BF-HMW615 contigs relative to the BF638R reference genome.Citation46 When needed, nucleotide or amino acid sequences were aligned using ClustalW.
Annotation
The sequences in GenBank were annotated by the Broad Institute as part of the genome sequencing but more than 80% of the proteins (122/144) were annotated as “hypothetical.” To augment this annotation, we separately annotated all three sequences using the RAST Server, which assigns proteins to subsystems. With the addition of the RAST information, 55 proteins remained annotated as hypothetical. Finally, we submitted the protein sequences to Phyre2-fold recognition analysisCitation47 and succeeded in obtaining a predicted protein function for all but 17 proteins (38 were annotated at greater than 50% confidence and 25/55 at greater than 90% confidence) (Table S2).
Mating of BF-HMW615 with B. fragilis 638R
BF638R and BF-HMW615 were grown overnight anaerobically at 37 °C in BHIS. 200 µl of each culture were mixed, centrifuged at 13000 g for 2 min and then reconstituted in 100 µl of BHIS. The mixed cells were then spread on BHI plates (Anaerobe systems, CA, USA) and incubated anaerobically overnight at 37 °C. The growth on the plate was harvested and then resuspended (mating mix) in ~1.5 ml 10% glycerol TSB storage media (Key Scientific Products Inc., TX, USA). A 150 µl aliquot of mating mix was plated on BHIS plates containing tetracycline (1 µg/ml) and rifampicin (10µg/ml). The frequency of the transfer was estimated to be 1 × 10−9 (number of transconjugants [BF638R-CtnHyb]/numbers of donors [BF HMW615]).
Potential tetracycline-mediated increase in mating frequency was measured by three methods with slight alterations to the regular mating procedure. 1) BF-HMW615 was grown overnight in presence of tetracycline (1 µg/ml) prior to regular mating. 2) BF-HMW615 was treated with tetracycline (1 µg/ml) for 1 h at 37 °C. The cells were then washed twice with equal volumes of BHIS and the regular mating procedure with BF638R was followed. 3) Overnight grown cells of BF638R and BF-HMW615 mix (200 µl of each) were each plated on BHIS plates containing a very low concentration of tetracycline (0.001 µg/ml), incubated overnight anaerobically at 37 °C, and then the regular mating procedure was followed.
Mating of BF-HMW615 with E. coli
200 µl of overnight cultures of BF-HMW615 and E. coli AG100 cells were mixed and plated on BHI plates and incubated overnight at 37 °C under anaerobic conditions. The cells were then harvested and pooled in 1 ml of broth and 50 µl of cells were plated on LB plate containing 40 µg/ml kanamycin and incubated aerobically at 37 °C.
Cloning of resistance genes contained within CTnHyb and introduction into E. coli
HMPREF1204_02969 (a gene coding for the aminoglycoside 3′-phosphotransferase that confers kanamycin resistance) and HMPREF1204_02965 (a mefA-like efflux transporter gene) were PCR amplified from BF-HMW615, ligated into pSportI (Invitrogen) and introduced into E. coli DH5α. Transformants were selected and purified on ampicillin containing LB plates and the presence of the introduced genes were confirmed by PCR and sequencing.
MIC determinations
MICs were determined using E-test technology (BioMerieux) according to the manufacturer’s directions. An inoculum of one McFarland unit was used on a Brucella Blood Agar plate (Anaerobe Systems, Morgan Hill, CA).
Transcription of genes contained within CTnHyb genes in BF-HMW615
RNA was prepared from cells using the Qiagen RNAeasy kit (Qiagen, CA, USA) according to manufacturer’s directions. The total RNA was enriched for mRNA by removing the majority of rRNA using the Ambion Microbe Express Kit (Life Technologies). cDNA was prepared using the Invitrogen Superscript kit (Life Technologies). cDNA was quantified by RNA-Seq analysis (Otogenetics, Norcross, USA). RNA-Seq files were analyzed using the Lasergene Genomics Suite (DNASTAR, Inc., Madison, USA).
Registration of CTnHyb
CTnHyb was registered in the transposon registry (http://www.ucl.ac.uk/eastman/research/departments/microbial-diseases/tn) as Transposon 6243. The complete sequence and annotation has been submitted to NCBI GenBank (Submission ID 1725586, to be released June 10, 2014).
Conclusion
CTnHyb represents a mechanism for a single Bacteroides isolate to become a reservoir for a variety of resistance genes, both from Bacteroides and other species; these genes can then be transferred both to other Bacteroides and unrelated bacteria. Identifying the factors that increase CTn accumulation within a strain as well as factors that increase transfer of CTns to other bacteria is critical information that could lead to therapeutic regimens against resistance dissemination.
Additional material
Download Zip (383.5 KB)Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Disclosure of Funding
This work was supported in part by the Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development, Biomedical Laboratory Research and Development. RB’s fellowship was funded by a Scholarship from Conselho Nacional de Desenvolvimento Científico e Tecnológico—“National Counsel of Technological and Scientific Development” grant number 237612/2012-7, Brazil.
Acknowledgments
We would like to acknowledge Diane Citron and Dr Ellie Goldstein and Dr Tomofuso Tsuchiya for providing us with strains BF HMW615 and E. coli KAM43, respectively.
References
- Dalke K. Mobile DNA: Genomic Studies Illuminate Antibiotic Resistance. Genome News Network 2003; http://www.genomenewsnetwork.org/articles/04_03/mobile.shtml.
- Mahowald MA, Rey FE, Seedorf H, Turnbaugh PJ, Fulton RS, Wollam A, Shah N, Wang C, Magrini V, Wilson RK, et al. Characterizing a model human gut microbiota composed of members of its two dominant bacterial phyla. Proc Natl Acad Sci U S A 2009; 106:5859 - 64; http://dx.doi.org/10.1073/pnas.0901529106; PMID: 19321416
- Nguyen M, Vedantam G. Mobile genetic elements in the genus Bacteroides, and their mechanism(s) of dissemination. Mob Genet Elements 2011; 1:187 - 96; http://dx.doi.org/10.4161/mge.1.3.18448; PMID: 22479685
- Salyers AA, Shoemaker NB, Stevens AM, Li LY. Conjugative transposons: an unusual and diverse set of integrated gene transfer elements. Microbiol Rev 1995; 59:579 - 90; PMID: 8531886
- Wexler HM. Bacteroides: the good, the bad, and the nitty-gritty. Clin Microbiol Rev 2007; 20:593 - 621; http://dx.doi.org/10.1128/CMR.00008-07; PMID: 17934076
- Pumbwe L, Chang A, Smith RL, Wexler HM. BmeRABC5 is a multidrug efflux system that can confer metronidazole resistance in Bacteroides fragilis.. Microb Drug Resist 2007; 13:96 - 101; http://dx.doi.org/10.1089/mdr.2007.719; PMID: 17650960
- Husain F, Veeranagouda Y, Hsi J, Meggersee R, Abratt V, Wexler HM. Two multidrug-resistant clinical isolates of Bacteroides fragilis carry a novel metronidazole resistance nim gene (nimJ). Antimicrob Agents Chemother 2013; 57:3767 - 74; http://dx.doi.org/10.1128/AAC.00386-13; PMID: 23716049
- Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, Formsma K, Gerdes S, Glass EM, Kubal M, et al. The RAST Server: rapid annotations using subsystems technology. BMC Genomics 2008; 9:75; http://dx.doi.org/10.1186/1471-2164-9-75; PMID: 18261238
- Overbeek R, Begley T, Butler RM, Choudhuri JV, Chuang HY, Cohoon M, de Crécy-Lagard V, Diaz N, Disz T, Edwards R, et al. The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucleic Acids Res 2005; 33:5691 - 702; http://dx.doi.org/10.1093/nar/gki866; PMID: 16214803
- Santagati M, Iannelli F, Cascone C, Campanile F, Oggioni MR, Stefani S, Pozzi G. The novel conjugative transposon tn1207.3 carries the macrolide efflux gene mef(A) in Streptococcus pyogenes.. Microb Drug Resist 2003; 9:243 - 7; http://dx.doi.org/10.1089/107662903322286445; PMID: 12959402
- Wang Y, Wang GR, Shelby A, Shoemaker NB, Salyers AA. A newly discovered Bacteroides conjugative transposon, CTnGERM1, contains genes also found in gram-positive bacteria. Appl Environ Microbiol 2003; 69:4595 - 603; http://dx.doi.org/10.1128/AEM.69.8.4595-4603.2003; PMID: 12902247
- Liu C, Finegold SM, Song Y, Lawson PA. Reclassification of Clostridium coccoides, Ruminococcus hansenii, Ruminococcus hydrogenotrophicus, Ruminococcus luti, Ruminococcus productus and Ruminococcus schinkii as Blautia coccoides gen. nov., comb. nov., Blautia hansenii comb. nov., Blautia hydrogenotrophica comb. nov., Blautia luti comb. nov., Blautia producta comb. nov., Blautia schinkii comb. nov. and description of Blautia wexlerae sp. nov., isolated from human faeces. Int J Syst Evol Microbiol 2008; 58:1896 - 902; http://dx.doi.org/10.1099/ijs.0.65208-0; PMID: 18676476
- Tobes R, Pareja E. Bacterial repetitive extragenic palindromic sequences are DNA targets for Insertion Sequence elements. BMC Genomics 2006; 7:62; http://dx.doi.org/10.1186/1471-2164-7-62; PMID: 16563168
- Mingoia M, Tili E, Manso E, Varaldo PE, Montanari MP. Heterogeneity of Tn5253-like composite elements in clinical Streptococcus pneumoniae isolates. Antimicrob Agents Chemother 2011; 55:1453 - 9; http://dx.doi.org/10.1128/AAC.01087-10; PMID: 21263055
- Schwarz FV, Perreten V, Teuber M. Sequence of the 50-kb conjugative multiresistance plasmid pRE25 from Enterococcus faecalis RE25. Plasmid 2001; 46:170 - 87; http://dx.doi.org/10.1006/plas.2001.1544; PMID: 11735367
- Kato N, Yamazoe K, Han CG, Ohtsubo E. New insertion sequence elements in the upstream region of cfiA in imipenem-resistant Bacteroides fragilis strains. Antimicrob Agents Chemother 2003; 47:979 - 85; http://dx.doi.org/10.1128/AAC.47.3.979-985.2003; PMID: 12604530
- Matsuo T, Nakamura K, Kodama T, Mikami T, Hiyoshi H, Tsuchiya T, Ogawa W, Kuroda T. Characterization of all RND-type multidrug efflux transporters in Vibrio parahaemolyticus.. Microbiologyopen 2013; 2:725 - 42; PMID: 23894076
- Sonnenburg JL, Angenent LT, Gordon JI. Getting a grip on things: how do communities of bacterial symbionts become established in our intestine?. Nat Immunol 2004; 5:569 - 73; http://dx.doi.org/10.1038/ni1079; PMID: 15164016
- Forsythe P, Sudo N, Dinan T, Taylor VH, Bienenstock J. Mood and gut feelings. Brain Behav Immun 2010; 24:9 - 16; http://dx.doi.org/10.1016/j.bbi.2009.05.058; PMID: 19481599
- Mazmanian SK, Liu CH, Tzianabos AO, Kasper DL. An immunomodulatory molecule of symbiotic bacteria directs maturation of the host immune system. Cell 2005; 122:107 - 18; http://dx.doi.org/10.1016/j.cell.2005.05.007; PMID: 16009137
- Mazmanian SK, Round JL, Kasper DL. A microbial symbiosis factor prevents intestinal inflammatory disease. Nature 2008; 453:620 - 5; http://dx.doi.org/10.1038/nature07008; PMID: 18509436
- Salyers AA, Gupta A, Wang Y. Human intestinal bacteria as reservoirs for antibiotic resistance genes. Trends Microbiol 2004; 12:412 - 6; http://dx.doi.org/10.1016/j.tim.2004.07.004; PMID: 15337162
- Vedantam G. Antimicrobial resistance in Bacteroides spp.: occurrence and dissemination. Future Microbiol 2009; 4:413 - 23; http://dx.doi.org/10.2217/fmb.09.12; PMID: 19416011
- Aminov RI. Horizontal gene exchange in environmental microbiota. Front Microbiol 2011; 2:158; http://dx.doi.org/10.3389/fmicb.2011.00158; PMID: 21845185
- Bonheyo G, Graham D, Shoemaker NB, Salyers AA. Transfer region of a bacteroides conjugative transposon, CTnDOT. Plasmid 2001; 45:41 - 51; http://dx.doi.org/10.1006/plas.2000.1495; PMID: 11319931
- Whittle G, Shoemaker NB, Salyers AA. Characterization of genes involved in modulation of conjugal transfer of the Bacteroides conjugative transposon CTnDOT. J Bacteriol 2002; 184:3839 - 47; http://dx.doi.org/10.1128/JB.184.14.3839-3847.2002; PMID: 12081954
- Bacic M, Parker AC, Stagg J, Whitley HP, Wells WG, Jacob LA, Smith CJ. Genetic and structural analysis of the Bacteroides conjugative transposon CTn341. J Bacteriol 2005; 187:2858 - 69; http://dx.doi.org/10.1128/JB.187.8.2858-2869.2005; PMID: 15805532
- Schlesinger DJ, Shoemaker NB, Salyers AA. Possible origins of CTnBST, a conjugative transposon found recently in a human colonic Bacteroides strain. Appl Environ Microbiol 2007; 73:4226 - 33; http://dx.doi.org/10.1128/AEM.00455-07; PMID: 17483268
- Wang GR, Shoemaker NB, Jeters RT, Salyers AA. CTn12256, a chimeric Bacteroides conjugative transposon that consists of two independently active mobile elements. Plasmid 2011; 66:93 - 105; http://dx.doi.org/10.1016/j.plasmid.2011.06.003; PMID: 21777612
- Ochman H, Lawrence JG, Groisman EA. Lateral gene transfer and the nature of bacterial innovation. Nature 2000; 405:299 - 304; http://dx.doi.org/10.1038/35012500; PMID: 10830951
- Burrus V, Pavlovic G, Decaris B, Guédon G. Conjugative transposons: the tip of the iceberg. Mol Microbiol 2002; 46:601 - 10; http://dx.doi.org/10.1046/j.1365-2958.2002.03191.x; PMID: 12410819
- Song B, Shoemaker NB, Gardner JF, Salyers AA. Integration site selection by the Bacteroides conjugative transposon CTnBST. J Bacteriol 2007; 189:6594 - 601; http://dx.doi.org/10.1128/JB.00668-07; PMID: 17616597
- Laprise J, Yoneji S, Gardner JF. IntDOT interactions with core sites during integrative recombination. J Bacteriol 2013; 195:1883 - 91; http://dx.doi.org/10.1128/JB.01540-12; PMID: 23335422
- Sommer MO, Dantas G, Church GM. Functional characterization of the antibiotic resistance reservoir in the human microflora. Science 2009; 325:1128 - 31; http://dx.doi.org/10.1126/science.1176950; PMID: 19713526
- Stokes HW, Gillings MR. Gene flow, mobile genetic elements and the recruitment of antibiotic resistance genes into Gram-negative pathogens. FEMS Microbiol Rev 2011; 35:790 - 819; http://dx.doi.org/10.1111/j.1574-6976.2011.00273.x; PMID: 21517914
- Gillings MR, Stokes HW. Are humans increasing bacterial evolvability?. Trends Ecol Evol 2012; 27:346 - 52; http://dx.doi.org/10.1016/j.tree.2012.02.006; PMID: 22459247
- Privitera G, Dublanchet A, Sebald M. Transfer of multiple antibiotic resistance between subspecies of Bacteroides fragilis. J Infect Dis 1979; 139:97 - 101; http://dx.doi.org/10.1093/infdis/139.1.97; PMID: 108340
- George AM, Levy SB. Gene in the major cotransduction gap of the Escherichia coli K-12 linkage map required for the expression of chromosomal resistance to tetracycline and other antibiotics. J Bacteriol 1983; 155:541 - 8; PMID: 6307967
- Matsuo T, Hayashi K, Morita Y, Koterasawa M, Ogawa W, Mizushima T, Tsuchiya T, Kuroda T. VmeAB, an RND-type multidrug efflux transporter in Vibrio parahaemolyticus.. Microbiology 2007; 153:4129 - 37; http://dx.doi.org/10.1099/mic.0.2007/009597-0; PMID: 18048926
- Pumbwe L, Ueda O, Yoshimura F, Chang A, Smith RL, Wexler HM. Bacteroides fragilis BmeABC efflux systems additively confer intrinsic antimicrobial resistance. J Antimicrob Chemother 2006; 58:37 - 46; http://dx.doi.org/10.1093/jac/dkl202; PMID: 16757501
- Wareham DW, Wilks M, Ahmed D, Brazier JS, Millar M. Anaerobic sepsis due to multidrug-resistant Bacteroides fragilis: microbiological cure and clinical response with linezolid therapy. Clin Infect Dis 2005; 40:e67 - 8; http://dx.doi.org/10.1086/428623; PMID: 15824978
- Sherwood JE, Fraser S, Citron DM, Wexler H, Blakely G, Jobling K, Patrick S. Multi-drug resistant Bacteroides fragilis recovered from blood and severe leg wounds caused by an improvised explosive device (IED) in Afghanistan. Anaerobe 2011; 17:152 - 5; http://dx.doi.org/10.1016/j.anaerobe.2011.02.007; PMID: 21376821
- Chen T, Dong H, Yong R, Duncan MJ. Pleiotropic pigmentation mutants of Porphyromonas gingivalis.. Microb Pathog 2000; 28:235 - 47; http://dx.doi.org/10.1006/mpat.1999.0338; PMID: 10764615
- Carver T, Berriman M, Tivey A, Patel C, Böhme U, Barrell BG, Parkhill J, Rajandream MA. Artemis and ACT: viewing, annotating and comparing sequences stored in a relational database. Bioinformatics 2008; 24:2672 - 6; http://dx.doi.org/10.1093/bioinformatics/btn529; PMID: 18845581
- Darling AE, Mau B, Perna NT. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 2010; 5:e11147; http://dx.doi.org/10.1371/journal.pone.0011147; PMID: 20593022
- Rissman AI, Mau B, Biehl BS, Darling AE, Glasner JD, Perna NT. Reordering contigs of draft genomes using the Mauve aligner. Bioinformatics 2009; 25:2071 - 3; http://dx.doi.org/10.1093/bioinformatics/btp356; PMID: 19515959
- Kelley LA, Sternberg MJ. Protein structure prediction on the Web: a case study using the Phyre server. Nat Protoc 2009; 4:363 - 71; http://dx.doi.org/10.1038/nprot.2009.2; PMID: 19247286