Abstract
A-type lamins provide a scaffold for tethering chromatin and protein complexes regulating nuclear
structure and function. Interest in lamins increased after mutations in the LMNA gene were found
to be associated with a variety of human disorders termed laminopathies. These include muscular
dystrophy, cardiomyopathy, lipodystrophy, peripheral neuropathy and premature aging syndromes
such as progeria. In addition, altered expression of A-type lamins is emerging as a contributing
factor to tumorigenesis. How different alterations in a gene that is ubiquitously expressed can
cause such an array of systemic as well as tissue specific diseases remains an enigma. Several
lines of evidence indicate that mutant forms of A-type lamins impact on genome function and
integrity. A current model suggests that genomic instability plays a major part in the
pathophysiology of some lamin-related diseases. However, this model remains to be fully
investigated. Here we discuss recent studies revealing novel functions for A-type lamins in the
maintenance of telomeres and in the DNA damage response (DDR) pathway. These findings
have shed some light ontotheputative molecular mechanisms by which alterations in A-type
lamins induce genomic instability and contribute to disease.
Introduction
Recent studies suggest that defects in the ability of cells to properly repair DNA damage contribute to the genomic instability of some laminopathies, especially premature aging diseases such as Hutchinson Gilford Progeria Syndrome (HGPS). Our findings demonstrate that the loss of A-type lamins in mouse cells leads to alterations of telomere structure, length and function, in addition to defects in the DDR pathway. These results suggest that deficiency in A-type lamins could contribute to the genomic instability that drives tumorigenesis and premature aging phenotypes. At the molecular level, loss of A-type lamins leads to the destabilization of 53BP1 protein, a key mediator in DDR, thus providing a putative mechanism by which loss of A-type lamins induces genomic instability.
A-type lamins.
Lamins A and C, members of the type V intermediate filaments family,Citation1 are main components of the nuclear laminaCitation2 and are also found as part of a nucleoplasmic network that associates tightly with chromatin.Citation3–Citation5 A-type lamins are primarily expressed in cells after the onset of differentiationCitation6 and are thought to exert specialized functions.Citation7 Synthesis of Lamin A entails unique post-translational processing events, converting a prelamin A precursor into the mature lamin A form. This processing includes farnesylation of prelamin A carboxy-terminal CAAX motif, cleavage of—AAX, carboxymethylation of the terminal cysteine, and a second cleavage of the last 15 amino acids by the metalloprotease Zmpste24.Citation8–Citation10 A mutation in the LMNA gene that abrogates the second cleavage site leads to the expression of a dominant-negative prelamin A isoform known as progerin, which causes HGPS, the most severe laminopathy.Citation11–Citation13 Accordingly, mice knockout for Zmpste24 exhibit similar phenotypes as human patients with HGPS.Citation10,Citation14 Hundreds of disease-associated mutations have been identified within the LMNA gene.Citation7,Citation15,Citation16 In addition, changes in the expression of lamins are observed in leukemia, lymphomas, small cell lung and ovarian cancer, as well as colon carcinoma. Interestingly, these changes are often associated with increased aggressiveness and poor prognosis.Citation17–Citation20 The broad range of diseases associated with either mutations in the LMNA gene or changes in the expression of A-type lamins has recently attracted much attention towards elucidating the functions of these structural nuclear proteins.
Telomere structure, length and function.
Alterations in telomere function are a hallmark of cancer and aging phenotypes. Proper telomere function relies on the formation of a specialized higher-order structure that shelters the ends of linear chromosomes from the attack of nuclear activities, preserving chromosome integrity and cellular proliferative potential.Citation21–Citation24 Formation of this structure requires a minimal length of telomeric DNA repeats and a number of telomere structural proteins. These include a multiprotein complex known as “shelterin” or “telosome” and a number of DNA repair factors.Citation25–Citation28 Telomeric DNA is maintained primarily by telomeraseCitation29 or by the Alternative Lengthening of Telomeres (ALT) mechanism.Citation30,Citation31 Telomeric chromatin modulating activities such as histone methyltransferases,Citation32–Citation34 Rb family membersCitation35,Citation36 and DNA methyltransferasesCitation37 also play a role in the maintenance of telomere length homeostasis.Citation38 In summary, the action of telomere length maintenance mechanisms, telomere binding proteins and telomere chromatin-modifying activities ensures the preservation of telomere length homeostasis and the end-capping function of telomeres.
Role for A-Type Lamins in the DDR
Mutations in the LMNA gene can induce genomic instability. Common defects in the DDR have been observed in the premature aging laminopathies HGPS and Mandibuloacral dysplasia type A (MADA) and in the Zmpste24−/− mouse model of progeria (). Fibroblasts from patients with MADA, caused by the homozygous R527H mutation in the LMNA gene, are impaired in their ability to repair DNA damage. This is demonstrated by reduced activation of p53 and its downstream targets, increased chromosome damage and residual γH2AX foci, which are characteristic of unrepaired DNA lesions.Citation39 Fibroblasts from HGPS patients and Zmpste24−/− mice have defective DNA repair pathways and increased basal DNA damage levels, exhibiting chromosome aberrations and increased sensitivity to DNA-damaging agents.Citation14,Citation40 These cells also show delayed recruitment of 53BP1 to γH2AX-labeled DNA repair foci after ionizing radiation and delayed resolution of these foci. Furthermore, progeroid fibroblasts exhibit impaired recruitment of double-strand break (DSB) repair factors Rad50 and Rad51 to sites of DNA damage, as well as aberrant accumulation of Xeroderma Pigmentosum group A (XPA) protein, which is in part responsible for the defects in DNA repair.Citation40,Citation41 Additional studies showed that HGPS fibroblasts display an activated DDR, as manifested by enhanced basal γH2AX and active p53.Citation42 Overall, these studies provide a strong correlation between proper processing of lamin A and the maintenance of genomic stability. In addition, they indicate that an altered ability to properly repair DNA damage could contribute to the pathophysiology of premature aging laminopathies.
Role for A-Type Lamins in Telomere Biology
Binding of lamins A/C to telomeric sequences in vitro was first reported in 1988 by Shoeman et al.Citation43 Subsequent studies revealed that human telomeres associate with the nuclear matrixCitation44 putatively through telomere binding proteins.Citation45 More recent studies have confirmed the binding of A-type lamins to mammalian telomeres in vivo by chromatin immunoprecipitation (ChIP).Citation46 Using live cell imaging and fluorescence microscopy, the binding of lamina-associated proteins such as LAP2α and BAF to telomeres during nuclear assembly was also demonstrated.Citation47 This suggests a putative role for these proteins in mediating the tethering of telomeres to A-type lamins. The extent of this tethering and the functional implications for telomere biology are only beginning to be uncovered.
The best evidence for a role of A-type lamins in telomere biology comes from HGPS fibroblasts, which exhibit faster telomere attrition than normal counterparts.Citation48 These results were confirmed by a study that monitored the length of individual telomeres in fibroblasts and hematopoietic cells from HGPS patientsCitation49 (). Interestingly, fibroblasts expressing progerin display telomere shortening and an increase in signal free ends while hematopoietic cells, which do not express detectable levels of lamin A, display normal telomere length. The authors concluded that the expression of progerin is necessary for the telomere shortening phenotype, and that the effect of A-type lamins on telomere length is direct. In clear contrast to these results, alterations of telomere length have not been observed in Zmpste24−/− fibroblastsCitation10 (and our unpublished results). This discrepancy might be due to differences in telomerase activity between mouse and human somatic cells. Despite normal maintenance of telomere length, it is possible that Zmpste24 deficiency induces telomere dysfunction. In another study, isogenic lines of skin fibroblasts were transfected with wild-type or mutant forms of lamin A previously associated with progeroid syndromes. Nuclear abnormalities, a faster rate of telomere attrition, and a shortened replicative lifespan were observed upon expression of either the wild-type protein or the mutants.Citation50 These results suggest that mutations in the LMNA gene and changes in the expression of A-type lamins can affect telomere homeostasis. Further evidence of crosstalk between A-type lamins and telomeres is the finding that the proliferative defects of human fibroblasts expressing lamin A mutants are rescued by telomerase.Citation51 In spite of the data indicating that A-type lamins play a role in the maintenance of telomeres the molecular mechanisms remain unknown. Similarly, whether or not alterations in telomere biology contribute to the phenotype of lamin-related diseases need to be determined.
Genomic Instability in Mouse Models of Laminopathies
Mouse models of laminopathies have become instrumental in the understanding of the molecular basis of these diseases.Citation52 The Lmna−/− and Zmpste24−/− mouse models have been the most extensively characterizedCitation10,Citation14,Citation53 (). At the cellular level, both models recapitulate some HGPS phenotypes such as ultrastructural defects of the nuclear envelope and loss of heterochromatin from the nuclear periphery.Citation10,Citation53,Citation54 Zmpste24−/− fibroblasts also feature defects in DNA repair;Citation40 observation that led investigators to propose that increased genomic instability contributes to the pathogenesis of progeria. The phenotype of Lmna−/− cells with respect to genomic instability is yet to be fully characterized. Molecular studies suggest that A-type lamins exert a role in tumor suppression. Loss of A-type lamins induces destabilization of Rb family membersCitation55,Citation56 and reduced levels and mislocalization of ING1,Citation57 which are proteins with well-established tumor suppressor functions. In the case of Rb family members, A-type lamins deficiency promotes their degradation by the proteasome, rendering cells unresponsive to Rb-mediated signals.Citation56 Hence, Lmna−/− fibroblasts exhibit similar characteristics to Rb-deficient cells, including increased pool of proliferating cells undergoing DNA replication, premature entry of cells into S-phase after release from a G1 arrest, smaller cellular size, and failure to undergo growth arrest in response to DNA damage.Citation55 The status of the Rb pathway in fibroblasts from HGPS patients is controversial. A recent study showed decreased Rb levels in lines of HGPS patients.Citation57 However, others have reported normal levels of Rb but a marked decrease in the inactive hyperphosphorylated form of the protein required for the G1 to S transition of the cell cycle.Citation58 Accordingly, reconstitution of mutant forms of lamin A associated with HGPS into Lmna−/− fibroblasts increased the cellular levels of Rb and rescued the ability of these cells to respond to Rb-mediated signals.Citation56 Based on these data, it is tempting to speculate that expression of progerin increases the pool of active Rb, limiting the proliferative potential of HGPS cells. In contrast, loss of A-type lamins and reduced Rb levels would contribute to the uncontrolled proliferation that characterizes tumor cells. Consistent with this model, a variety of human tumors silence the LMNA gene via hypermethylation of its promoter.Citation17–Citation20 In most cases, downregulation of A-type lamins was associated with increased tumor aggressiveness and poor prognosis.
Our recent data indicate that complete loss of A-type lamins also leads to genomic instability. In particular, alterations of telomere structure, length and function, and destabilization of a key factor (53BP1) in the DDR was observed in Lmna−/− mouse fibroblasts (). These results suggest that the loss of A-type lamins, which characterizes certain human tumors, could contribute to the genomic instability that drives malignancy. Future studies are needed to address if the loss of A-type lamins in human cells recapitulates the defects in telomere biology, DNA repair and overall genomic instability reported for mouse cells.
Alterations of Telomere Metabolism in the Lmna−/− Mouse Model
Loss of A-type lamins induces a modest telomere shortening phenotype and a pronounced increased frequency of signal-free ends (loss of telomeric signals)Citation59 ( and ). The fact that telomere shortening is rescued by reconstitution of either lamin A or lamin C indicates that both forms of A-type lamins participate in telomere length control.Citation60 An in vitro assay showed no differences in the overall telomerase activity between Lmna+/+ and Lmna−/− fibroblasts. In addition, similar telomere-bound levels of shelterin complex components TRF1 and TRF2 were found in both genotypes.Citation59 These results suggest that an alternative mechanism is responsible for the telomere shortening/loss in Lmna−/− cells.
Expression of lamin A mutants has been shown to impact DNA replication and repair.Citation61,Citation62 In addition, a study demonstrating that proliferative defects of human fibroblasts expressing lamin A mutants are rescued by telomeraseCitation51 suggests that telomerase exerts a protective function against telomere defects induced by alterations in A-type lamins. This protective function of telomerase has also been shown in cells deficient in other factors participating in telomere replication and repair, such as the RecQ helicase WernerCitation63 and the flap endonuclease FEN1.Citation64 Replication of telomeres presents unique problems due to the linear nature of chromosomal DNA and the formation of unusual structures (G-quadruplexes and T-loops) that slow or even stall fork progression. Recruitment of factors involved in DNA repair/recombination to telomeres is thought to facilitate the repair of DNA lesions that occur during telomere replication, as well as contribute to the formation of the higher-order structure of telomeres after completion of replication.Citation28 Telomere replication and telomerase-dependent telomere elongation are highly coordinated processes. They require proper functioning of the DNA replication machinery, binding of the shelterin complex components and DNA repair factors, and the accessibility of telomerase. Alterations in any of these processes can impact telomere metabolism. We speculate that the loss of A-type lamins affects the proficient coordination of telomere replication/repair and telomerase-dependent telomere elongation and that the overexpression of telomerase overcomes some of these defects.
Alternatively or concomitantly, aberrant recombination events involving telomeres could contribute to the telomere shortening/loss phenotype upon loss of A-type lamins. Three types of recombination events involving telomeres have been described: (1) telomeric sister chromatid exchanges (T-SCE) which often result in the elongation of one telomere at the expense of shortening the sister telomere and are characteristic of ALT-positive tumor cells;Citation65 (2) formation of telomeric circles (T-circles)Citation66 that result from recombination at the level of the T-loop; and (3) formation of telomeric DNA-containing double minute chromosomes (TDMs), which are extrachromosomal elements resulting from either recombination of telomeres with interstitial telomere-related sequencesCitation67 or DSBs at fragile sites.Citation68 Chromosome Orientation FISH (CO-FISH)Citation69 showed no evidence of increased T-SCE events in Lmna−/− cells with respect to wild-type controls.Citation59 However, we cannot rule out that loss of A-type lamins induces other recombination events which contribute to telomere instability.
Several lines of evidence indicate that the acquisition of a heterochromatic structure at telomeres is also critical for the maintenance of telomere length homeostasis. Trimethylation of histone H3 at lysine 9 (H3K9me3) and histone H4 at lysine 20 (H4K20me3) are characteristic heterochromatic marks of telomeres.Citation38 In addition, non-coding RNAs (TERRAs) accumulate at mammalian telomeres.Citation70,Citation71 Our studies demonstrated that loss of A-type lamins impacts telomeric chromatin structureCitation59 ( and ). Specifically, a decrease in H4K20me3 and TERRAs levels was observed in Lmna−/− fibroblasts. Previous studies demonstrated that Rb family members and histone methyltransferases Suv4-20 h1/h2 stabilize the levels of H4K20me3.Citation32,Citation36,Citation72 Mouse embryonic fibroblasts triple knockout for Rb family members or double knockout for Suv4-20 h1/h2 exhibit reduced levels of H4K20me3 at telomeric and pericentric heterochomatin. Thus, the decrease in H4K20me3 levels in Lmna−/− fibroblasts at these domains is consistent with destabilization of Rb family members. Intriguingly, loss of Rb family members and decreased levels of H4K20me3 were previously associated with telomere elongation.Citation32,Citation35 In Lmna−/− fibroblasts however, the combined loss of A-type lamins and Rb family members and the concomitant decrease in H4K20me3 levels leads to a telomere shortening phenotype. These results suggest that A-type lamins might be necessary for the elongation of telomeres upon Rb- and H4K20me3-deficiency. Future studies are needed to identify the mechanism by which loss of Rb function leads to telomere elongation and determine whether or not A-type lamins deficiency hinders this process. An interesting observation that came out of these studies is that the defects in heterochromatic marks upon loss of A-type lamins are different from those observed upon expression of progerin in HGPS fibroblasts. A decrease in H3K9me3 and an increase in H4K20me was observed in these cells, demonstrating distinct functional consequences of complete loss of A-type lamins and expression of lamin A mutants, with respect to epigenetic defects.Citation59,Citation73 Overall, our study demonstrates that complete loss of A-type lamins impacts telomere metabolism, although the specific mechanisms remain unknown.
A-type lamins have been ascribed a major role in the tethering of chromatin to specific subnuclear compartments. We found that mouse telomeres associate with A-type lamins and that loss of A-type lamins leads to a shift of telomere distribution towards the nuclear periphery and away from the nuclear centerCitation59 ( and ). In addition, a recent study demonstrated that the D4Z4 human subtelomeric repeat localizes the nearby telomere to the nuclear periphery in an A-type lamins-dependent manner.Citation74 Overall, these studies indicate that A-type lamins aid in the recruitment of specific sequences to the nuclear periphery as well as participate in the distribution of telomeres throughout the nucleoplasm. The functional consequences of the loss of tethering of telomeres to A-type lamins remain to be characterized.
Defects in DDR and DNA Repair in the Lmna−/− Mouse Model
Lmna−/− fibroblasts exhibit aneuploidy, increased frequency of chromosome and chromatid breaks, increased basal levels of DNA damage, and defects in the non-homologous end-joining (NHEJ) of dysfunctional telomeresCitation59 ( and ). Collectively, these data indicate that loss of A-type lamins hinders some steps during the sensing, signaling or repair of DNA damage. Monitoring of the levels of different DDR factors revealed a marked decrease in the levels of 53BP1 protein. 53BP1 is a key mediator in the cellular response to DNA damage, proposed to function at the interface of DNA replication, recombination, and repair. In contrast, the levels of other proteins involved in DDR and DNA repair—ATM, DNA-PK, Mre11, Nbs1, Ku70, RAD51, MDC1 and ERCC1—were not altered, implying a preferential effect on 53BP1. Importantly, reconstitution of either lamin A or lamin C rescued the levels of 53BP1, supporting a role for these proteins in the stabilization of 53BP1. Similarly, treatment of Lmna+/+ and Lmna−/− fibroblasts with a proteasome inhibitor stabilized 53BP1 levels, implicating the proteasome in the degradation of 53BP1. Given the known role of A-type lamins in preventing the degradation of Rb family members by the proteasome,Citation55,Citation56 we envision that the stabilization of 53BP1 and Rb proteins by A-type lamins might be achieved by a similar mechanism. The decrease in 53BP1 could explain some of the phenotypes observed upon loss of A-type lamins. A recent study demonstrated that loss of 53BP1 hinders the processing of dysfunctional telomeres by NHEJ.Citation75 The authors showed that the loss of 53BP1 restricts the mobility of dysfunctional telomeres. This led to the proposal that 53BP1 has an active role in chromatin dynamics that facilitate the association and fusion of dysfunctional telomeres that might be far away within the nucleus. 53BP1 deficiency provides a putative mechanism by which alteration of A-type lamins function impacts on this process. However, a clear demonstration is pending. The defects in NHEJ of dysfunctional telomeres in Lmna−/− fibroblasts could be solely due to destabilization of 53BP1. Alternatively, A-type lamins could play an active role in the 53BP1-mediated regulation of mobility and NHEJ of dysfunctional telomeres, other than stabilization of 53BP1. A functional relationship between 53BP1 and A-type lamins during the processing of dysfunctional telomeres requires further investigation.
53BP1 knockout mice exhibit a phenotype consistent with defects in DNA repair, such as increased radiosensitivity, immunodeficiency and cancer susceptibility. Several lines of evidence indicate the participation of 53BP1 in NHEJ of DNA double-strand breaks at internal sites of chromosomes.Citation76–Citation78 Putative roles for A-type lamins in these processes await future characterization.
A role for 53BP1 in DNA replication has also been described. 53BP1 is required for efficient accumulation of the Bloom (BLM) helicase and p53 at sites of stalled replication.Citation79 Additional studies have shown that 53BP1, BLM and RAD51 interact during stalled replication and that loss of 53BP1 decreases cell survival and enhances chromosomal aberrations upon replication arrest.Citation80 We envision that the decrease in 53BP1 upon loss of A-type lamins could affect telomere replication, contributing to the defects in telomere metabolism observed in Lmna−/− fibroblasts.
Summary
Previous studies showed that cells from premature aging laminopathies are hindered in their ability to properly deal with DNA damage, leading to genomic instability. Our studies revealed that complete loss of A-type lamins also affects the cellular response to DNA damage as well as the maintenance of telomere stability, which could represent a second source for genomic instability in laminopathies. In addition, our data suggest that 53BP1 deficiency may be a key factor in the genomic instability observed in Lmna−/− cells. Future studies will need to determine if 53BP1 deficiency and alterations of telomere biology are phenocopied in human laminopathies and if they contribute to the pathophysiology of these diseases. In addition, determining the molecular mechanisms by which the loss of A-type lamins impacts on the different mechanisms regulating telomere metabolism and DNA repair will be fundamental for the development of novel therapeutic strategies to treat these diseases.
Figures and Tables
Figure 1 Loss of A-type lamins induces genomic instabilitySummary of the recently identified alterations in telomere biology and in the DNA damage response pathway upon loss of A-type lamins in mouse cells.Citation5,Citation9 Lmna−/− fibroblasts exhibit alterations in the structure of telomere chromatin Specifically, a decrease in H4K20me3 and TERRAs levels was observed. The decrease in H4K20me3 is consistent with destabilization of Rb family members in these cells. In addition, loss of A-type lamins leads to alterations in the nuclear distribution of telomeres as determined by 3D FISH, and in telomere metabolism. Telomere shortening—as shown by TRF and Q-FISH assays—and telomere dysfunction—increased number of chromosomes that feature undetectable telomere tracks—were observed. Furthermore, Lmna−/− fibroblasts feature aneuploidy, chromosome and chromatid breaks, and basal γH2AX foci indicative of unrepaired DNA damage. Loss of A-type lamins also hinders the processing of dysfunctional telomeres by NHEJ. The concomitant destabilization of 53BP1 protein could be in part responsible for the increased genomic instability and the defects in NHEJ upon loss of A-type lamins. Collectively, these data indicate that A-type lamins function in the maintenance of genome stability.
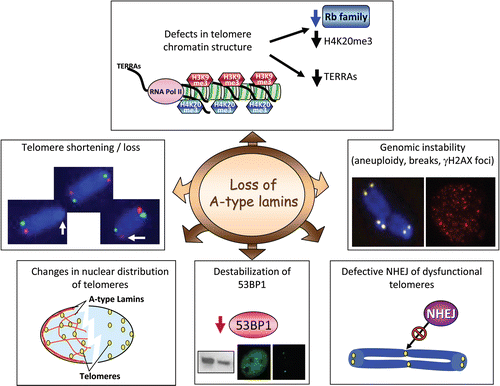
Table 1 Comparison of the most relevant phenotypes present in cells from Lmna K.O. and Zmpste24 K.O. mice and HGPS patients
References
- McKeon FD, Kirschner MW, Caput D. Homologies in both primary and secondary structure between nuclear envelope and intermediate filament proteins. Nature 1986; 319:3 - 5
- Gerace L, Blum A, Blobel G. Immunocytochemical localization of the major polypeptides of the nuclear pore complex-lamina fraction. Interphase and mitotic distribution. J Cell Biol 1978; 79:546 - 566
- Bridger JM, Kill IR, O'Farrell M, Hutchison CJ. Internal lamin structures within G1 nuclei of human dermal fibroblasts. J Cell Sci 1993; 104:297 - 306
- Goldman AE, Moir RD, Montag-Lowy M, Stewart M, Goldman RD. Pathway of incorporation of microinjected lamin A into the nuclear envelope. J Cell Biol 1992; 119:725 - 735
- Hozak P, Sasseville AM, Raymond Y, Cook PR. Lamin proteins form an internal nucleoskeleton as well as a peripheral lamina in human cells. J Cell Sci 1995; 108:635 - 644
- Harborth J, Elbashir SM, Bechert K, Tuschl T, Weber K. Identification of essential genes in cultured mammalian cells using small interfering RNAs. J Cell Sci 2001; 114:4557 - 4565
- Broers JL, Ramaekers FC, Bonne G, Yaou RB, Hutchison CJ. Nuclear lamins: laminopathies and their role in premature ageing. Physiol Rev 2006; 86:967 - 1008
- Beck LA, Hosick TJ, Sinensky M. Isoprenylation is required for the processing of the lamin A precursor. J Cell Biol 1990; 110:1489 - 1499
- Bergo MO, Gavino B, Ross J, Schmidt WK, Hong C, Kendall LV, et al. Zmpste24 deficiency in mice causes spontaneous bone fractures, muscle weakness, and a prelamin A processing defect. Proc Nat Acad Sci USA 2002; 99:13049 - 13054
- Pendas AM, Zhou Z, Cadinanos J, Freije JM, Wang J, Hultenby K, et al. Defective prelamin A processing and muscular and adipocyte alterations in Zmpste24 metalloproteinase-deficient mice. Nat Genet 2002; 31:94 - 99
- De Sandre-Giovannoli A, Bernard R, Cau P, Navarro C, Amiel J, Boccaccio I, et al. Lamin a truncation in Hutchinson-Gilford progeria. Science 2003; 300:2055
- Eriksson M, Brown WT, Gordon LB, Glynn MW, Singer J, Scott L, et al. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature 2003; 423:293 - 298
- Hennekam RC. Hutchinson-Gilford progeria syndrome: review of the phenotype. Am J Med Genet 2006; 140:2603 - 2624
- Varela I, Cadinanos J, Pendas AM, Gutierrez-Fernandez A, Folgueras AR, Sanchez LM, et al. Accelerated ageing in mice deficient in Zmpste24 protease is linked to p53 signalling activation. Nature 2005; 437:564 - 568
- Capell BC, Collins FS. Human laminopathies: nuclei gone genetically awry. Nat Rev Genet 2006; 7:940 - 952
- Prokocimer M, Davidovich M, Nissim-Rafinia M, Wiesel-Motiuk N, Bar D, Barkan R, et al. Nuclear lamins: key regulators of nuclear structure and activities. J Cell Mol Med 2009; In press
- Agrelo R, Setien F, Espada J, Artiga MJ, Rodriguez M, Perez-Rosado A, et al. Inactivation of the lamin A/C gene by CpG island promoter hypermethylation in hematologic malignancies, and its association with poor survival in nodal diffuse large B-cell lymphoma. J Clin Oncol 2005; 23:3940 - 3947
- Broers JL, Raymond Y, Rot MK, Kuijpers H, Wagenaar SS, Ramaekers FC. Nuclear A-type lamins are differentially expressed in human lung cancer subtypes. Am J Pathol 1993; 143:211 - 220
- Prokocimer M, Margalit A, Gruenbaum Y. The nuclear lamina and its proposed roles in tumorigenesis: projection on the hematologic malignancies and future targeted therapy. J Struct Biol 2006; 155:351 - 360
- Willis ND, Cox TR, Rahman-Casans SF, Smits K, Przyborski SA, van den Brandt P, et al. Lamin A/C is a risk biomarker in colorectal cancer. PLoS One 2008; 3:2988
- Blackburn EH. Switching and signaling at the telomere. Cell 2001; 106:661 - 673
- de Lange T. Protection of mammalian telomeres. Oncogene 2002; 21:532 - 540
- Griffith JD, Comeau L, Rosenfield S, Stansel RM, Bianchi A, Moss H, de Lange T. Mammalian telomeres end in a large duplex loop. Cell 1999; 97:503 - 514
- Harley CB, Futcher AB, Greider CW. Telomeres shorten during ageing of human fibroblasts. Nature 1990; 345:458 - 460
- de Lange T. Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev 2005; 19:2100 - 2110
- Slijepcevic P. The role of DNA damage response proteins at telomeres—an “integrative” model. DNA Repair 2006; 5:1299 - 1306
- Stewart SA, Weinberg RA. Telomeres: Cancer to Human Aging. Annu Rev Cell Dev Biol 2006;
- Verdun RE, Karlseder J. The DNA damage machinery and homologous recombination pathway act consecutively to protect human telomeres. Cell 2006; 127:709 - 720
- Greider CW, Blackburn EH. A telomeric sequence in the RNA of Tetrahymena telomerase required for telomere repeat synthesis. Nature 1989; 337:331 - 337
- Dunham MA, Neumann AA, Fasching CL, Reddel RR. Telomere maintenance by recombination in human cells. Nat Genet 2000; 26:447 - 450
- Muntoni A, Reddel RR. The first molecular details of ALT in human tumor cells. Hum Mol Genet 2005; 14:191 - 196
- Benetti R, Gonzalo S, Jaco I, Schotta G, Klatt P, Jenuwein T, Blasco MA. Suv4-20 h deficiency results in telomere elongation and derepression of telomere recombination. J Cell Biol 2007; 178:925 - 936
- Garcia-Cao M, O'Sullivan R, Peters AH, Jenuwein T, Blasco MA. Epigenetic regulation of telomere length in mammalian cells by the Suv39 h1 and Suv39 h2 histone methyltransferases. Nat Genet 2004; 36:94 - 99
- Jones B, Su H, Bhat A, Lei H, Bajko J, Hevi S, et al. The histone H3K79 methyltransferase Dot1L is essential for mammalian development and heterochromatin structure. PLoS Genet 2008; 4:1000190
- Garcia-Cao M, Gonzalo S, Dean D, Blasco MA. A role for the Rb family of proteins in controlling telomere length. Nat Genet 2002; 32:415 - 419
- Gonzalo S, Garcia-Cao M, Fraga MF, Schotta G, Peters AH, Cotter SE, et al. Role of the RB1 family in stabilizing histone methylation at constitutive heterochromatin. Nat Cell Biol 2005; 7:420 - 428
- Gonzalo S, Jaco I, Fraga MF, Chen T, Li E, Esteller M, Blasco MA. DNA methyltransferases control telomere length and telomere recombination in mammalian cells. Nat Cell Biol 2006; 8:416 - 424
- Blasco MA. The epigenetic regulation of mammalian telomeres. Nat Rev Genet 2007; 8:299 - 309
- di Masi A, D'Apice MR, Ricordy R, Tanzarella C, Novelli G. The R527H mutation in LMNA gene causes an increased sensitivity to ionizing radiation. Cell Cycle 2008; 7:2030 - 2037
- Liu B, Wang J, Chan KM, Tjia WM, Deng W, Guan X, et al. Genomic instability in laminopathy-based premature aging. Nat Med 2005; 11:780 - 785
- Liu Y, Wang Y, Rusinol AE, Sinensky MS, Liu J, Shell SM, Zou Y. Involvement of xeroderma pigmentosum group A (XPA) in progeria arising from defective maturation of prelamin A. Faseb J 2008; 22:603 - 611
- Scaffidi P, Misteli T. Lamin A-dependent nuclear defects in human aging. Science 2006; 312:1059 - 1063
- Shoeman RL, Wadle S, Scherbarth A, Traub P. The binding in vitro of the intermediate filament protein vimentin to synthetic oligonucleotides containing telomere sequences. J Biol Chem 1988; 263:18744 - 18749
- de Lange T. Human telomeres are attached to the nuclear matrix. EMBO J 1992; 11:717 - 724
- Luderus ME, van Steensel B, Chong L, Sibon OC, Cremers FF, de Lange T. Structure, subnuclear distribution and nuclear matrix association of the mammalian telomeric complex. J Cell Biol 1996; 135:867 - 881
- Raz V, Vermolen BJ, Garini Y, Onderwater JJ, Mommaas-Kienhuis MA, Koster AJ, et al. The nuclear lamina promotes telomere aggregation and centromere peripheral localization during senescence of human mesenchymal stem cells. J Cell Sci 2008; 121:4018 - 4028
- Dechat T, Gajewski A, Korbei B, Gerlich D, Daigle N, Haraguchi T, et al. LAP2alpha and BAF transiently localize to telomeres and specific regions on chromatin during nuclear assembly. J Cell Sci 2004; 117:6117 - 6128
- Allsopp RC, Vaziri H, Patterson C, Goldstein S, Younglai EV, Futcher AB, et al. Telomere length predicts replicative capacity of human fibroblasts. Proc Nat Acad Sci USA 1992; 89:10114 - 10118
- Decker ML, Chavez E, Vulto I, Lansdorp PM. Telomere length in Hutchinson-Gilford progeria syndrome. Mech Ageing Dev 2009; 130:377 - 383
- Huang S, Risques RA, Martin GM, Rabinovitch PS, Oshima J. Accelerated telomere shortening and replicative senescence in human fibroblasts overexpressing mutant and wild-type lamin A. Exp Cell Res 2008; 314:82 - 91
- Kudlow BA, Stanfel MN, Burtner CR, Johnston ED, Kennedy BK. Suppression of proliferative defects associated with processing-defective lamin A mutants by hTERT or inactivation of p53. Mol Biol Cell 2008; 19:5238 - 5248
- Stewart CL, Kozlov S, Fong LG, Young SG. Mouse models of the laminopathies. Exp Cell Res 2007; 313:2144 - 2156
- Sullivan T, Escalante-Alcalde D, Bhatt H, Anver M, Bhat N, Nagashima K, et al. Loss of A-type lamin expression compromises nuclear envelope integrity leading to muscular dystrophy. J Cell Biol 1999; 147:913 - 920
- Oberdoerffer P, Sinclair DA. The role of nuclear architecture in genomic instability and ageing. Nat Rev 2007; 8:692 - 702
- Johnson BR, Nitta RT, Frock RL, Mounkes L, Barbie DA, Stewart CL, et al. A-type lamins regulate retinoblastoma protein function by promoting subnuclear localization and preventing proteasomal degradation. Proc Nat Acad Sci USA 2004; 101:9677 - 9682
- Nitta RT, Jameson SA, Kudlow BA, Conlan LA, Kennedy BK. Stabilization of the retinoblastoma protein by A-type nuclear lamins is required for INK4A-mediated cell cycle arrest. Mol Cell Biol 2006; 26:5360 - 5372
- Han X, Feng X, Rattner JB, Smith H, Bose P, Suzuki K, et al. Tethering by lamin A stabilizes and targets the ING1 tumour suppressor. Nat Cell Biol 2008; 10:1333 - 1340
- Dechat T, Shimi T, Adam SA, Rusinol AE, Andres DA, Spielmann HP, et al. Alterations in mitosis and cell cycle progression caused by a mutant lamin A known to accelerate human aging. Proc Nat Acad Sci USA 2007; 104:4955 - 4960
- Gonzalez-Suarez I, Redwood AB, Perkins SM, Vermolen B, Lichtensztejin D, Grotsky DA, et al. Novel roles for A-type lamins in telomere biology and the DNA damage response pathway. EMBO J 2009; 28:2414 - 2427
- Gonzalez-Suarez I, Redwood AB, Gonzalo S. Loss of A-type lamins and genomic instability. Cell Cycle 2009; 8:3860 - 3865
- Moir RD, Spann TP, Herrmann H, Goldman RD. Disruption of nuclear lamin organization blocks the elongation phase of DNA replication. J Cell Biol 2000; 149:1179 - 1192
- Spann TP, Moir RD, Goldman AE, Stick R, Goldman RD. Disruption of nuclear lamin organization alters the distribution of replication factors and inhibits DNA synthesis. J Cell Biol 1997; 136:1201 - 1212
- Crabbe L, Verdun RE, Haggblom CI, Karlseder J. Defective telomere lagging strand synthesis in cells lacking WRN helicase activity. Science 2004; 306:1951 - 1953
- Saharia A, Guittat L, Crocker S, Lim A, Steffen M, Kulkarni S, Stewart SA. Flap endonuclease 1 contributes to telomere stability. Curr Biol 2008; 18:496 - 500
- Henson JD, Neumann AA, Yeager TR, Reddel RR. Alternative lengthening of telomeres in mammalian cells. Oncogene 2002; 21:598 - 610
- Wang RC, Smogorzewska A, de Lange T. Homologous recombination generates T-loop-sized deletions at human telomeres. Cell 2004; 119:355 - 368
- Zhu XD, Niedernhofer L, Kuster B, Mann M, Hoeijmakers JH, de Lange T. ERCC1/XPF removes the 3′ overhang from uncapped telomeres and represses formation of telomeric DNA-containing double minute chromosomes. Mol Cell 2003; 12:1489 - 1498
- Coquelle A, Rozier L, Dutrillaux B, Debatisse M. Induction of multiple double-strand breaks within an hsr by meganucleaseI-SceI expression or fragile site activation leads to formation of double minutes and other chromosomal rearrangements. Oncogene 2002; 21:7671 - 7679
- Bailey SM, Goodwin EH, Cornforth MN. Strand-specific fluorescence in situ hybridization: the CO-FISH family. Cytogenet Genome Res 2004; 107:14 - 17
- Azzalin CM, Reichenbach P, Khoriauli L, Giulotto E, Lingner J. Telomeric repeat containing RNA and RNA surveillance factors at mammalian chromosome ends. Science 2007; 318:798 - 801
- Schoeftner S, Blasco MA. Developmentally regulated transcription of mammalian telomeres by DNA-dependent RNA polymerase II. Nat Cell Biol 2008; 10:228 - 236
- Gonzalo S, Blasco MA. Role of Rb family in the epigenetic definition of chromatin. Cell Cycle 2005; 4:752 - 755
- Shumaker DK, Dechat T, Kohlmaier A, Adam SA, Bozovsky MR, Erdos MR, et al. Mutant nuclear lamin A leads to progressive alterations of epigenetic control in premature aging. Proc Nat Acad Sci USA 2006; 103:8703 - 8708
- Ottaviani A, Schluth-Bolard C, Rival-Gervier S, Boussouar A, Rondier D, Foerster AM, et al. Identification of a perinuclear positioning element in human subtelomeres that requires A-type lamins and CTCF. EMBO J 2009; 28:2428 - 2436
- Dimitrova N, Chen YC, Spector DL, de Lange T. 53BP1 promotes non-homologous end joining of telomeres by increasing chromatin mobility. Nature 2008; 456:524 - 528
- Manis JP, Morales JC, Xia Z, Kutok JL, Alt FW, Carpenter PB. 53BP1 links DNA damage-response pathways to immunoglobulin heavy chain class-switch recombination. Nat Immunol 2004; 5:481 - 487
- Ward IM, Reina-San-Martin B, Olaru A, Minn K, Tamada K, Lau JS, Cascalho M, et al. 53BP1 is required for class switch recombination. J Cell Biol 2004; 165:459 - 464
- Difilippantonio S, Gapud E, Wong N, Huang CY, Mahowald G, Chen HT, et al. 53BP1 facilitates long-range DNA end-joining during V(D)J recombination. Nature 2008; 456:529 - 533
- Sengupta S, Robles AI, Linke SP, Sinogeeva NI, Zhang R, Pedeux R, et al. Functional interaction between BLM helicase and 53BP1 in a Chk1-mediated pathway during S-phase arrest. J Cell Biol 2004; 166:801 - 813
- Tripathi V, Nagarjuna T, Sengupta S. BLM helicase-dependent and -independent roles of 53BP1 during replication stress-mediated homologous recombination. J Cell Biol 2007; 178:9 - 14