
Abstract
Multipotent neural stem/progenitor cells (NSPCs) produce various types of neurons and glial cells during the development of the central nervous system (CNS); however, NSPCs are not always able to generate all types of neural cells. The formation of complex neural networks relies on proper cytogenesis from NSPCs, which is under strict spatiotemporal regulation by intrinsic and extrinsic mechanisms. The neurogenesis-to-gliogenesis switch, a major event during CNS development, is largely dependent on the cell-autonomous temporal specification of NSPCs. The results of several previous studies suggest that developmental stage-dependent changes in the epigenetic status of proneural and astrocytic genes correlate with the temporal identity transition of developing NSPCs. These changes are related to alterations in the responsiveness of NSPCs to extrinsic neurogenic or gliogenic factors. Here, we discuss our recent findings that microRNA-mediated regulation of competence and relationships between multi-layered molecular regulatory systems control the temporal specification of NSPCs during CNS development.
Keywords: :
Competence Regulation by Epigenetic Modifications
In the canonical model of the fate determination of neural stem/progenitor cells (NSPCs), competitive sequestration of the Smad/CBP/p300 transcriptional co-activator complex by neurogenin (Ngn) or signal transducer and activator of transcription-3 (Stat3) drives a neurogenic or gliogenic fate by recruitment of the complex to neuronal or astrocytic genes, respectively.Citation1,Citation2 However, several lines of evidence regarding epigenetic regulation of Stat3 binding sites in glia-associated genes have suggested that the molecular machinery involved in the fate determination of NSPCs functions in a context-dependent manner.Citation3,Citation4 Cardiotrophin-1 (CT-1), which is derived from differentiated neurons, activates the JAK-STAT pathway and induces glial differentiation of late developmental NSPCs;Citation5 however, epigenetic silencing of the STAT3 binding sites in the promoters of glial genes prevents the response to this signaling pathway in early developmental NSPCs (, 1).Citation6,Citation7 Responsiveness to the neurogenic Wnt signal is also developmental stage-dependent; in late developmental NSPCs, epigenetic modification of the proneural Ngn gene prevents binding of the β-catenin/Tcf transcriptional complex (, 2).Citation8
Figure 1. (A) Molecular mechanisms underlying the regulation of the neurogenic-to-gliogenic transition of developing NSPCs by epigenetic modulation and miR-17/106. (1) Epigenetic silencing of the STAT3 binding sites in the promoters of glial genes such as Gfap prevents the response to the activated JAK-STAT signaling pathway in early developmental NSPCs. (2) Epigenetic modification of the proneural Ngn gene in late developmental NSPCs prevents binding of the Wnt signal-induced β-catenin/Tcf transcriptional complex. (3) High expression levels of miR-17/106 in early developmental NSPCs prevents activation of the p38 MAPK pathway and the initiation of neurogenic-to-gliogenic competence transition. (B) An overview of the multi-layered regulatory systems and molecular mediators involved in the neurogenic-to-gliogenic transition of NSPCs. In early developmental stages, miR-17/106 represses p38 expression and prevents the initiation of competence transition. Sequential expression of a number of transcription factors, including Foxg1, Onecut1/2, Nurr1, and multiple other proteins, regulates neuron-subtype specifications. Expression of Coup-tfI/II triggers the neurogenic-to-gliogenic competence transition of NSPCs. In late developmental NSPCs, upregulation of p38 caused by a decline in miR-17/106 expression and changes in the epigenetic statuses of proneural and astrocytic genes induce changes in the responsiveness to extrinsic signals, and BMP, Notch, JAK-STAT, and MEK signals promote gliogenesis. Regulators of the timing of differentiation and subsequent maturation also coordinate the neurogenic-to-gliogenic transition of NSPCs.
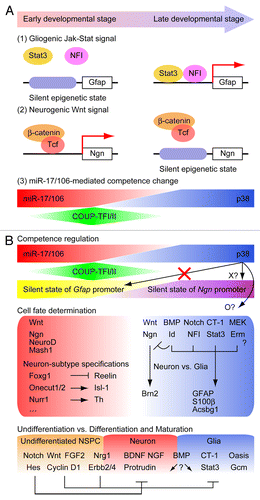
In a previous study, we demonstrated that chicken ovalbumin upstream promoter-transcription factors I and II (Coup-tfI and Coup-tfII), which are both nuclear orphan receptors, are involved in the neurogenic-to-gliogenic transition of NSPCs.Citation9 Remarkably, the results of the study suggested that Coup-tfI and Coup-tfII neither repress neuronal differentiation nor promote glial differentiation but, instead, alter the competence of NSPCs. Double knockdown of Coup-tfI/II inhibited the induction of gliogenesis and sustained the generation of early-born neurons by promoting the resistance of NSPCs to gliogenic cytokines such as CT-1, leukemia inhibitory factor (LIF), and bone morphogenetic protein (BMP). Moreover, the promoter of the gene encoding glial fibrillary acidic protein (Gfap) was maintained in an early developmental silent epigenetic state in the double knockdown NSPCs.Citation9 These observations indicate that Coup-tfI and Coup-tfII function as the trigger of the neurogenic-to-gliogenic competence transition of NSPCs.
Competence Regulation by miR-17/106
The expression of Coup-tfI/II in NSPCs peaks at the mid-gestation stage and then declines during the gliogenic phase. Therefore, we hypothesized that epigenetic modulations of the target genes of Coup-tfI/II are involved in the competence transition of NSPCs. However, our recent functional screening analysis revealed that microRNAs (miRNAs) miR-17 and miR-106a/b, which have redundant functions and are encoded in paralogous miR-17–92, miR-106b-25, and miR-106a-363 clusters, are the downstream effectors of Coup-tfI/II that are responsible for the competence transition of NSPCs (, 3).Citation10 In this study, similar to the effect of Coup-tfI/II knockdown, overexpression of miR-17 inhibited gliogenesis; however, unlike Coup-tfI/II knockdown, it did not affect late developmental epigenetic de-silencing of the Gfap promoter. In addition, overexpression of miR-17 restored the neuropotency of late developmental gliogenic NSPCs, even after de-silencing of the Gfap promoter.Citation10 In the same study, we used proteomics analyses in combination with in silico predictions of miRNA-binding sites to identify p38 (also known as Mapk14) as a direct target of miR-17/106 that controls the competence transition of NSPCs. The expression level of p38 in NSPCs increases during development and inhibition of p38 by a short hairpin RNA or the specific inhibitor SB203580 restored the neurogenic competence of late developmental NSPCs.Citation10 We also investigated the function of p38 using a miR-17/106-resistant form of the cDNA clone that lacked the miR-17-binding site. In these experiments, overexpression of p38 abolished the neurogenic effect of miR-17 and induced the generation of astrocytes from early developmental NSPCs in the presence of LIF and BMP2. Overall, these results suggest that p38 plays a critical role in regulating the temporal identity transition of NSPCs; however, elucidation of the upstream and downstream molecular events is required to understand the mechanisms involved in competence regulation fully.
In addition to miR-17/106, various other miRNAs are also associated with NSPC-driven cytogenesis. The expression levels of miR-124 and let-7 family members increase upon differentiation of NSPCs into neurons. In a previous mouse study, loss of miR-124 induced ectopic gliogenesis at the expense of neurogenesis.Citation11 However, another study suggested that miR-124 plays a role in the axonal maturation of hippocampal neurons by repressing the gene (Lhx2) that encodes LIM-homeodomain transcription factor, a critical regulator of early forebrain development.Citation12 Along with let-7 and miR-124, miR-9 and its star strand (miR-9*) control the proliferation and commitment of NSPCs by repressing the expression levels of the nuclear receptor TLX and a SWI/SNF-like BAF chromatin remodeling complexes BAF53a (also known as Actl6a), respectively.Citation13-Citation15 Furthermore, the role of the miR-17–92 cluster in cortical development has also been investigated.Citation16 The cluster members miR-19 and miR-92 regulate the proliferation and transition of radial glial cells to intermediate progenitors by targeting the tumor suppressor gene Pten and the transcription factor Tbr2 gene, respectively. As described above, these miR-124, miR9/9*, and let-7 seem to regulate the timing of the differentiation and maturation of NSPCs rather than the neurogenic-to-gliogenic competence transition.
Relationships between Multi-Layered Regulatory Systems
Although the miR-17/106-mediated competence regulatory system is involved in the neurogenesis-to-gliogenesis transition, the relationships between this and other systems are unclear. One can argue that there are three major hierarchical regulatory layers: competence regulation, cell fate determination, and differentiation/maturation (). As mentioned above, overexpression of miR-17 did not alter the epigenetic state of the Gfap promoter and delayed overexpression of miR-17 restored the neuropotency of gliogenic NSPCs. Therefore, the miR-17/106-p38 axis appears to function independently of epigenetic de-silencing of glial genes. Although we have not examined the influence of overexpression of miR-17 on the epigenetic status of proneural genes, the results of the delayed overexpression experiments suggest that miR-17 abolishes epigenetic silencing of proneural genes even in highly gliogenic NSPCs. Similarly, overexpression of the high mobility group AT-hook 2 (HMGA2) protein also restores the neuropotency of late developmental gliogenic NSPCs.Citation17 HMGA2 is a chromatin remodeling factor that modulates the global chromatin state to generate transcriptionally permissive euchromatin, suggesting that epigenetic regulation plays an important role in the neurogenic-to-gliogenic transition of NSPCs. Moreover, let-7b regulates HMGA2 translation and controls the self-renewal of NSPCs.Citation18 Therefore, investigation of the relationships between miRNA-mediated competence changes and epigenetic regulation, including the crosstalk between the miR-17/106-p38 and let7-HMGA2 axes, should lead to a more complete understanding of the regulatory system underlying NSPC-driven cytogenesis.
Several gliogenic signals have been identified, including the BMP-Id,Citation19 Notch-nuclear factor I (NFI),Citation20 CT-1-STAT3,Citation5 and MEK-ErmCitation21 pathways (). Overexpression of a constitutively active form of Mek1 induces an ectopic increase in the number of GFAP-positive astrocytes and reduces neurogenesis in the mouse brain.Citation21 Furthermore, deletion of Mek1/2 results in a severe loss of gliogenesis and prolonged neurogenesis at late embryonic stages. Therefore, it will be interesting to investigate the interactions between the Raf-Mek-Erk and p38 MAPK pathways. On the other hand, other evidence suggests that the coordinate action of the BMP, Notch, and JAK-STAT pathways is central to the differentiation of astrocytes. BMP signaling induces the expression of negative helix-loop-helix Id proteins that inhibit Ngn and cause astrocytic differentiation of NSPCs in combination with the activation of the JAK-STAT pathway.Citation19 In the early developmental stages, Notch signaling induces the expression of the basic helix-loop-helix transcription factors Hes-1/5 and maintains the undifferentiated state of NSPCs by inhibiting neuronal differentiation. In later stages of development, Notch signaling induces the expression of the gliogenic transcription factor NFIA, which is a key activator of gliogenesis and a repressor of Hes-1.Citation20,Citation22,Citation23 However, we found that overexpression of NFIA in neurogenic NSPCs caused only a limited increase in the number of GFAP-positive astrocytes (unpublished data), suggesting that it functions in a context-dependent manner. In support of this concept, NFIA is necessary for not only gliogenesis, but also neuronal differentiation and axonal guidance.Citation23-Citation25 Moreover, Sox9 is an essential inducer of NFIA expression in embryonic spinal cord and forms a complex with NFIA that binds to cis-elements to regulate a subset of target genes.Citation26 The molecular mechanisms underlying the multiple functions of NFI are still unclear; therefore, unveiling the relationships between temporal NFI functions and the p38 MAPK pathway is an important requirement. In our recent study, overexpression of p38 induced precocious gliogenesis in neurogenic NSPCs only when gliogenic factors were supplied.Citation10 Moreover, treatment of NSPCs with the p38-specific inhibitor SB203580 or knockdown of p38 with a specific short hairpin RNA repressed gliogenesis. These observations suggest that, rather than acting as a negative regulator of neurogenesis, p38 positively but permissively regulates gliogenesis (). Downstream targets of p38 may require the gliogenic function of NFI, and miR-17/106-mediated regulation of competence may therefore be essential for the neurogenic-to-gliogenic transition of NSPCs.
Concluding Remarks
The expression levels of multiple miRNAs are altered during development; at least some of these miRNAs should be associated with the temporal competence of NSPCs. Elucidation of the functions of various miRNAs in developing NSPCs should lead to a more complete understanding of the regulation of competence and the roles of the multi-layered molecular machinery involved in the development of the central nervous system.
| Abbreviations: | ||
| BMP | = | bone morphogenetic protein |
| CNS | = | central nervous system |
| CT-1 | = | cardiotrophin-1 |
| LIF | = | leukemia inhibitory factor |
| miRNA | = | microRNA |
| Ngn | = | neurogenin |
| NSPC | = | neural stem/progenitor cell |
| Stat | = | signal transducer and activator of transcription |
Disclosure of Potential Conflicts of Interest
H.O. is a scientific consultant for San Bio, Inc., Eisai Co., Ltd., and Daiichi Sankyo Co., Ltd.
References
- NakashimaK, YanagisawaM, ArakawaH, KimuraN, HisatsuneT, KawabataM, MiyazonoK, TagaT. Synergistic signaling in fetal brain by STAT3-Smad1 complex bridged by p300. Science1999; 284:479 - 82; http://dx.doi.org/10.1126/science.284.5413.479; PMID: 10205054
- SunY, Nadal-VicensM, MisonoS, LinMZ, ZubiagaA, HuaX, FanG, GreenbergME. Neurogenin promotes neurogenesis and inhibits glial differentiation by independent mechanisms. Cell2001; 104:365 - 76; http://dx.doi.org/10.1016/S0092-8674(01)00224-0; PMID: 11239394
- ShenQ, WangY, DimosJT, FasanoCA, PhoenixTN, LemischkaIR, IvanovaNB, StifaniS, MorriseyEE, TempleS. The timing of cortical neurogenesis is encoded within lineages of individual progenitor cells. Nat Neurosci2006; 9:743 - 51; http://dx.doi.org/10.1038/nn1694; PMID: 16680166
- OkanoH, TempleS. Cell types to order: temporal specification of CNS stem cells. Curr Opin Neurobiol2009; 19:112 - 9; http://dx.doi.org/10.1016/j.conb.2009.04.003; PMID: 19427192
- Barnabé-HeiderF, WasylnkaJA, FernandesKJ, PorscheC, SendtnerM, KaplanDR, MillerFD. Evidence that embryonic neurons regulate the onset of cortical gliogenesis via cardiotrophin-1. Neuron2005; 48:253 - 65; http://dx.doi.org/10.1016/j.neuron.2005.08.037; PMID: 16242406
- TakizawaT, NakashimaK, NamihiraM, OchiaiW, UemuraA, YanagisawaM, FujitaN, NakaoM, TagaT. DNA methylation is a critical cell-intrinsic determinant of astrocyte differentiation in the fetal brain. Dev Cell2001; 1:749 - 58; http://dx.doi.org/10.1016/S1534-5807(01)00101-0; PMID: 11740937
- SongMR, GhoshA. FGF2-induced chromatin remodeling regulates CNTF-mediated gene expression and astrocyte differentiation. Nat Neurosci2004; 7:229 - 35; http://dx.doi.org/10.1038/nn1192; PMID: 14770186
- HirabayashiY, SuzkiN, TsuboiM, EndoTA, ToyodaT, ShingaJ, KosekiH, VidalM, GotohY. Polycomb limits the neurogenic competence of neural precursor cells to promote astrogenic fate transition. Neuron2009; 63:600 - 13; http://dx.doi.org/10.1016/j.neuron.2009.08.021; PMID: 19755104
- NakaH, NakamuraS, ShimazakiT, OkanoH. Requirement for COUP-TFI and II in the temporal specification of neural stem cells in CNS development. Nat Neurosci2008; 11:1014 - 23; http://dx.doi.org/10.1038/nn.2168; PMID: 19160499
- Naka-KanedaH, NakamuraS, IgarashiM, AoiH, KankiH, TsuyamaJ, TsutsumiS, AburataniH, ShimazakiT, OkanoH. The miR-17/106-p38 axis is a key regulator of the neurogenic-to-gliogenic transition in developing neural stem/progenitor cells. Proc Natl Acad Sci U S A2014; 111:1604 - 9; http://dx.doi.org/10.1073/pnas.1315567111; PMID: 24474786
- ÅkerblomM, SachdevaR, BardeI, VerpS, GentnerB, TronoD, JakobssonJ. MicroRNA-124 is a subventricular zone neuronal fate determinant. J Neurosci2012; 32:8879 - 89; http://dx.doi.org/10.1523/JNEUROSCI.0558-12.2012; PMID: 22745489
- SanukiR, OnishiA, KoikeC, MuramatsuR, WatanabeS, MuranishiY, IrieS, UneoS, KoyasuT, MatsuiR, et al. miR-124a is required for hippocampal axogenesis and retinal cone survival through Lhx2 suppression. Nat Neurosci2011; 14:1125 - 34; http://dx.doi.org/10.1038/nn.2897; PMID: 21857657
- ZhaoC, SunG, LiS, ShiY. A feedback regulatory loop involving microRNA-9 and nuclear receptor TLX in neural stem cell fate determination. Nat Struct Mol Biol2009; 16:365 - 71; http://dx.doi.org/10.1038/nsmb.1576; PMID: 19330006
- ZhaoC, SunG, LiS, LangMF, YangS, LiW, ShiY. MicroRNA let-7b regulates neural stem cell proliferation and differentiation by targeting nuclear receptor TLX signaling. Proc Natl Acad Sci U S A2010; 107:1876 - 81; http://dx.doi.org/10.1073/pnas.0908750107; PMID: 20133835
- YooAS, StaahlBT, ChenL, CrabtreeGR. MicroRNA-mediated switching of chromatin-remodelling complexes in neural development. Nature2009; 460:642 - 6; PMID: 19561591
- BianS, HongJ, LiQ, SchebelleL, PollockA, KnaussJL, GargV, SunT. MicroRNA cluster miR-17-92 regulates neural stem cell expansion and transition to intermediate progenitors in the developing mouse neocortex. Cell Rep2013; 3:1398 - 406; http://dx.doi.org/10.1016/j.celrep.2013.03.037; PMID: 23623502
- KishiY, FujiiY, HirabayashiY, GotohY. HMGA regulates the global chromatin state and neurogenic potential in neocortical precursor cells. Nat Neurosci2012; 15:1127 - 33; http://dx.doi.org/10.1038/nn.3165; PMID: 22797695
- NishinoJ, KimI, ChadaK, MorrisonSJ. Hmga2 promotes neural stem cell self-renewal in young but not old mice by reducing p16Ink4a and p19Arf Expression. Cell2008; 135:227 - 39; http://dx.doi.org/10.1016/j.cell.2008.09.017; PMID: 18957199
- NakashimaK, TakizawaT, OchiaiW, YanagisawaM, HisatsuneT, NakafukuM, MiyazonoK, KishimotoT, KageyamaR, TagaT. BMP2-mediated alteration in the developmental pathway of fetal mouse brain cells from neurogenesis to astrocytogenesis. Proc Natl Acad Sci U S A2001; 98:5868 - 73; http://dx.doi.org/10.1073/pnas.101109698; PMID: 11331769
- NamihiraM, KohyamaJ, SemiK, SanosakaT, DeneenB, TagaT, NakashimaK. Committed neuronal precursors confer astrocytic potential on residual neural precursor cells. Dev Cell2009; 16:245 - 55; http://dx.doi.org/10.1016/j.devcel.2008.12.014; PMID: 19217426
- LiX, NewbernJM, WuY, Morgan-SmithM, ZhongJ, CharronJ, SniderWD. MEK Is a Key Regulator of Gliogenesis in the Developing Brain. Neuron2012; 75:1035 - 50; http://dx.doi.org/10.1016/j.neuron.2012.08.031; PMID: 22998872
- DeneenB, HoR, LukaszewiczA, HochstimCJ, GronostajskiRM, AndersonDJ. The transcription factor NFIA controls the onset of gliogenesis in the developing spinal cord. Neuron2006; 52:953 - 68; http://dx.doi.org/10.1016/j.neuron.2006.11.019; PMID: 17178400
- PiperM, BarryG, HawkinsJ, MasonS, LindwallC, LittleE, SarkarA, SmithAG, MoldrichRX, BoyleGM, et al. NFIA controls telencephalic progenitor cell differentiation through repression of the Notch effector Hes1. J Neurosci2010; 30:9127 - 39; http://dx.doi.org/10.1523/JNEUROSCI.6167-09.2010; PMID: 20610746
- ShuT, ButzKG, PlachezC, GronostajskiRM, RichardsLJ. Abnormal development of forebrain midline glia and commissural projections in Nfia knock-out mice. J Neurosci2003; 23:203 - 12; PMID: 12514217
- PlachezC, LindwallC, SunnN, PiperM, MoldrichRX, CampbellCE, OsinskiJM, GronostajskiRM, RichardsLJ. Nuclear factor I gene expression in the developing forebrain. J Comp Neurol2008; 508:385 - 401; http://dx.doi.org/10.1002/cne.21645; PMID: 18335562
- KangP, LeeHK, GlasgowSM, FinleyM, DontiT, GaberZB, GrahamBH, FosterAE, NovitchBG, GronostajskiRM, et al. Sox9 and NFIA coordinate a transcriptional regulatory cascade during the initiation of gliogenesis. Neuron2012; 74:79 - 94; http://dx.doi.org/10.1016/j.neuron.2012.01.024; PMID: 22500632