Abstract
The nuclear envelope and lamina define the nuclear periphery and are implicated in many nuclear processes, including chromatin organization, transcription and DNA replication. Mutations in lamin A proteins, major components of the lamina, interfere with these functions and cause a set of phenotypically diverse diseases referred to as laminopathies. The phenotypic diversity of laminopathies is thought to be the result of alterations in specific protein- and chromatin interactions due to lamin A mutations. Systematic identification of lamin A-protein and -chromatin interactions will be critical to uncover the molecular etiology of laminopathies. Here we summarize and critically discuss recent technology to analyze lamina protein- and chromatin interactions.
Introduction
The nuclear periphery is marked by the nuclear envelope (NE), which is composed of an outer and inner nuclear membrane (ONM, INM, respectively) interrupted by nuclear pore complexes (NPC).Citation1 The nuclear lamina lines the NE and consists of a large collection of proteins, most prominently the intermediate filament A-type (Lamin A, C) and B-type lamins (Lamin B1, B2), INM proteins anchoring the lamina to the NE (including Emerin, MAN1, LAP2, Nesprin) and proteins that modulate and interact with chromatin such as BAF, HP1 and histone deacetylase 3 ().Citation2
Mutations in the lamina's major constituent, particularly the A-type lamins, cause a diverse set of human diseases collectively referred to as laminopathies; these include several types of muscular dystrophies, lipodystrophies, cardiomyopathies, neurological disorders and premature aging syndromes.Citation3 The phenotypic diversity of laminopathies is hypothesized to be caused by lamin A mutations affecting specific lamina-protein and -chromatin-interactions, thereby compromising nuclear integrity, higher-order chromatin organization, gene expression and/or various other nuclear processes.Citation4 In line with this notion are the observations that Dunnigan familial partial lipodystrophy (FPLD)-associated lamin A mutations specifically disrupt interaction with the adipogenic transcription factor Srebp1Citation5 and that failure of progerin, a lamin A mutant that causes premature aging, to interact with the NURD chromatin remodeling complex contributes to loss of peripheral heterochromatin, a hallmark of the premature aging disorder Hutchinson-Gilford Progeria syndrome (HGPS).Citation6 Another progeria-associated lamin A mutation, E145K, leads to aberrant interaction of the lamina with telomeres.Citation7
A key challenge in the field of lamin biology is to identify all protein and chromatin interactions at the nuclear periphery. Over the past years, several approaches have been developed and applied in order to systematically map the complete spectrum of lamina-protein and -chromatin interactions. Such approaches are crucial to pinpoint the biological function of the lamina and pinpoint molecular defects for specific laminopathies. In this review we provide an overview of current technologies aimed at identifying protein- and chromatin-interactions at the lamina. We focus on proteome- and genome-wide unbiased approaches, with particular emphasis on technical advantages and potential pitfalls in the context of subsequent mass spectrometry,Citation8,Citation9 high-throughput microscopy and mass sequencing analysis.Citation10 The methods discussed, each with its strengths and weaknesses, are all complementary but they all contribute to increasing our knowledge of function of lamins and the nuclear periphery.
Protein Interactions
Visual screens.
A basic approach to identifying lamina proteins is by visual inspection of the localization of candidate proteins. In visual screens putative lamina proteins are expressed using specific tags and their localization is then detected either using live cell imaging or indirect immunofluorescence. The use of fluorescent or other epitope tags has made it possible to systematically visualize the localization of a large number of putative lamina proteins and lamin-interacting proteins by high-throughput microscopy. The most commonly used tags are GFP and Myc. The foremost advantage of visual screens is the instant registration of dynamic or abnormal changes in subcellular protein-complex localization in the context of changed cell physiology or protein mutation, respectively (). Capture of such dynamic behavior has been successfully appliedCitation11 to the study of the nuclear lamina; lamin A interactions with Rb, c-FOS and SMAD2 occur only under specified conditions, like proliferation, differentiation and TGFβ stimulation.Citation12,Citation13 Sensitivity, specificity and spatial resolution in cell-based imaging are somewhat confounded by the necessity to co-stain for relevant subcellular domains as well as low cellular abundance of NE proteins.Citation14–Citation17 Bioinformatic approaches can assist in visual screens by selecting for predicted membrane proteinsCitation18 localized at the NE.Citation19
A powerful and more sophisticated variation of a visual screen to identify novel lamin interaction partners is the use of GFP-fusion protein libraries. In this approach collections of GFP-tagged fusion proteins are expressed, screened by high-throughput microscopy for subcellular localization, and clones of interest are subsequently sequenced to determine the identity of the expressed protein. Rolls et al. successfully applied a heterologous promoter-driven GFP-fusion library to screen 40,000 clones and test in an unbiased fashion whether they localized to the nuclear periphery.Citation20 In doing so they discovered the NE membrane protein Nurim, a six-transmembrane spanning INM protein with potential isoprenylcysteine carboxymethyltransferase enzymatic activity for Caax-motifs.Citation20,Citation21 A drawback of protein overexpression is possible mis-localization of the fusion protein; there is an obvious need for analysis of the effect of protein expression levels on its localization and the validation of each hit by analysis of the endogenous protein.Citation15,Citation22 A key advantage of visual screens is that interactors which may associate with the small, but significant pool of lamin components present in the nucleoplasm, can be distinguished from those that interact with the peripheral pool of lamins. In a more physiologically relevant approach, Bickmore and colleagues identified the NE associated protein Lyric/AEG-1, an apoptosis and cell growth implicated transcriptional regulator, by using gene trapping to insert 1,350 reporters into active genes.Citation11,Citation23 In this approach, endogenously expressed genes were spliced onto a genomically integrated LacZ cassette, which allowed visualization of the resulting fusion proteins by X-gal staining and β-galactosidase immunohistochemistry.Citation11 Although such visual screens are becoming increasingly feasible, they are relatively labor intensive and slow.
Biochemical fractionation.
NE proteins are highly lipophylic and lamina proteins are strongly resistant to high concentrations of salts and detergents. These biochemical characteristics are exploited in fractionation studies to separate NE and lamina proteins from other subnuclear domains. Such isolation drastically increases the frequency of detecting NE and lamina proteins by unbiased biochemical methods e.g., mass spectrometry (MS) (). Combined with recent MS advances in complex protein mixture analysis,Citation9,Citation24 such fractionation studies have the potential to contribute significantly to the identification of the full lamina proteome. As MS analysis itself cannot distinguish lamina proteins from contaminants, fraction purity is crucial. Various assays have been developed to fractionate NE and lamina proteome subsets, each with a specific trade-off between purity and the amount of background proteins.
Protein Correlation Profiling (PCP) was developed to determine subcellular protein localization in crude extracts, separated by rate-zonal centrifugation into fractions which are subsequently analyzed by MS analysis ().Citation25 In essence, the technology relies on co-detection of proteins known to reside in the organelle of interest and novel proteins.Citation25 The main advantage of PCP is its ability to detect multiple co-segregating proteins in a complex mixture without the need to fully isolate and highly purify subcellular fractions. However, PCP comes at a price: as many subcellular domains are only partially separated by centrifugation, non-specific interactors co-purify and separation of true interactors relies strongly on computational analysis. Follow-up studies to characterize the properties of identified proteins are imperative. Although PCP analysis has not been applied yet to distinguish and define nuclear lamina/envelope protein domains, the fact that differences in rate-zonal resolving properties have previously been used to purify NE fractions,Citation26 makes PCP a promising technique to identify NE and lamina proteins.
In comparison to PCP, differential extraction assays use more highly purified fractions for MS analysis and consequently reduce non-specific co-purification (). In general, biochemical fractionation of the NE starts with isolation of nuclei, removal of non-NE membrane fractions by centrifugation and digestion of chromatin to remove nucleoplasmic contents. Crude NE and lamina fractions are subsequently extracted in salt, detergent and chaotrope or alkaline buffers to further remove different types of proteins. Salt preferentially dissolves chromatin-bound proteins and other non-membrane, weakly attached lamina proteins, but not lamina proteins (). Detergents (Octyl glucoside, Trx-100, Empigen BB) mimic the lipid bilayer environment and particularly dissolve membrane-associated proteins, except those that are anchored to the detergent resistant lamina (). In this manner Cronshaw et al. successfully identified 6 novel NPC components, among which ALADIN, the gene mutated in the triple-A or Allgrove syndrome.Citation27 Chaotropes (urea, thiourea) and alkalines (NaOH) are used to solubilize cytoskeletal, chromatin and lamina components, while leaving integral transmembrane proteins embedded in the insoluble membrane fraction (). As such lamina and lamina-anchored INM/NE proteins are extracted by combined application of salt and detergent (; reviewed in ref. Citation29 and Citation30), whereas integral INM-, ONM- and ER-membrane proteins are purified by chaotrope or alkaline extraction ().
Despite increased sample purity by applying differential extractions, these fractionation studies are still hampered by co-purification of non-specific interactors, in particular from the peripheral ER, which is continuous with the ONM and therefore difficult to separate from the NE and lamina. To further reduce false positive hits, Dreger et al. compared salt, detergent and chaotrope/alkaline extractions, and were able to eliminate ER contaminants in chaotrope/alkaline resistant fractions.Citation28 This strategy identified 19 previously unknown and putative integral INM proteins including Unc84a (Sun1), LUMA and two LAP2 isoforms.Citation28 A disadvantage of this comparative approach is that selection is based on the assumption that all INM and lamina proteins have similar biochemical extraction characteristics. However, several lamina anchored INM proteins, like emerin, LBR and LAP1, behave biochemically very different in detergent or chaotrope-based extraction.Citation28
In an extenstion of purification methods, subtractive approaches can be used to filter out ER residing proteins. In these methods proteins identified in non-NE/lamina fractions, thus enriched for background, are subtracted from proteins detected in differential NE/lamina extracts. Schirmer and colleagues used microsomal membrane fractions as a source of background proteins, as these ER-rich fractions are easily obtained and can be prepared free of nuclear membranes.Citation29 By combining differential fractionation and subtractive proteomics, they identified 67 previously unidentified NE transmembrane proteins. The disadvantage of using a reference background source is that proteins that reside in both the ER and NE, like AEG-1/Lyric, Sec13 and Torsin A, are inadvertently discarded.Citation30–Citation32 This is a serious concern, since it is now estimated that one third of cellular proteins have multiple organellar localizations.Citation25
Affinity purification.
An alternative approach to identifying lamina interactions is affinity purification. In these approaches a protein of interest is epitope tagged and the bait protein is affinity purified using antibodies against the tag (). Affinity purification can identify and distinguish bound protein complexes from each other by co-elution and MS analysis. Such gradual elution was used for example to separate emerin-interacting RNA processing, signaling and chromatin remodeling complexes.Citation33 In addition, it is possible to study interactions in the context of post-translational modifications by using specific antibodies directed against them, for example phosphorylation-dependent LBR-p32/p34,Citation34 lamin A-Rb and -Smad2,Citation12 interactions. In contrast to biochemical fractionation, in which fractions are generated under denaturing conditions, the main challenge in affinity purification assays is to extract as many NE and lamina proteins while leaving protein interactions intact. Overly stringent solubilization dissolves many proteins at the cost of disrupting complexes, while excessively mild conditions do not dissolve all relevant interactors. Various strategies have been applied to find a good balance for this trade-off.
In classic immunoprecipitations (IPs) solubilization conditions are optimized for the protein of interest (). Low amounts of detergents and salt preferentially solubilize nucleoplasmic pools of proteins (), as described to exist for lamin A,Citation35 and were mainly applied to study easy extractable, weakly bound NE interactors (Smad2, PP2A, Rb, Ubc9, hnRNP1, EGF1, SREBP1; See ).Citation5,Citation12,Citation36–Citation38 Increasing amounts of detergents, salts and the solvent glycerolCitation39 successfully solubilize protein complexes of well-anchored NE and lamina components (LAP2β, Emerin, Nesprin2, Lamin B; ), although sometimes at the cost of disrupting interactions (LaminA/B1/B2-LAP2β)Citation40 (). Highly stringent conditions were applied when studying NPC proteins as they were assumed to be highly stable structures ().Citation41–Citation43 Even though NPCs apparently better withstand stringent extraction, increased stringency of washing buffers disrupts interactions ().Citation41 An additional disadvantage to be accounted for is that lysis buffers also can affect the antibody/epitope-interaction. Various groups therefore prefer to dilute buffer compositions after initial lysis, which combines increased solubilization with the ability of protein complexes to reassemble and antibodies to bind under sequentially milder conditions. This strategy was used to identify unknown interactors for BAF and Emerin ().Citation33,Citation44 Other limitations of antibody-based methods include the unavailability of IP-able antibodies,Citation36 antibodies that recognize multiple epitopes (MAN1 antiserum),Citation36 antibodies that cross-react undesirably with non-mature forms of a protein (prelamin A versus lamin A)Citation38 or even disrupt protein interactions (laminA/B2-LAP2β).Citation40
To avoid the use of antibodies, precipitations can be performed using bacterially expressed and purified baits, conjugated to beads prior to incubation with solubilized protein extracts. Fusing the bait to an epitope tag contributes to high quality purification of the bait and efficient precipitation of interactors from protein extracts (). This approach was combined with mild lysis, for BAF and emerin interactors,Citation33,Citation44 or more stringent buffers, for lamin-LAP2β and -nesprin 2 interactions ().Citation40,Citation45 In accordance with mild solubilization, identified BAF and emerin interactors represented many proteins that also reside outside the nuclear periphery (PARP, HP1gamma, RBBP4,7).Citation40,Citation45 Another advantage of using a bait is the ability to pinpoint interactions to relevant protein domains, as the bait does not have to be incorporated in vivo and therefore cannot mislocalize, as described to occur for LAP2β constructs.Citation40 The main disadvantages of IPs is that solubilization issues still remain and interactions formed in vitro do not necessarily occur in vivo.
A major step forward in overcoming solubilization problems is the OneSTrEP (OST) pull-down assay,Citation6,Citation46 which combines the use of a biotin resembling OST-tagCitation47,Citation48 with mild cross-linking of cells prior to solubilization (). Cross-linking allows extraction of the total lamin A/C poolCitation46 () while leaving protein interactions intact. The OST-tagged protein and its interactors can then be highly efficiently precipitated under denaturing conditions using a high-affinity, engineered streptavidin analogue (). Stringent washes reduce background, especially relevant for A-type lamins, known to be “sticky” proteins and reported to precipitate in negative pull-down controls as well.Citation44 The main advantages of the OST pull-down are that it can be used to identify a full in vivo interactome of a protein regardless of its subnuclear position, detect the effect mutations have on these interactions and identify weak interactors.Citation46 OST pull-downs have been used to compare protein interactions for lamin A and progerin, an HGPS causing lamin A mutant and detected a decreased interaction for progerin with NPC components.Citation44 The main disadvantages of this approach are that it is not possible to study endogenous proteins and that cross-linking does not allow gradual elution and thereby separation of interacting protein complexes ().
Yeast two-hybrid.
An alternative to affinity purifications is yeast two-hybrid (Y2H) (), in which a direct interaction between a DNA-binding domain fused bait and a co-expressed transcriptional activating domain fused prey allows growth under restrictive conditions. When the primary interest is to identify weak and direct interactors, Y2H is useful as bait and prey are expressed by strong exogenous promoters, protein solubilization is not required and weak interactions are sufficient to allow restrictive growth. The focus on direct protein interactions, which might best reflect the core activities of the protein of interest, restricts the mapping of a complete interactome. Protein fragments can easily be used as bait since they don't have to be incorporated in vivo, and have been applied to describe interactions between the specific domains of the nuclear envelope proteins otefin, lamin A, nesprin.Citation45,Citation49,Citation50 The benefit of choosing the exact bait composition can further be exploited by choosing domains involved in disease mechanisms, like the 50 amino acid deleted region in progerin, shown in a Y2H screen to interact with the NURD chromatin remodeling complex component Rbbp4.Citation6 Disadvantages of Y2H assays are the lack of information on protein complex composition, the inability to study posttranslational modifications and the large amount of false positives identified. The large amount of background can be caused by endogenous transcriptional activity of bait or prey proteins as reported for cFOS domains used to map lamin A interaction,Citation13 bait or prey proteins affecting yeast growth under restrictive conditions, and the fact that investigated interactions may never occur in vivo (). In addition, Y2H approaches for lamin proteins are particularly difficult since expression of lamin-fusion proteins in S. Cerevisiae has detrimental effects on the organism.
Chromatin-Interactions
In addition to protein-protein interactions, the importance of interactions between chromatin and the lamina is increasingly appreciated. In particular, many lamin proteins are now known to directly or indirectly interact with chromatin and chromatin defects are a hallmark of several laminopathies.Citation51–Citation53 These observations have catalyzed the development of unbiased screening techniques for chromatin interactions at the NE. A broad distinction can be made between assays using affinity purification and those based on enzymatic activity ( and ).
Affinity based approaches: ChIP & OST pull-down.
Chromatin-protein interactions are most commonly interrogated using chromatin-immunoprecipitation (ChIP) methods. In this approach, a protein of interest is cross-linked to chromatin and immunoprecipitated using a specific antibody against the protein. The DNA is then identified either by targeted PCR methods or by genome-wide microarray or sequencing approaches. The major difference between conventional IPs and ChIP is the addition of a cross-linking step prior to solubilization of intact protein-chromatin complexes. Cross-linking provides the advantage of combining ultra-sonication and stringent lysis, to shear DNA and dissolve NE proteins (), with good preservation of protein-chromatin interactions (). Just as for classic IPs, lysis buffers still need to be adjusted to the strength of the epitope-antigen interaction. For this reason, initial ChIP studies were performed on Myc-tagged NPC proteins in S. Cerevisae,Citation54 as NPCs are easily dissolved in the absence of nuclear lamina and high quality ChIP-suited Myc antibodies are commercially available. For the INM protein Src1, a MAN1 resembling protein, interactions with (sub)telomeric regions were identified in yeast using a high affinity protA-system.Citation55,Citation56 Silver et al. used endogenous Nup93 in HeLa cells by dialyzing the initial lysis buffer to a milder variant prior to incubation with antibodies.Citation57 The foremost advantage of using antibodies is the ability to study endogenous proteins and chromatin interactions in the context of posttranslational modifications ().
A modification of the classical ChIP approach is the use of the OneSTrEP tag (OST) pull-down which enables high affinity precipitation of OST-tagged proteins under denaturing conditions completely dissolving A-type lamins, comparable to the use of OST tags used for pull-down of proteins ().Citation46 The OST pull-down for identification of chromatin interactions is highly similar to that for detecting protein interactions and only includes slight changes in sonication and washing conditions.Citation46 Although OST pull-downs have the advantage of easy solubilization and high affinity pull-down without the use of antibodies, which in the case of lamin A have not been ideal in ChIP experiments, a limitation is the inability to directly study endogenous proteins and posttranslational modifications ().
Enzymatic activity based approaches: DamID, in vivo CHeC, ChIC.
DamID is an enzyme-based method for the in vivo mapping of chromatin-protein interactions. In DamID a protein of interest is fused to a DNA adenine methyltransferase (Dam) and expressed. Upon binding of the fusion protein to chromatin, the Dam activity marks in the vicinity bound chromatin by methylation, thereby enabling selective DpnI restriction in vitro. The marked sites can then be identified by targeted PCR or, more commonly, by genome-wide microarray analysis and deep-sequencingCitation58 (). The main advantage of using a tag that enzymatically marks DNA is that only isolation of DNA, not of intact protein/chromatin complexes, is required, thus eliminating any issues related to interaction stability. In addition, there is no need for cross-linking, thereby avoiding potential fixation artifacts. These characteristics made DamID the first technique to characterize and compare chromatin interactions for the relative insoluble lamin B and emerin proteins in a genome-wide fashion and resulted in the characterization of lamin associated domains (LADs) which define regions of the genome that preferentially interact with the lamina.Citation58,Citation59 Disadvantages of DamID include the inability to study posttranslational modifications, a slightly reduced resolution compared to alternative assays and potential interference of the tag with protein localization or function ().Citation60 In addition, since DamID relies on the expression of an enzymatically active fusion protein, it monitors chromatin interactions over a relative long period of time (app. 24 hours) and therefore is less useful to detect rapid interaction changes and dynamic reorganization of chromatin. Due to the DNA binding activities and high enzymatic activity of Dam, tagged proteins can only be expressed in trace amounts in order to prevent saturation of non-targeted DNA methylation.Citation61 This makes it not possible to study chromatin interactions in a dosage dependent manner, which could be relevant for diseases in which phenotypes are dependent on the amount of protein, such as HGPS.Citation62
An alternative method to measure protein-chromatin interactions is ChEC (chromatin endogenous cleavage). In this approach, micrococcal nuclease (MNase) is fused to a protein of interest and expressed. The fusion protein is recruited to its endogenous sites on chromatin where the MNase introduces double strand breaks at nuclease hypersensitive sites (HS).Citation63,Citation64 The MNase tag remains inactive under physiological Ca2+ concentrations, which provides the ability to selectively turn its activity on in vivo by addition of calcium chloride to mildly permeabilized cells. Cleaved chromatin can either be directly used to map HS sites by indirect end-labeling and Southern blotting, or is first selectively amplified by ligation-mediated PCR prior to genome-wide microarray analysis and deep-sequencing ().Citation63 Laemmli and colleagues used this approach to map chromatin interactions of the nuclear pore complex protein Nup2 and found that Nup2-gene promoter interactions typically are an early event of gene activation and are independent of transcription.Citation63 Control over MNase activity and relative short times needed for digestion make this assay suitable for detection of rapid changes in interactions. The major strength of in vivo ChEC is that there is no need to dissolve intact protein/chromatin complexes and information on chromatin structure is obtained by mapping HS (). In comparison to DamID higher expression levels of MNase-tagged proteins can be used, although at very high expression levels background issues were reported.Citation64 ChEC can also be modified to study posttranslational modifications as the MNase tag can also be conjugated to an antibody of interest. This in vitro method is referred to as chromatin immunocleavage (ChIC). In ChIC cross-linked cells are lysed and incubated with MNase-coupled antibodies that bind to the epitope of interest after induction of DNA cleavage by Ca2+ (). ChIC is a hybrid between affinity- and enzyme-based approaches in that it uses cross-linking and antibodies, but does not need to fully dissolve and precipitate intact protein-chromatin complexes due to the use of enzymatic activity, which specifically marks bound DNA ().
Concluding Remarks
The characterization of structural and other functional components in the nuclear lamina is vital for our understanding of higher-order chromatin organization, transcription, DNA replication and various other nuclear processes. Recent development of powerful techniques to map protein and chromatin interactions has begun to reveal these roles.
Several approaches to identify the interaction network at the nuclear periphery are now available. These methods are all complementary and each has its own usefulness and limitations. Ideally, one would map the interactions of proteins and chromatin using multiple, complementary techniques. At present this is practically often not feasible, however, as interaction-detection methods are improved, it should become possible to interrogate interactions by multiple means. For now, the choice of method often relies on the particular question to be addressed. When it is important to identify multiple sub-cellular localizations of a protein, visual screens are the best option. Biochemical fractionation studies best assist in revealing a full proteome. A more detailed impression of an individual protein's interactome can be obtained by classic IPs to study endogenous proteins, by OST pull-down to identify weak and relative insoluble proteins and by IPs using a bait to distinguish individual protein complexes. Mapping interactions of direct and weak interactors to protein domains can best be done by Y2H. For DNA interactions a careful choice has to be made between the need for studying endogenous proteins and posttranslational modifications (ChIP, ChIC), obtaining extra information on chromatin structure (In vivo ChEC, ChIC), full protein solubilization and obtaining an instant snapshot of interactions (OST pull-down), or not dissolving protein/chromatin complexes and capturing interactions over a longer period of time (DamID).
Even though the overlap between various chromatin techniques is slightly bigger than for protein techniques, in both fields a combinatorial or comparative use of techniques, as well as the target proteins they are applied on, will lead to more reliable results and provide a better understanding of the NE. These methods are becoming increasingly routinely used in many laboratories and there is no doubt that proteome and genome-wide mapping method and screening for mutation-induced interaction changes will play a key role in unraveling nuclear lamina function and laminopathy disease mechanism.
Abbreviations
| ChEC | = | chromatin endogenous cleavage |
| ChIC | = | chromatin immunocleavage |
| FPLD | = | Dunnigan familial partial lipodystrophy |
| HGPS | = | Hutchinson-Gilford Progeria syndrome |
| INM | = | inner nuclear membrane |
| MS | = | mass spectrometry |
| NE | = | nuclear envelope |
| NET | = | nuclear envelope transmembrane protein |
| NPC | = | nuclear pore complex |
| ONM | = | outer nuclear membrane |
| OST | = | OneSTrEP |
| PCP | = | protein correlation profiling |
| Srebp1 | = | adipocyte differentiation factor sterol element binding protein 1 |
| Y2H | = | yeast two-hybrid |
Figures and Tables
Figure 1 A schematic view of lamina and lamina-interacting protein fractions purified by various techniques. The nuclear periphery consists of an inner nuclear membrane (INM), outer nuclear membrane (ONM) and is connected to the endoplasmic reticulum (ER). (Left) Salt solubilizes weakly attached lamina proteins, but not the nuclear lamina. Detergents preferentially dissolve membrane proteins that are not anchored in the detergent resistant lamina.Citation28,Citation65 Chaotropes and alkaline extraction generate an insoluble fraction mainly consisting of integral membrane proteins.Citation65 Immunoprecipitations (right) with an antibody directed against lamin A/C, using mild lysis conditions (for example 0.1% NP-40, 250 mM NaClCitation12), preferentially dissolve and precipitate nucleosoluble A-type lamins and protein interactors.Citation35 For a lamin A OST pull-down assay,Citation46 cross-linking, indicated by crosses, captures protein-protein and protein-chromatin interactions and allows solubilization of the total lamin A/C pool while preserving interactions.
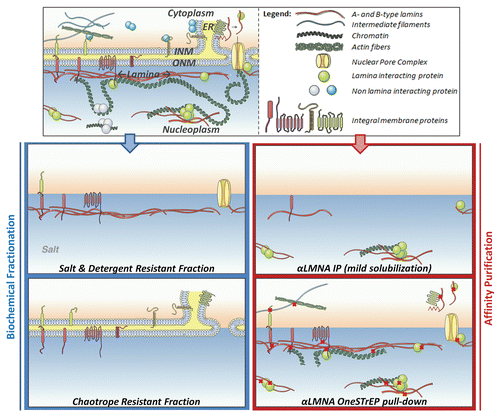
Figure 2 Schematic overview of techniques to identify chromatin interactions, which are categorized in enzymatic- and affinity-based approaches. For DamIDCitation58 a DNA adenine methyltransferase (Dam) tag (ball on stick) is fused to the protein of interest and adenylates (star) bound chromatin in vivo, enabling in vitro selective DpnI (scissor) restriction and subsequent amplification of restricted chromatin by ligation mediated PCR (LMPCR). For in vivo chromatin endogenous cleavage (ChEC)Citation63 a protein of interest is fused to a micrococcal nuclease (MNase) tag, which introduces DNA double strand breaks (scissors) upon introduction of calcium chloride to weakly permeabilized cells. Due to the mild permeabilization of cells prior to addition of calcium chloride for activation, the MNase digestion step is indicated as being partially in vitro and in vivo. Restricted DNA is amplified by LMPCR. For chromatin immunocleavage (ChIC)Citation63 cells are cross-linked (crosses). In vitro, MNase-conjugated antibody interacts with the epitope of interest and induces DNA breaks enabling LMPCR amplification of cleaved chromatin. For chromatin immunoprecipitation (ChIP) chromatin-protein interactions are cross-linked and chromatin is randomly sheared, typically by ultrasonication, (lightning arrow and stripes). Antibodies are used to precipitate the endogenous protein of interest with the help of antibody binding beads (big ball). In a OneSTrEP (OST) pull-down a OST-tagged protein is expressed.Citation58 Cells are cross-linked and ultrasonicated. The OST-protein is highly efficiently precipitated by a streptactin matrix (big square).
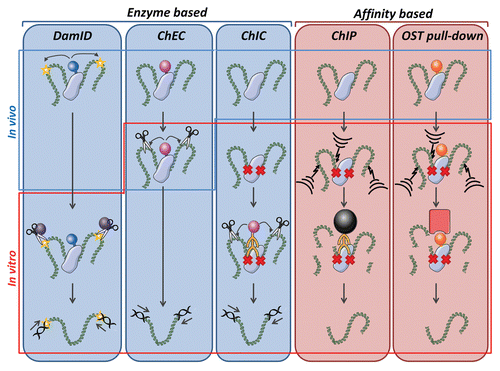
Table 1 Techniques to identify protein interactions at the nuclear lamina
Table 2 Solubilization conditions to identify protein interactions at the nuclear lamina.
Table 3 Techniques to identify chromatin interactions at the nuclear lamina
Additional material
Download Zip (2.1 MB)Related Research Data
References
- Foisner R. Collas P. Dynamic connections of nuclear envelope proteins to chromatin and the nuclear matrix. Nuclear Envelope Dynamics in Embryos and Somatic Cells 2003; 4:43 - 59
- Mattout-Drubezki A, Gruenbaum Y. Dynamic interactions of nuclear lamina proteins with chromatin and transcriptional machinery. Cell Mol Life Sci 2003; 60:2053 - 2063
- Broers JL, Hutchison CJ, Ramaekers FC. Laminopathies. J Pathol 2004; 204:478 - 488
- Broers JL, Ramaekers FC, Bonne G, Yaou RB, Hutchison CJ. Nuclear lamins: laminopathies and their role in premature ageing. Physiol Rev 2006; 86:967 - 1008
- Lloyd DJ, Trembath RC, Shackleton S. A novel interaction between lamin A and SREBP1: implications for parial lipodystrophy and other laminopathies. Human Molecular Genetics 2002; 11:769 - 777
- Pegoraro G, Kubben N, Wickert U, Gohler H, Hoffmann K, Misteli T. Ageing-related chromatin defects through loss of the NURD complex. Nat Cell Biol 2009; 11:1261 - 1267
- Taimen P, Pfleghaar K, Shimi T, Moller D, Ben-Harush K, Erdos MR, et al. A progeria mutation reveals functions for lamin A in nuclear assembly, architecture and chromosome organization. Proc Natl Acad Sci USA 2009; 106:20788 - 20793
- Wu CC, Yates JR 3rd. The application of mass spectrometry to membrane proteomics. Nat Biotechnol 2003; 21:262 - 267
- Yates JR, Ruse CI, Nakorchevsky A. Proteomics by mass spectrometry: approaches, advances and applications. Annu Rev Biomed Eng 2009; 11:49 - 79
- Werner T. Next generation sequencing in functional genomics. Brief Bioinform 2010; 11:499 - 511
- Sutherland HG, Mumford GK, Newton K, Ford LV, Farrall R, Dellaire G, et al. Large-scale identification of mammalian proteins localized to nuclear sub-compartments. Hum Mol Genet 2001; 10:1995 - 2011
- Van Berlo JH, Voncken JW, Kubben N, Broers JL, Duisters R, van Leeuwen RE, et al. A-type lamins are essential for TGFbeta1 induced PP2A to dephosphorylate transcription factors. Hum Mol Genet 2005; 14:2839 - 2849
- Ivorra C, Kubicek M, Gonzalez JM, Sanz-Gonzalez SM, Alvarez-Barrientos A, O'Connor JE, et al. A mechanism of AP-1 suppression through interaction of c-Fos with lamin A/C. Genes Dev 2006; 20:307 - 320
- Ross-Macdonald P, Coelho PS, Roemer T, Agarwal S, Kumar A, Jansen R, et al. Large-scale analysis of the yeast genome by transposon tagging and gene disruption. Nature 1999; 402:413 - 418
- Kumar A, Agarwal S, Heyman JA, Matson S, Heidtman M, Piccirillo S, et al. Subcellular localization of the yeast proteome. Genes Dev 2002; 16:707 - 719
- Simpson JC, Wellenreuther R, Poustka A, Pepperkok R, Wiemann S. Systematic subcellular localization of novel proteins identified by large-scale cDNA sequencing. EMBO Rep 2000; 1:287 - 292
- Simpson JC, Pepperkok R. Localizing the proteome. Genome Biol 2003; 4:240
- Kahsay RY, Gao G, Liao L. An improved hidden Markov model for transmembrane protein detection and topology prediction and its applications to complete genomes. Bioinformatics 2005; 21:1853 - 1858
- Shen HB, Chou KC. Nuc-PLoc: a new web-server for predicting protein subnuclear localization by fusing PseAA composition and PsePSSM. Protein Eng Des Sel 2007; 20:561 - 567
- Rolls MM, Stein PA, Taylor SS, Ha E, McKeon F, Rapoport TA. A visual screen of a GFP-fusion library identifies a new type of nuclear envelope membrane protein. J Cell Biol 1999; 146:29 - 44
- Hofemeister H, O'Hare P. Analysis of the localization and topology of nurim, a polytopic protein tightly associated with the inner nuclear membrane. J Biol Chem 2005; 280:2512 - 2521
- Huh WK, Falvo JV, Gerke LC, Carroll AS, Howson RW, Weissman JS, et al. Global analysis of protein localization in budding yeast. Nature 2003; 425:686 - 691
- Thirkettle HJ, Mills IG, Whitaker HC, Neal DE. Nuclear LYRIC/AEG-1 interacts with PLZF and relieves PLZF-mediated repression. Oncogene 2009; 28:3663 - 3670
- Wu CC, MacCoss MJ, Howell KE, Yates JR 3rd. A method for the comprehensive proteomic analysis of membrane proteins. Nat Biotechnol 2003; 21:532 - 538
- Foster LJ, de Hoog CL, Zhang Y, Zhang Y, Xie X, Mootha VK, et al. A mammalian organelle map by protein correlation profiling. Cell 2006; 125:187 - 199
- Clawson GA, Moody DE, James J, Smuckler EA. Nuclear envelope alterations accompanying thioacetamide-related enlargement of the nucleus. Cancer Res 1981; 41:519 - 526
- Cronshaw JM, Krutchinsky AN, Zhang W, Chait BT, Matunis MJ. Proteomic analysis of the mammalian nuclear pore complex. J Cell Biol 2002; 158:915 - 927
- Dreger M, Bengtsson L, Schoneberg T, Otto H, Hucho F. Nuclear envelope proteomics: novel integral membrane proteins of the inner nuclear membrane. Proc Natl Acad Sci USA 2001; 98:11943 - 11948
- Korfali N, Fairley EA, Swanson SK, Florens L, Schirmer EC. Use of sequential chemical extractions to purify nuclear membrane proteins for proteomics identification. Methods Mol Biol 2009; 528:201 - 225
- Sutherland HG, Lam YW, Briers S, Lamond AI, Bickmore WA. 3D3/lyric: a novel transmembrane protein of the endoplasmic reticulum and nuclear envelope, which is also present in the nucleolus. Exp Cell Res 2004; 294:94 - 105
- Siniossoglou S, Wimmer C, Rieger M, Doye V, Tekotte H, Weise C, et al. A novel complex of nucleoporins, which includes Sec13p and a Sec13p homolog, is essential for normal nuclear pores. Cell 1996; 84:265 - 275
- Goodchild RE, Dauer WT. Mislocalization to the nuclear envelope: an effect of the dystonia-causing torsinA mutation. Proc Natl Acad Sci USA 2004; 101:847 - 852
- Holaska JM, Wilson KL. An emerin “proteome”: purification of distinct emerin-containing complexes from HeLa cells suggests molecular basis for diverse roles including gene regulation, mRNA splicing, signaling, mechanosensing and nuclear architecture. Biochemistry 2007; 46:8897 - 8908
- Nikolakaki E, Simos G, Georgatos SD, Giannakouros T. A nuclear envelope-associated kinase phosphorylates arginine-serine motifs and modulates interactions between the lamin B receptor and other nuclear proteins. J Biol Chem 1996; 271:8365 - 8372
- Muralikrishna B, Thanumalayan S, Jagatheesan G, Rangaraj N, Karande AA, Parnaik VK. Immunolocalization of detergent-susceptible nucleoplasmic lamin A/C foci by a novel monoclonal antibody. J Cell Biochem 2004; 91:730 - 739
- Lin F, Morrison JM, Wu W, Worman HJ. MAN1, an integral protein of the inner nuclear membrane, binds Smad2 and Smad3 and antagonizes transforming growth factor-beta signaling. Hum Mol Genet 2005; 14:437 - 445
- Zhong N, Radu G, Ju W, Brown WT. Novel progerininteractive partner proteins hnRNP E1, EGF, Mel 18 and UBC9 interact with lamin A/C. Biochem Biophys Res Commun 2005; 338:855 - 861
- Capanni C, Mattioli E, Columbaro M, Lucarelli E, Parnaik VK, Novelli G, et al. Altered pre-lamin A processing is a common mechanism leading to lipodystrophy. Hum Mol Genet 2005; 14:1489 - 1502
- Farnum M, Zukoski C. Effect of glycerol on the interactions and solubility of bovine pancreatic trypsin inhibitor. Biophys J 1999; 76:2716 - 2726
- Lang C, Krohne G. Lamina-associated polypeptide 2beta (LAP2beta) is contained in a protein complex together with A- and B-type lamins. Eur J Cell Biol 2003; 82:143 - 153
- Grandi P, Dang T, Pane N, Shevchenko A, Mann M, Forbes D, et al. Nup93, a vertebrate homologue of yeast Nic96p, forms a complex with a novel 205-kDa protein and is required for correct nuclear pore assembly. Mol Biol Cell 1997; 8:2017 - 2038
- Zhang SX, Garcia-Gras E, Wycuff DR, Marriot SJ, Kadeer N, Yu W, et al. Identification of direct serum-response factor gene targets during Me2SO-induced P19 cardiac cell differentiation. J Biol Chem 2005; 280:19115 - 1926
- Doufexis M, Storr HL, King PJ, Clark AJ. Interaction of the melanocortin 2 receptor with nucleoporin 50: evidence for a novel pathway between a G-protein-coupled receptor and the nucleus. FASEB J 2007; 21:4095 - 4100
- Montes de Oca R, Shoemaker CJ, Gucek M, Cole RNW. Barrier-to-Autointegration Factor Proteome Reveals Chromatin-Regulatory Partners. PLoS One 2009; 4:7050
- Libotte T, Zaim H, Abraham S, Padmakumar VC, Schneider M, Lu W, et al. Lamin A/C-dependent localization of Nesprin-2, a giant scaffolder at the nuclear envelope. Mol Biol Cell 2005; 16:3411 - 3424
- Kubben N, Voncken J, Demmers JA, Calis C, van Almen G, Misteli T, et al. Identification of differential protein interactiors of lamin A and progerin. Nucleus 2010; 1:460 - 471
- Junttila MR, Saarinen S, Schmidt T, Kast J, Westermarck J. Single-step Strep-tag purification for the isolation and identification of protein complexes from mammalian cells. Proteomics 2005; 5:1199 - 1203
- Witte CP, Noel LD, Gielbert J, Parker JE, Romeis T. Rapid one-step protein purification from plant material using the eight-amino acid StrepII epitope. Plant Mol Biol 2004; 55:135 - 147
- Sakaki M, Koike H, Takahashi N, Sasagawa N, Tomioka S, Arahata K, et al. Interaction between emerin and nuclear lamins. J Biochem 2001; 129:321 - 327
- Goldberg M, Lu H, Stuurman N, Ashery-Padan R, Weiss AM, Yu J, et al. Interactions among Drosophila nuclear envelope proteins lamin, otefin and YA. Mol Cell Biol 1998; 18:4315 - 4323
- Maraldi NM, Lattanzi G, Capanni C, Columbaro M, Mattioli E, Sabatelli P, et al. Laminopathies: a chromatin affair. Adv Enzyme Regul 2006; 46:33 - 49
- Ruault M, Dubarry M, Taddei A. Re-positioning genes to the nuclear envelope in mammalian cells: impact on transcription. Trends Genet 2008; 24:574 - 581
- Schneider R, Grosschedl R. Dynamics and interplay of nuclear architecture, genome organization and gene expression. Genes Dev 2007; 21:3027 - 3043
- Casolari JM, Brown CR, Komili S, West J, Hieronymus H, Silver PA. Genome-wide localization of the nuclear transport machinery couples transcriptional status and nuclear organization. Cell 2004; 117:427 - 439
- Partridge JF, Borgstrom B, Allshire RC. Distinct protein interaction domains and protein spreading in a complex centromere. Genes Dev 2000; 14:783 - 791
- Grund SE, Fischer T, Cabal GG, Antunez O, Perez-Ortin JE, Hurt E. The inner nuclear membrane protein Src1 associates with subtelomeric genes and alters their regulated gene expression. J Cell Biol 2008; 182:897 - 910
- Brown CR, Kennedy CJ, Delmar VA, Forbes DJ, Silver PA. Global histone acetylation induces functional genomic reorganization at mammalian nuclear pore complexes. Genes Dev 2008; 22:627 - 639
- Pickersgill H, Kalverda B, de Wit E, Talhout W, Fornerod M, van Steensel B. Characterization of the Drosophila melanogaster genome at the nuclear lamina. Nat Genet 2006; 38:1005 - 1014
- Guelen L, Pagie L, Brasset E, Meuleman W, Faza MB, Talhout W, et al. Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature 2008; 453:948 - 951
- Vogel MJ, Peric-Hupkes D, van Steensel B. Detection of in vivo protein-DNA interactions using DamID in mammalian cells. Nat Protoc 2007; 2:1467 - 1478
- Kurshakova MM, Krasnov AN, Kopytova DV, Shidlovskii YV, Nikolenko JV, Nabirochkina EN, et al. SAGA and a novel Drosophila export complex anchor efficient transcription and mRNA export to NPC. EMBO J 2007; 26:4956 - 4965
- Scaffidi P, Misteli T. Reversal of the cellular phenotype in the premature aging disease Hutchinson-Gilford progeria syndrome. Nat Med 2005; 11:440 - 445
- Schmid M, Arib G, Laemmli C, Nishikawa J, Durussel T, Laemmli UK. Nup-PI: the nucleopore-promoter interaction of genes in yeast. Mol Cell 2006; 21:379 - 391
- Schmid M, Durussel T, Laemmli UK. ChIC and ChEC; genomic mapping of chromatin proteins. Mol Cell 2004; 16:147 - 157
- Schirmer EC, Gerace L. The nuclear membrane proteome: extending the envelope. Trends Biochem Sci 2005; 30:551 - 558
- Fink JL, Karunaratne S, Mittal A, Gardiner DM, Hamilton N, Mahony D, et al. Towards defining the nuclear proteome. Genome Biol 2008; 9:R15
- Miller BR, Forbes DJ. Purification of the vertebrate nuclear pore complex by biochemical criteria. Traffic 2000; 1:941 - 951
- Schirmer EC, Florens L, Guan T, Yates JR 3rd, Gerace L. Nuclear membrane proteins with potential disease links found by subtractive proteomics. Science 2003; 301:1380 - 1382
- Batrakou DG, Kerr AR, Schirmer EC. Comparative proteomic analyses of the nuclear envelope and pore complex suggests a wide range of heretofore unexpected functions. J Proteomics 2009; 72:56 - 70
- Maison C, Pyrpasopoulou A, Theodoropoulos PA, Georgatos SD. The inner nuclear membrane protein LAP1 forms a native complex with B-type lamins and partitions with spindle-associated mitotic vesicles. EMBO J 1997; 16:4839 - 4850
- Dreuillet C, Tillit J, Kress M, Ernoult-Lange M. In vivo and in vitro interaction between human transcription factor MOK2 and nuclear lamin A/C. Nucleic Acids Res 2002; 30:4634 - 4642
- Wagner N, Weber D, Seitz S, Krohne G. The lamin B receptor of Drosophila melanogaster. J Cell Sci 2004; 117:2015 - 2028
- Rasala BA, Orjalo AV, Shen Z, Briggs S, Forbes DJ. ELYS is a dual nucleoporin/kinetochore protein required for nuclear pore assembly and proper cell division. Proc Natl Acad Sci USA 2006; 103:17801 - 17806
- Apel ED, Lewis RM, Grady RM, Sanes JR. Syne-1, a dystrophin- and Klarsicht-related protein associated with synaptic nuclei at the neuromuscular junction. J Biol Chem 2000; 275:31986 - 31995
- Menon BB, Sarma NJ, Pasula S, Deminoff SJ, Willis KA, Barbara KE, et al. Reverse recruitment: the Nup84 nuclear pore subcomplex mediates Rap1/Gcr1/Gcr2 transcriptional activation. Proc Natl Acad Sci USA 2005; 102:5749 - 5754
- Casolari JM, Brown CR, Drubin DA, Rando OJ, Silver PA, et al. Developmentally induced changes in transcriptional program alter spatial organization across chromosomes. Genes Dev 2005; 19:1188 - 1198
- Cabal GG, Genovesio A, Rodriguez-Navarro S, Zimmer C, Gadal O, Lesne A, et al. SAGA interacting factors confine sub-diffusion of transcribed genes to the nuclear envelope. Nature 2006; 441:770 - 773
- Schirmer EC. The epigenetics of nuclear envelope organization and disease. Mutat Res 2008; 647:112 - 121