Abstract
The DNA damage response (DDR) is comprised of a network of proteins that respond to DNA damage. Mediator of DNA Damage Checkpoint 1 (MDC1) plays an early and important role in the DDR. Recent data show that MDC1 binds multiple proteins that participate in various aspects of the DDR, positioning it at the core of the DDR. Furthermore, interactions with non-DDR proteins were also revealed, suggesting novel roles for MDC1. In this review we provide a comprehensive overview of all known MDC1-binding proteins and discuss their role. We present these binding partners according to their function, thereby providing the reader with a detailed and updated overview of the cellular response to DNA damage. We discuss more recent findings in detail and conclude by presenting the challenges the field faces in the future.
Introduction
The DNA damage response.
DNA damage can ultimately lead to genomic instability and carcinogenesis if not properly addressed. It is therefore not surprising that an elaborate network of proteins, termed the DNA damage response (DDR), has evolved to protect our genome (for more on the importance of an intact DDR see box 1).Citation1–Citation3
DDR proteins are classified into several groups according to their role: Sensors, transducers, mediators and effectors. The DDR cascade starts with the sensors that detect the damage and convey the initial signal to the transducers. The transducers, aided by the mediators, amplify the signal and transmit it to the effectors, which carry out diverse roles such as repair, checkpoint activation and if necessary—apoptosis.Citation4
One of the hallmarks of the DDR is the local accumulation of proteins at sites of double-strand breaks (DSBs). In a manner of minutes, multiple DDR proteins assemble at break sites. The accumulation of these proteins is not restricted to the break site itself, but rather spreads across large megabase domains flanking the DSB, forming microscopically-visible foci.Citation5 It has recently been shown that the local concentration of DDR proteins at one genomic location, even in the absence of an actual break, is sufficient to activate the DDR.Citation6
For most DDR proteins, focus formation seems to be a linear and step-wise process with upstream proteins showing faster kinetics. For example, MDC1 and RNF8 arrive very early to DSBs (in less than a minute) and are required for recruitment of BRCA1 and 53BP1.Citation7–Citation10 Accordingly, BRCA1 and 53BP1 show slower kinetics and are not required for the recruitment of MDC1 or RNF8 to break sites (See and later in text). Thus, focus formation is clearly arranged as a hierarchical cascade and it is of major interest to understand the exact molecular details of this cascade.
MDC1—a brief introduction.
MDC1, also known as NFBD1 (Nuclear Factor with BRCT Domains 1), was independently identified as an important player in the DDR by several groups approximately seven years ago. It was established that MDC1 is not only recruited to break sites but is also important for recruitment of many other DDR proteins to sites of damage, such as BRCA1, 53BP1 and the Mre11/Rad50/NBS1 (MRN) complex.Citation11–Citation15 It was shown that downregulation of MDC1 causes multiple phenotypes, the major ones being (1) hypersensitivity of cells to DSB inducing conditions such as exposure to ionizing radiation (IR), (2) improper activation of two DNA damage specific cell cycle checkpoints—the G2/M and the intra-S-phase checkpoints, (3) aberrant activation of DNA damage induced apoptosisCitation12 and (4) inefficient phosphorylation of several DDR regulators (such as the effector kinase ChkCitation11,Citation16). It should be noted that there are conflicting data regarding the role of MDC1 in the phosphorylation of some DDR proteins (e.g., Chk2 phosphorylationCitation11,Citation17) and future work is required in this aspect. From these early works it was obvious that MDC1 has a central role in the DDR but it was not clear how exactly it carries out these multiple tasks. It was suggested that MDC1 mainly acts as an adaptor protein that recruits DDR proteins to sites of damage. This hypothesis has been well substantiated by the large amount of data generated in recent years. Furthermore, additional roles have been ascribed to MDC1, including DNA repair via the non-homologous end joining (NHEJ)Citation18 and homologous recombination (HR)Citation19,Citation20 pathways, activation of the decatenation checkpointCitation21 and mitosis.Citation22
MDC1 is a large protein, spanning 2,089 residues in human. Preliminary analysis of its sequence revealed three distinct domains (): an N-terminal FHA domain, a central Pro/Ser/Thr-rich repeat domain (PST repeat) and a C-terminal tandem BRCT (tBRCT) domain (for more on the FHA and tBRCT domains see box 2).Citation11,Citation12,Citation14 Early work has focused on the FHA and tBRCT domains of MDC1, as they were easily identified and were known to mediate protein-protein interactions. The PST repeat is a unique domain, with an imperfect repetitive motif of ∼41 residues. Human MDC1 contains 13 full PST repeats, with a few partial repeats flanking both sides of these central repeats (see ). The FHA and tBRCT domains of MDC1 are fairly conserved and the main difference between human and murine MDC1 is in the number of PST repeats—human MDC1 contains approximately 5 additional PST repeats. The PST motif does not appear in other proteins, making it hard to predict a role for it.Citation11,Citation12,Citation14 However, one report has demonstrated that the PST repeat can act as a protein-binding module.Citation23 In addition to these three domains, recent work revealed novel motives in MDC1 that also mediate protein-protein interactions (see and later in text). MDC1 is therefore a multi-faceted mediator, allowing multiple protein-protein interactions that are crucial for an intact DDR (see ).
The making of a focus.
As noted earlier, multiple DDR proteins assemble quickly and efficiently at large chromatin regions that surround sites of breaks. Once a DSB is formed, it is detected and converted into a molecular signal, which is substantially amplified, allowing the recruitment, retention and activation of downstream DDR proteins at sites of damage. These local aggregations of proteins can be visualized by standard microscopy and are seen as discrete foci. Although the exact role of these foci is unclear, current models suggest that they direct repair of DSBs via the local accumulation of repair factors while allowing the conversion of a fairly small amount of DSBs into a strong enough signal that will trigger efficient activation of cell cycle checkpoints.Citation4–Citation6,Citation26
Although much progress has been made in the field of DNA damage, one important question remains largely unanswered—how is a DSB detected and who is the primary sensor? Although a clear answer remains elusive, the most prominent candidate to date is a complex of three proteins—the MRN complex.Citation26 Mre11 and Rad50 form a hetero-dimer structure that is capable of tethering DNA ends. Mre11 also possesses a nuclease activity that is suggested to act in resection of complex DSBs.Citation27 NBS1 seems to be the regulatory subunit of the complex with an important role in recruiting the entire MRN complex to sites of damage. It has been shown that the MRN complex is important for activation and recruitment of the transducer kinase ATM to sites of damage.Citation28 More recently, NBS1 was found to contain a conserved C-terminal motif that binds ATM, thus revealing the molecular mechanism for the role of the MRN complex in ATM recruitment.Citation29
One of the most studied proteins in the DDR is histone H2AX,Citation5 a variant of histone H2A. Histone H2AX is an integral part of the nucleosome and constitutes approximately 10–15% of the histone H2A pool. Histone H2AX differs from histone H2A by a longer C-terminal tail that contains a conserved SQEY-COOH motif. The phosphorylation of the serine residue of this motif (Ser-139) is an early and important event in the DDR. The phosphorylated form of histone H2AX, known as γ-H2AX, appears within minutes after the formation of a DSB, tagging large chromatin domains flanking the break.Citation30 Phosphorylation of histone H2AX is carried out by one of three phospho-inositide-3-kinase-related protein kinases (PIKKs)—ATM, ATR or DNA-PK. A rough generalization is that H2AX phosphorylation is mediated by ATM and DNA-PK in response to DSBs in a partially redundant way,Citation31,Citation32 and by ATR in response to ultra-violet (UV) damage or replication stress.Citation33
Although histone H2AX is not essential for DSB repair, studies of histone H2AX knockout mice show that it is important for focus formation by many DDR proteins and that it has a role in DSB repair fidelity and efficiency, checkpoint activation and tumor suppression.Citation34–Citation36 If histone H2AX has such an important role in recruiting DDR proteins to sites of damage, why is it not essential for repair? First of all, DSBs vary in their severity. Simple DSBs are repaired quickly whereas complex DSBs seem to require processing and thus take more time to repair. This is evident by the fact that irradiated cells first show many small foci. As cells recover, the number of foci decreases (signifying repair) and the remaining foci get larger.Citation4,Citation37 Additionally, several key DDR proteins—NBS1, BRCA1 and 53BP1—are recruited to DSBs even in the absence of histone H2AX.Citation38 These proteins show early DSB recruitment kinetics in WT or histone H2AX null cells (minutes) but fail to accumulate later on in histone H2AX null cells (hours). Histone H2AX is therefore required for the sustained accumulation of these DDR factors at DSBs but not for their initial recruitment. Taken together, these observations suggest a model in which most DSBs are simple, repaired quickly and do not require histone H2AX. More complex DSBs would require histone H2AX in order to facilitate the local accumulation of additional repair factors as well as activation of cell cycle checkpoints.Citation4 It should also be noted that histone H2AX null cells fail to properly activate cell cycle checkpoints in response to IR doses that induce physiological amounts of DSBs, whereas non-physiological doses of IR can lead to activation of these checkpoints even in the absence of histone H2AX.Citation39 Thus, histone H2AX is required for activation of cell cycle checkpoints even in the event of a few DSBs, by participating in signal amplification and recruitment of downstream DDR proteins. In the following sections we will take a closer look into the earliest steps of focus formation, as well as downstream events, by describing key interactions that involve MDC1.
The DDR Described Through an Account of MDC1 and its Interacting Proteins
Focus formation.
γ-H2AX binds the tBRCT domain. Although it was clear early on that γ-H2AX serves as a very important epigenetic marker for DSBs, it was not known how it mediates the recruitment of downstream DDR proteins. A major advance in our understanding of how a focus is formed was the discovery that MDC1 directly binds γ-H2AX.Citation16,Citation40
Early experiments, which employed phospho-Ser oriented peptide library screening, determined that the tBRCT domain of MDC1 shows strong selectivity for a Tyr or Phe residue at the +3 position, yielding a pS-X-X-Y/F consensus binding motif.Citation41 Histone H2AX contains a C-terminal pSQEY motif, which fits this consensus sequence and was indeed demonstrated to be a docking platform for the tBRCT domain of MDC1. The tBRCT domain of MDC1 shows high specificity for the C-terminus of histone H2AX only in its phosphorylated form, thus allowing MDC1 to function as a specific sensor for γ-H2AX. Interestingly, it was shown that the tBRCT domain of MDC1 binds to a motif that requires an adjacent free carboxyl-terminus, leading to the definition of a more specific binding consensus sequence of pS-X-X-Y/F-COOH.Citation40 Analysis of the crystal structure of the tBRCT domain of MDC1 bound to a γ-H2AX peptide provided structural insights into how this domain recognizes the free carboxyl-terminus of γ-H2AX.Citation40,Citation42 Two phosphatases, PP2A and PP4, have been implicated in dephosphorylation of γ-H2AX.Citation43,Citation44 It was proposed that MDC1 protects γ-H2AX from dephosphorylation by its strong binding, thereby not only mediating downstream γ-H2AX signals but also preserving γ-H2AX itself.Citation40
Since the phenotype of MDC1 knockout miceCitation16 was very similar to that of histone H2AX knockout mice,Citation36,Citation45 it was proposed that the major physiological role of MDC1 is mediating histone H2AX-dependent signaling.
Recently, a novel regulatory phosphorylation was identified on Tyr-142 of histone H2AX, which is its most C-terminal residue. Tyr-142 phosphorylation is mediated by a newly identified kinase, WSTF,Citation46 and its dephosphorylation is mediated by a known tyrosine phosphatase—EYA.Citation47 The phosphorylation of Tyr-142 occurs on the tBRCT-binding motif of γ-H2AX and blocks binding of MDC1 to γ-H2AX. It was therefore suggested to act as an additional regulatory mechanism to prevent the formation and spreading of γ-H2AX in the absence of a break.
Phospho-ATM binds the FHA domain. ATM is the main transducer involved in the response to DSBs.Citation48–Citation50 ATM is activated after DSB induction and its activation is accompanied by induction of several post-translational modifications such as several autophosphorylationsCitation51,Citation52 and at least one acetylation.Citation53,Citation54 Autophosphorylation of Ser-1981, the most characterized of these events, leads to activation of ATM via the dissociation of the inactive ATM dimer.Citation52 However, the role of these autophosphorylations is still under dispute, as mutations in several such sites in mice, including Ser-1987 (which corresponds to Ser-1981 in humans), do not affect ATM activity.Citation55,Citation56 Following DSB induction, a fraction of ATM is rapidly recruited to damage sites.Citation57 As noted earlier, NBS1 recruits ATM to break sites via a conserved C-terminal motif. Subsequently, many ATM substrates are phosphorylated at the damaged sites.Citation48–Citation50
Analysis of pSer-1981-ATM (pATM) levels after DSB induction in MDC1 null cells indicated that MDC1 does not affect the activation of ATM itself. However, the recruitment of pATM to sites of damage and its chromatin retention were impaired in the absence of MDC1.Citation16 Since MDC1 binds γ-H2AX, this implied that MDC1 allows accumulation of pATM at sites of damage by bridging between pATM and γ-H2AX. This work showed that MDC1 indeed binds ATM and mapped the interaction to the FHA domain of MDC1. Furthermore, ATM and MDC1 interact directly, as the study employed purified proteins.Citation16 A very recent report has substantiated and extended these findings and demonstrated that the interaction between the FHA domain of MDC1 and ATM requires phosphorylation of Ser-1981.Citation58 In this study, So et al. demonstrate that the initial recruitment of ATM to DSBs requires the MRN complex but does not depend on MDC1 or Ser-1981. However, the prolonged retention of ATM at DSBs requires both phosphorylation of Ser-1981 and the presence of MDC1. Collectively, their data show that activated ATM is retained at DSBs by its phospho-dependent interaction with MDC1. Importantly, this prolonged retention of activated ATM at DSBs is crucial for the phosphorylation of downstream effectors and resistance to IR.Citation58 The most important insight gained from this study is that MDC1 acts not only as a sensor of damaged chromatin (via γ-H2AX), but also as a sensor for activated ATM. It is worth noting that the FHA domain of MDC1 deviates from the typical specificity of FHA domains for phospho-Thr motives, as it binds a phospho-Ser motif in ATM.Citation59 Structural analysis of the FHA domain of MDC1 should highlight the structural features that differentiate this domain from other FHA domains.
Together, these studies provide a mechanism for the positive feedback loop of focus formation as follows—when a DSB is formed, the MRN complex is quickly recruited to the break site (). ATM is initially recruited by the MRN complex, leading to its activation and subsequent phosphorylation of adjacent histone H2AX molecules forming γ-H2AX ().Citation29 MDC1 directly binds γ-H2AX via its tBRCT domainCitation16,Citation40 and pSer-1981-ATM via its FHA domain. This allows a local concentration of activated ATM, which in turn phosphorylates more distal histone H2AX molecules, thus creating a positive feedback loop ().
NBS1 binds the phospho-SDT repeats. NBS1 appears at DSBs very quickly, even after 20–30 seconds after damage induction.Citation60 This initial recruitment of NBS1 to DSBs, which does not require histone H2AX,Citation38 is also MDC1-independent. This was shown by depletion of MDC1 in human cellsCitation60 and by knockout of MDC1 in mice.Citation16 However, the prolonged retention of NBS1 at DSBs requires both histone H2AX and MDC1,Citation16,Citation60 much like the retention of activated ATM. The mechanism by which MDC1 regulates NBS1 retention at DSBs was poorly understood until recent independent reports by four groups which demonstrated that MDC1 directly binds NBS1.Citation61–Citation64 Since NBS1 also binds ATM,Citation29 these results suggest that MDC1 can recruit multiple ATM molecules to DSBs, both directly through its FHA domain as well as indirectly through NBS1, thus allowing a more efficient and quick positive feedback loop ().
Mapping of the interaction between NBS1 and MDC1 revealed that it required both the FHA domain and the adjacent tBRCT domain of NBS1 and a novel conserved motif in the N-terminus of MDC1. This motif, designated as the SDT repeat, appears six times in MDC1 and consists of highly conserved serine and threonine residues surrounded by acidic residues with a consensus sequence of SDTD-[V/A/D]-[D/E]-[D/E] (). The SDT repeat fits the consensus phosphorylation site for the acidophilic casein kinase 2 (CK2). All groups found that CK2 constitutively phosphorylates MDC1 on its SDT repeats. The phosphorylation of both serine and threonine residues was required for binding of NBS1 to MDC1. Inhibition of CK2 activity (by chemical inhibitors or siRNA) prevented phosphorylation of MDC1, thus preventing binding to NBS1 and localization of NBS1 to DSBs. Similar results were obtained when mutations or deletions were introduced in either the SDT repeats of MDC1 or the FHA-tBRCT domains of NBS1.Citation61–Citation64
Interesting questions arise from the data discussed above. Why does MDC1 contain multiple SDT repeats? And are all required for binding to NBS1? Results show that even a single repeat can bind NBS1, as a doubly phosphorylated SDT peptide (pSDpT) could directly bind the purified MRN complexCitation64 or a purified FHA-tBRCT fragment of NBS1.Citation61 However, progressively mutating more serine and threonine residues of the SDT repeats led to gradually lower affinity of MDC1 to NBS1 and to impaired redistribution of NBS1 to foci, suggesting that the existence of multiple SDT repeats in MDC1 allows cooperative and/or redundant binding to NBS1.Citation62,Citation63
Another interesting issue is the unique mode of binding between the FHA and tBRCT domains of NBS1 and the phospho-SDT motif of MDC1. FHA and tBRCT domains can each mediate protein-protein interactions on their own, showing specificity for pThr or pSer, respectively (see box 2). However, NBS1 seems to have combined both domains to yield a third domain. Two very recent structural and functional studies shed some light on this matter.Citation65,Citation66 The solved structures of the FHA-tBRCT domain of fission yeast and human NBS1 show a conserved structure in which the FHA and tBRCT domains each retained their respective domain fold. However, the FHA domain became fused to the first BRCT domain. The distance between the two binding sites of the FHA and tBRCT domains of NBS1 is such that a single molecule would only be able to bind both sites at once if at least 22 amino acids separate its interacting sites. It is therefore not possible for a single SDT repeat to bind both domains simultaneously, suggesting that two different SDT repeats bind the FHA and tBRCT domains. The distance between adjacent SDT repeats in MDC1 is quite variable (ranging from 26 to 81 residues) but is above this threshold, allowing any two SDT repeats to bind NBS1. Additionally, a combination of peptide binding experiments and mutational analyses demonstrate that while the FHA and tBRCT domains can each bind a phospho-SDT repeat to a certain degree in vitro, both are required for a functionally stable interaction in vivo. Overall, these observations provide a good explanation for the duplication of the SDT motif in MDC1 and suggest that the FHA domain of NBS1 binds the pThr of one SDT repeat while the tBRCT domain of NBS1 binds the pSer of a second SDT repeat.Citation65,Citation66 This bipartite mode of binding between MDC1 and NBS1 seems unique. Two other proteins that contain SDT-like motives only bind the FHA domain of NBS1—Lif1 in budding yeast (ortholog of mammalian XRCC4) and Ctp1 in fission yeast (ortholog of Sae2 in budding yeast and CtIP in mammals).Citation65,Citation66
RNF8 binds the phospho-TQXF repeats. Recently, a new factor in the DDR has been identified—RNF8.Citation7–Citation10 This protein contains an FHA domain as well as a RING finger domain, which acts as the catalytic core of certain E3 ubiquitin (Ub) ligases.Citation8 Analysis of RNF8 after damage induction shows that it is rapidly recruited to DSBs. RNF8 recruitment was dependent on histone H2AX and MDC1, but not on BRCA1 and 53BP1. On the other hand, BRCA1 and 53BP1 recruitment did require RNF8. An important observation was that the localization of RNF8 to sites of damage depended on its FHA domain while its RING domain was important for recruitment of BRCA1 and 53BP1.Citation7–Citation9
FHA domains exhibit specificity for phosphorylated threonine residues. Peptide library screening revealed that the FHA domain of RNF8 shows specificity for a pT-X-X-Y/F motif,Citation7 which bears a striking similarity to the pS-X-X-Y/F-COOH binding motif of the tBRCT domain of MDC1.Citation40 Sequence analysis revealed that human MDC1 contains four TQXF motifs (three in murine MDC1) that fit the binding preference of the FHA domain of RNF8 ().Citation7,Citation9 As expected, the FHA domain of RNF8 binds MDC1 through these TQXF repeats. Phosphorylation of these repeats occurs in response to DNA damage in an ATM-dependent manner, allowing a phospho-specific interaction between MDC1 and RNF8 only when DNA damage is induced.Citation7–Citation10 The TQXF repeats bind RNF8 in a redundant manner, as a single phospho-TQXF peptide could bind RNF8Citation9 and mutations of at least three of the four TQXF repeats were needed to prevent RNF8 binding.Citation8 This redundant mode of binding is very similar to that seen with NBS1 and its interaction with the SDT repeats of MDC1.
These results established that RNF8 is recruited to DSBs by a direct interaction with phospho-MDC1. But how does RNF8 control the accumulation of BRCA1 and 53BP1 at DSBs? Mutating or deleting the RING domain of RNF8 prevented BRCA1 and 53BP1 recruitment. This suggested that the E3 Ub ligase activity of RNF8 is required for BRCA1 and 53BP1 recruitment to DSBs. Exploring this possibility led to the discovery that RNF8, together with the E2 Ub activating enzyme UBC13, promotes poly-ubiquitination of histones H2A and H2AX at DSBs (). These ubiquitination events are crucial for recruitment of BRCA1 and 53BP1 and for an intact DDR (see box 3 for more on ubiquitination events at DSBs).Citation7–Citation10
53BP1 binds the tBRCT domain. p53 Binding Protein 1 (53BP1) is a large mediator protein which, like MDC1, contains a C-terminal tBRCT domain. In addition, 53BP1 contains a tandem Tudor (tTudor) domain that is essential for its localization to DSBs. The tTudor domain was shown to bind methylated lysine 79 of histone H3 and dimethylated lysine 20 of histone H4.Citation67–Citation70 These are constitutive histone modifications that lie within the histone core and current models suggest that they become accessible following formation of a break. A mechanism was suggested by which the MDC1- and RNF8-dependent ubiquitination events at DSBs promote chromatin relaxation which exposes these modifications, although the details of such a model remain to be established.Citation7–Citation10 While it is generally accepted that the tTudor is crucial for targeting of 53BP1 to sites of damage, it is not yet clear whether it is sufficient. Some reports suggest that the tTudor domain is enough for focus formation by 53BP1 while others show that it exhibits only partial recruitment to DSBs.Citation68–Citation72
We have recently shown that MDC1 directly binds 53BP1.Citation72 Analysis of the interaction between MDC1 and 53BP1 revealed that it is mediated by the tBRCT domain of MDC1. As mentioned earlier, the tBRCT domain of MDC1 shows specificity for a phosphorylated C-terminal motif.Citation40 However, such a motif does not exist in 53BP1 and detailed mapping revealed that MDC1 binds a central area of 53BP1. The interaction is modulated by phosphorylation but not strictly dependent on it. Deletion of the MDC1-binding region in 53BP1 led to aberrant recruitment of 53BP1 to foci, with a pattern similar to that of the tTudor domain alone, a result that indicates a possible role for the MDC1-53BP1 interaction in the recruitment of 53BP1 to DSBs.Citation72 Interestingly, a recent report demonstrated that a similar area in 53BP1 mediates homo-oligomerization of 53BP1 that is important for its focus formation.Citation71 While it is clear that MDC1 is required for 53BP1 recruitment to DSBs due to its role in RNF8-dependent histone ubiquitination, these recent studies show that additional events are likely to affect focus formation by 53BP1.
Interestingly, the interaction between MDC1 and 53BP1 is much stronger in mitotic cells.Citation15,Citation72 This result, coupled with the recent finding that MDC1 has a role in normal mitosis (discussed later),Citation22,Citation73 suggests that the interplay between MDC1 and 53BP1 might affect mitotic progression.
DNA repair.
DNA-PK binds the PST repeats. The PST repeats of MDC1 do not show any homology to other known sequences.Citation11,Citation12,Citation14 Since MDC1 is an adaptor protein, it was proposed that the PST repeats might also mediate protein-protein interactions. An attempt to discover PST repeat-binding proteins identified DNA-PK.Citation23 DNA-PK is composed of the large catalytic subunit, DNA-PKcs, and the heterodimer Ku (composed of Ku70 and Ku80). DNA-PK plays a central role in DNA repair via the non-homologous end joining (NHEJ) pathway.Citation74 This work suggested that MDC1 is important for IR-induced autophosphorylation of human DNA-PKcs on Thr-2609. This phosphorylation site has been previously demonstrated to be important for activation of DNA-PK and for subsequent DSB repair.Citation75 DSB repair via NHEJ was faulty in MDC1-depleted cells, assessed using a plasmid-based repair assay and a plasmid integration assay. This phenotype depended on the PST repeat domain of MDC1, which implies that MDC1 affects repair via the NHEJ pathway through its interaction with DNA-PK.Citation23 It is not known how MDC1 exerts its effect on DNA-PK. A direct interaction between MDC1 and DNA-PK was not established and it is not clear which part of the DNA-PK complex (i.e., DNA-PKcs or one of the Ku subunits) mediates the interaction.
Another supportive evidence for the role of MDC1 in NHEJ was demonstrated by the analysis of the fusion of dysfunctional telomeres as a model for DSBs. Loss of telomeric capping proteins leads to deprotection of telomeres, which are then recognized as DSBs and fused via repair by the NHEJ pathway. Inhibition of MDC1 itself, or its recruitment to chromatin, led to lower levels of telomere fusion, suggesting a positive role for MDC1 in NHEJ.Citation18 Two recent papers describe a role for 53BP1 in NHEJ of either dysfunctional telomeresCitation76 or distant breaks that form during V(D)J recombination.Citation77 Since MDC1 is required for recruitment of 53BP1 to DSBs, it is likely that a key role for MDC1 in NHEJ stems from its effect on 53BP1.
Rad51 binds the FHA domain. Apart from a role in the NHEJ pathway, there is evidence that MDC1 has a role in the homologous recombination (HR) repair pathway. According to work by Zhang et al. knockdown of MDC1 resulted in impaired repair by HR, determined by an assay that employs a GFP reporter that is only expressed when repaired via HR.Citation19
Rad51 is an early and central component of the HR pathway. Absence of MDC1 led to a general decrease in the levels of nuclear Rad51 and to lower levels of Rad51 foci in response to IR. Since MDC1 does not co-localize with Rad51 following IR,Citation11 it is unlikely that MDC1 directly affects recruitment of Rad51 to DSBs. The lower levels of Rad51 in the nucleus following MDC1 depletion suggested that MDC1 might affect Rad51 focus formation by regulating its stability. Pulse-chase experiments and analysis of Rad51 protein levels after cycloheximide treatment (blocks protein translation) were performed in MDC1 versus control knockdown cells. Results from these experiments show that Rad51 is less stable in the absence of MDC1. Therefore, the main mechanism of action of MDC1 in HR is mediated by the regulation of Rad51 stability, which affects its recruitment to DSBs and subsequent repair.Citation19
It was discovered that there is an interaction between the FHA domain of MDC1 and Rad51.Citation19 The interaction between MDC1 and Rad51 is direct and constitutive, as it is not influenced by IR. It also does not seem to require phosphorylation, a puzzling observation since FHA domains act as pThr-binding modules.Citation59 Reconstituting MDC1-depleted cells with an FHA-deleted form of MDC1 could partially rescue their HR-defective phenotype, suggesting that, while the FHA domain probably has a major role in HR by binding of Rad51 and allowing its stabilization, other parts of MDC1 may also be important.Citation19
Cell cycle checkpoints and apoptosis.
Phospho-Chk2 binds the FHA domain. An early report by Lou et al.Citation12 demonstrated a direct interaction between the activated form of Chk2 and MDC1. Chk2 is a central effector kinase, responsible for activation of cell cycle checkpoints and/or apoptosis via phosphorylation of multiple targets, including Ser-20 of p53.Citation78 Human Chk2 is phosphorylated on Thr-68 in an ATM-dependent manner at DSBs. This activated form of Chk2 does not accumulate in DSB-related foci but rather spreads throughout the nucleoplasm to perform its effector activities.Citation24
The interaction between Chk2 and MDC1 was found to be IR-dependent and a direct interaction was seen in vitro between a phospho-T68 peptide and a purified FHA domain of MDC1. Deleting the FHA domain of MDC1 or introducing a T68A mutation in Chk2 prevented their association. Thus, phosphorylation of Chk2 after IR on Thr-68 enables its binding to the FHA domain of MDC1.
Since Chk2 has a role in activating cell cycle checkpoints and apoptosis following IR, the authors explored the role of MDC1 in these pathways. Downregulation of MDC1 disrupted the activation of the intra-S-phase checkpoint and affected IR-induced apoptosis. Normal cells exhibit elevated levels of apoptosis following IR exposure. However, cells that lack Chk2 or MDC1 did not properly activate IR-induced apoptosis. Additionally, p53 accumulation and its phosphorylation on Ser-20 following IR were reduced when MDC1 was downregulated.Citation12
It is not clear how MDC1 regulates Chk2 and this effect remains in dispute. Some groups have reported that knockdown of MDC1 leads to a defect in phosphorylation of Chk2 on Thr-68,Citation17 and Ser-33/35,Citation13 while others have not seen such an effect.Citation11 If MDC1 does play a role in Chk2 activation, it is possible that it does so by bringing ATM and Chk2 into close proximity. Since both ATM and Chk2 bind the FHA domain of MDC1, it is unlikely that MDC1 directly brings them together. However, as mentioned earlier, MDC1 binds NBS1,Citation61–Citation64 which in turn binds ATM.Citation29 It is therefore possible for MDC1 to indirectly bridge between ATM and Chk2. This indirect interaction might be weak and transient enough to allow release of phospho-Chk2, which would then be free to diffuse away from the break and phosphorylate its targets.
p53 and Mdm2 bind the tBRCT domain. Two recent reports have suggested that MDC1 binds both p53 and Mdm2, thus affecting apoptosis.Citation79,Citation80 p53, a key tumor suppressor, is a transcription factor that is stabilized and phosphorylated in response to genotoxic stress. This leads to transcription of multiple target genes and allows activation of cell cycle checkpoints and/or apoptosis.Citation81 p53 protein levels are normally kept low, in part by ubiquitination of p53 by Mdm2 which leads to its degradation.Citation82,Citation83
The results presented in these studies show that MDC1 inhibits p53 activation and subsequent apoptosis.Citation79,Citation80 The authors observed that MDC1 protein and mRNA levels decrease within hours after DSB induction, using various chemical agents. This correlated with accumulation of p53. The inverse correlation suggested a functional link between MDC1 and p53. Immunofluorescent staining for MDC1 and p53 showed that after genotoxic stress, p53-positive cells do not exhibit MDC1 staining. Thus, MDC1 might somehow negatively regulate p53.
Overexpression of MDC1, while not affecting the initial accumulation and activation of p53, led to lower levels of p53 protein and lower expression of p53 target genes at later stages of the DDR (48 hours or more). Accordingly, downregulation of MDC1 led to accumulation of p53 and higher expression of p53 target genes.
The authors also explored the role of MDC1 in activation of apoptosis. Overexpression of MDC1 led to a lower level of apoptosis following damage induction, whereas MDC1 downregulation led to higher levels of apoptosis. Additionally, MDC1 levels in p53-proficient cells affected cell viability following genotoxic stress. More cells survived when MDC1 was highly expressed, whereas survival rate was lower when MDC1 was silenced. This phenotype was not observed in a p53-deficient cell line, suggesting that MDC1 has a p53-dependent role in promoting cell survival following DNA damage induction, presumably by preventing activation of apoptosis.Citation79
These results were indicative of a possible physical interaction between MDC1 and p53. Indeed, MDC1 and p53 interact and rough mapping by co-IP showed that the tBRCT domain of MDC1 binds the trans-activation domain of p53.Citation79 However, a direct interaction was not demonstrated. Interestingly, p53 phosphorylated on Ser-15 could not be detected in anti-MDC1 co-IP experiments. This is the first evidence that binding to the tBRCT domain of MDC1 is inhibited, rather than augmented, as a result of phosphorylation. The interaction was detectable in undisturbed cells, and was lost a few hours after damage induction, just as p53 and its Ser-15 phosphorylation are upregulated.Citation79
The results presented by Nakanishi et al.Citation79 contradict those observed by Lou et al.Citation12 While Nakanishi et al. suggested that MDC1 inhibits p53 activity, Lou et al. observed a positive role for MDC1 in p53 activation. It should be noted that Nakanishi et al. employed prolonged treatments with chemicals to induce DNA damage (mainly adriamycin),Citation79 whereas the most common DSB inducing agent, also employed by Lou et al.Citation12 is a single exposure to IR. It is thus possible that the conflicting results arose from the different methods for damage induction. Using a chemical leads to constant damage induction, which might lead to an aberrant DDR. Further studies are required to reconcile these discrepancies and determine the exact role of MDC1 in the regulation of p53.
In a short follow-up paper, Inoue et al.Citation80 suggest that MDC1 also regulates and interacts with Mdm2. MDC1 and Mdm2 were shown to bind each other by co-IP experiments and the tBRCT domain of MDC1 could be retrieved by anti-Mdm2 antibodies. Overexpression of MDC1 led to stabilization of Mdm2 protein, but not mRNA. More detailed work is required to establish a clear role for MDC1 in the regulation of Mdm2 in the context of the DDR. As p53 and Mdm2 interact with each other,Citation82,Citation83 it should be interesting to reveal whether these two studiesCitation79,Citation80 actually describe the same interaction or whether they report two separate ones.
The APC/C binds MDC1. In a search for proteins that interact with MDC1, the Anaphase Promoting Complex/Cyclosome (APC/C) has been identified as yet another tBRCT-binding partner.Citation73 The APC/C is a multi-subunit E3 Ub ligase with a central regulatory role in the progression of the cell cycle. The APC/C tags various targets for proteasomal degradation only during specific phases of the cell cycle, thus allowing their cyclic accumulation and degradation. The specificity of the APC/C, both for substrates and the timing of their degradation, is mainly achieved by association of the APC/C with one of two co-activators—Cdc20 and Cdh1.Citation84
In this study, we have shown that the entire APC/C is in complex with MDC1. An important observation was that the co-activator Cdh1 was also retrieved with the APC/C, suggesting that an active form of the APC/C binds MDC1. The interaction between MDC1 and the APC/C is mediated by the tBRCT domain of MDC1 and the phosphorylated form of the Cdc27 subunit of the APC/C. The C-terminal motif of Cdc27 (pSDEF-COOH) is very similar to that of γ-H2AX (pSQEY-COOH), suggesting that both proteins bind MDC1 via the same mechanism. Peptide competition experiments confirmed that the tBRCT domain of MDC1 binds Cdc27 or γ-H2AX in a mutually exclusive manner.Citation73
A very recent report has extended our work and demonstrated that MDC1, through its interaction with the APC/C, plays a role in normal mitosis.Citation22 While confirming our binding results, this work surprisingly shows that MDC1 also directly binds several other components of the APC/C, via the FHA, tBRCT and PST repeat domains of MDC1. This study shows that MDC1 has a role in the normal transition of cells from metaphase to anaphase, via a mechanism that is distinct from the DNA damage or spindle assembly checkpoints. Knockdown of MDC1 resulted in weaker binding of the co-activator Cdc20 to the APC/C and in lower activity of the APC/C in vitro. It was therefore suggested that MDC1 carries out its mitotic role by affecting APC/C activity.Citation22
Phospho-TopoIIα binds the tBRCT domain. A recent study has revealed an unexpected role for MDC1 and Topoisomerase IIα (TopoIIα) in the decatenation checkpoint.Citation21 This checkpoint is responsible for preventing entry into mitosis when sister chromatids are still entangled after replication, thus avoiding DNA breakage during chromosome separation. Using the tBRCT domain as bait, three proteins were retrieved from lysates and identified by mass spectrometry as TopoIIα, DNA ligase III and XRCC1.
The interaction between MDC1 and TopoIIα was verified by co-IP and GST pull-down experiments. Mapping of the interaction revealed that the tBRCT domain of MDC1 binds a motif surrounding Ser-1524 of TopoIIα. It is important to note that this motif does not conform to the established consensus binding sequence for the tBRCT domain (pS-X-X-Y/F-COOH).Citation40 It has an Asp residue rather than a Tyr/Phe at position +3 from the pSer and is not located at the C-terminus of TopoIIα.
Mutation of Ser-1524 of TopoIIα, deletion of the tBRCT domain of MDC1 or phosphatase treatment prevented the binding of TopoIIα to MDC1, establishing that phospho-Ser-1524 of TopoIIα binds the tBRCT domain of MDC1. Furthermore, a phospho-peptide derived from the sequence surrounding pSer-1524 could bind a purified GST-tBRCT fragment, suggesting a direct interaction between these proteins.
Treatment of cells with catalytic TopoII inhibitors, such as ICRF-193, leads to the stabilization of TopoII in its closed form and to unresolved catenated chromatids. Downregulation of either MDC1 or TopoIIα prevented the proper activation of the decatenation checkpoint, as assessed by scoring for mitotic cells after ICRF-193 treatment. Reconstituting TopoIIα deficient cells with a S1524A mutant failed to rescue them, supporting a model by which the interaction between MDC1 and TopoIIα has a role in the decatenation checkpoint.Citation21
The phosphorylation state of TopoIIα was analyzed, as it is required for the interaction between MDC1 and TopoIIα. Phosphorylation of Ser-1524 changes throughout the cell cycle, being higher during G2/M. However, treatment of cells with ICRF-193, which does lead to activation of the decatenation checkpoint, did not change the levels of Ser-1524 phosphorylation. The authors propose that ICRF-193 treatment might cause a conformational change in TopoIIα that exposes pSer-1524 for binding of MDC1. As all of the experiments in this study relied solely on ICRF-193 treatment, it remains to be established how MDC1 and TopoIIα activate this checkpoint during a normal cell cycle.
Summary and Challenges Ahead
In the recent years we have learned much about the molecular mechanisms that protect our genome from DSBs. However, key questions still remain unanswered. What is the molecular characteristic of a DSB that allows its detection? Is it a change in chromatin structure, the existence of a free double-stranded DNA end or maybe the occurrence of both? Another important question is how DSB detection gets converted into a signal. Is it merely binding of the MRN complex that is enough? Although we do not know enough about this very early step in DSB recognition, the following events are fairly well defined. We now know how the first set of DDR proteins are recruited to a DSB and have learned that damage-induced phosphorylation and ubiquitination of histones, as well as exposure of constitutive histone methylations, serve as important epigenetic marks of the break site. Still, we do not fully understand how these chromatin modifications play a role in recruitment of downstream effectors such as 53BP1 and BRCA1. Current models offer two possible explanations that are not mutually exclusive. One is that these modifications allow local changes in chromatin structure, thereby changing accessibility. Another option is that histone ubiquitination, much like phosphorylation, might serve as a specific binding platform for Ub-binding proteins. Understanding the role of these epigenetic marks is important and future work will undoubtedly uncover the molecular details of these events.
The emerging picture is that MDC1 plays a central role in the DDR, acting as a scaffold protein that binds multiple partners. Some of its interactions have been extensively studied, while others require more rigorous analysis to fully understand both mechanism and role. The FHA and tBRCT domain can each bind more than one protein and most of these interactions are probably mutually exclusive. It is thus likely that MDC1 resides in various complexes, both before and after induction of DSBs, each carrying out a different role.
A quick overview of MDC1-binding proteins shows that most of them bind MDC1 through its tBRCT domain (see ). This domain seems to have acquired multiple modes of binding with a broader spectrum of binding capabilities than was previously appreciated.Citation40 Additional biochemical and structural work is required to help us understand how the tBRCT domain of MDC1 shows specificity for different motives.
The FHA domain of MDC1 also shows a wider specificity than expected. Its binding to Rad51 does not seem to require phosphorylationCitation19 whereas its binding to ATM occurs via a phospho-Ser motif rather than an expected phospho-Thr motif.Citation58 Structural analysis coupled with oriented peptide library screening experiments based on both pSer and pThr peptides should help define the binding specificity of the FHA domain of MDC1.
A recurring theme in the architecture of MDC1 is the repetitive nature of some of its domains. The TQXF, SDT and PST domains are all comprised of imperfect repeats and the TQXF and SDT repeats share very similar characteristics. Both require phosphorylation to interact and a single motif is sufficient for binding. However, the SDT repeats have adopted a bipartite mode of binding to NBS1, which means that the six SDT repeats may constitute up to three potential NBS1-binding sites. If a single TQXF motif or a double SDT motif can bind their target, why were they duplicated several times? At least two possible explanations can be thought of. One is to allow a high stoichiometric ratio between MDC1 and its binding partners. Multiple repeats might allow binding of several RNF8 and NBS1 molecules onto a single MDC1 molecule, which would lead to a higher concentration of RNF8 and NBS1 at the break site. This in turn would allow faster and more efficient signal amplification. However, it is not clear whether steric considerations at all allow MDC1 to accommodate more than one molecule of each. Another option is that the local concentration of several binding sites increases the likelihood of a single molecule to bind MDC1, thus enhancing their binding affinities. Analyzing the stoichiometry of MDC1-containing complexes should resolve this issue.
Another challenge is to understand the function of the PST repeats, which have duplicated so much that they comprise about a third of human MDC1. It is tempting to speculate that the PST repeats exhibit a mode of binding similar to that of the TQXF and SDT repeats, e.g., redundant/cooperative and phospho-specific.
Apart from a role in the DDR, there is increasing evidence that MDC1 is involved in other biological processes. These include its recently demonstrated roles in the decatenation checkpointCitation21 and in the metaphase to anaphase transition in mitosis.Citation22,Citation73 Additionally, the fact that male MDC1 knockout mice exhibit defective spermatogenesisCitation16 implies a role in meiosis. Therefore, MDC1 emerges not only as a mediator of the DNA damage response, but also as a mediator of more general cell cycle related signals, and these two roles are most probably intertwined.
Box 1—The Importance of the DNA Damage Response
Among the different types of DNA lesions, DNA double-strand breaks (DSBs) are the most deleterious type. DSBs in the cells occur from endogenous causes, such as metabolic products and collapsed replication forks, and from exogenous factors, such as chemicals and ionizing radiation (IR). A defect in the ability to sense or repair DSBs can promote chromosomal rearrangements or loss of chromosome fragments. Consequently this can lead to apoptosis and more harmfully, promote genomic instability that can trigger carcinogenesis. Thus, eukaryotic cells have evolved different mechanisms that sense and repair DSBs to ensure the integrity of their genome.
The important role of the DNA damage response (DDR) is evident by the fact that mutations in many DDR proteins result in genomic instability syndromes. For example, mutations in the gene encoding for the transducer kinase ATM lead to the cancer-predisposing genetic disorder ataxia telangiectasia (AT). AT is characterized by genomic instability, a striking predisposition to lymphoreticular malignancies, cerebellar degeneration, thymic and gonadal atrophy, immunodeficiency and extreme sensitivity to DSB-inducing agents.Citation85 Similar disorders occur due to mutations in Mre11 [AT-like disease (ATLD)]Citation86 and NBS1 [Nijmegen breakage syndrome (NBS)].Citation87,Citation88 ATLD patients develop most of the hallmarks of AT, albeit with an onset at a later age and with slower progression.Citation86 The phenotype of NBS patients shows significant overlap with that of AT, except that NBS patients do not exhibit cerebellar degeneration.Citation87,Citation88 Another example is BRCA1. Mutations in BRCA1 result in gross genomic instability and are associated with hereditary breast and ovarian cancer.Citation89 For a comprehensive and recent review see reference Citation90.
Box 2—The FHA and BRCT Domains
Different protein-protein interaction motives have important roles in human diseases, particularly in relation to the cellular response to DNA damage and cancer. Among these domains are the forkhead associated (FHA) and the BRCA1 C-terminal (BRCT) domains.
FHA domains.
FHA domains mediate phospho-protein interactions. The domain was originally discovered by bioinformatics studies of the Forkhead family of transcription factors.Citation91 FHA domains are phospho-threonine (pThr)-binding modules that are found in many prokaryotic and eukaryotic proteins (examples for proteins discussed in this review—RNF8, Chk2, MDC1 and NBS1). Proteins containing FHA domains have roles in transcription, the DDR and cell cycle regulation. FHA domains also appear in several kinesin-like motors and regulators of small G proteins.Citation92 The pThr-containing motifs recognized by different FHA domains have been defined by peptide-library experiments revealing selection for particular residues around the pThr, located from four amino acids N-terminal to the pThr to three amino acids C-terminal to the pThr.Citation93,Citation94
The BRCT domain.
The BRCT domain was first identified as tandem repeats at the C-terminus of BRCA1,Citation95 a protein in which mutations were found in many hereditary breast and ovarian cancers.Citation96 A large number of proteins, all implicated in the DDR, contain single or multiple BRCT domains. BRCT domains consist of 85–95 amino acid residues that comprise several distinct clusters of conserved hydrophobic residues that together form the core of the BRCT fold. The role of the BRCT domains is probably to mediate protein-protein interactions, as many proteins containing BRCT domains mediate either BRCT-non BRCT or BRCT-BRCT interactions.Citation97
In several proteins the BRCT repeats appear in tandem, with variable linker regions, ranging from 0 to 24 residues in length, that separate the single domains.Citation97 A subset of tBRCT domains exhibits a phospho-serine binding capability.Citation98–Citation101
Many members of the DDR contain a phospho-specific interacting module, either tBRCT repeats or an FHA domain, and thus can mediate phospho-dependent interactions. As early events in the DDR involve crucial regulatory phosphorylation events, these phospho-dependent interactions facilitate the formation of signaling complexes, resulting in focus formation of multiple complexes at sites of DNA damage and activation of cell cycle checkpoints.
Box 3—Ubiquitination Events at DSBs
Recently, it was demonstrated that the response to DSBs involves a regulatory ubiquitination system that modifies the chromatin surrounding the DSBs (reviewed in refs. Citation102–Citation104).
Ubiquitination is a post-translational modification that covalently attaches Ubiquitin (Ub), a small protein, to a lysine residue on a target protein. It involves a Ub-activating enzyme (E1), a Ub-conjugating enzyme (E2), and a Ub ligase (E3) that confers substrate specificity.Citation105 Ubiquitination results in the attachment of a single Ub (mono-ubiquitination) or in the formation of poly-Ub chains. These chains are formed when Ub is conjugated to a lysine residue residing on another Ub molecule. Different types of chains can form, depending on the lysine residue of Ub that is further ubiquitinated. Ubiquitination provides either a signal for degradation (e.g., K48-poly-ubiquitination) or a regulatory signal (e.g., mono-ubiquitination or K63-poly-ubiquitination).Citation106 It forms a protein interaction interface recognized by specific Ub-binding domains, such as the Ub-interacting motif (UIM) and motif interacting with Ub (MIU).Citation107
The first evidence that ubiquitination plays an important role in the response to DSBs came from studies that implicated RAP80, a protein that contains two UIMs, as a DDR member that is required for focus formation by BRCA1.Citation108–Citation110 BRCA1 interacts with phosphorylated Abraxas that acts as an adaptor between BRCA1, RAP80 and the deubiquitinating enzyme BRCC36.Citation110 The UIMs of RAP80, which bind K6- and K63-linked poly-Ub chains, and their Ub-binding ability are required for the recruitment of BRCA1-RAP80 to DSBs.Citation108–Citation110 In addition, it was found that the E2 Ub-enzyme UBC13 is required for BRCA1 focus formation.Citation115 These results suggested that BRCA1 is recruited to DSBs via a Ub-dependent regulatory mechanism and implied for the first time, that the response to DSBs is not only dependent on phosphorylation events but also on ubiquitination.
The second evidence that ubiquitination has a major role in the DDR came from the finding that RNF8, an E3 Ub ligase, catalyzes ubiquitination events on chromatin at DNA lesions. RNF8 forms foci upon DSB induction, and its focus formation depends on MDC1 and γH2AX. RNF8 is recruited to sites of damage through its interaction with MDC1. RNF8 interacts with UBC13 to catalyze K63-linked Ub chains. Both histone H2A and H2AX were demonstrated to be ubiquitinated in response to DSBs in an RNF8-dependent manner. RNF8 Ub ligase activity is critical for focus formation by BRCA1 and 53BP1.Citation8,Citation116,Citation117
Recently, an additional E3 Ub ligase, RNF168, has been implicated in the DDR.Citation102,Citation118 RNF168 forms foci upon DSB induction in an RNF8-dependent manner. Cells downregulated for RNF168 show a similar phenotype to that of RNF8-depleted cells. Like RNF8, RNF168 contains a RING domain and works with UBC13. It contains two MIUs, which can bind K48- or K63-linked Ub chains. It was found that RNF168 binds ubiquitinated H2A-type histones, probably ubiquitinated by RNF8-UBC13, at sites of damage. RNF168 further ubiquitinates H2A-type histones at DSBs to form poly-Ub chains, and its Ub ligase activity is essential for focus formation by BRCA1 and 53BP1.Citation102,Citation118
Although focus formation by BRCA1 and 53BP1 relies on ubiquitination events at DSBs, their recruitment mechanism is different. The Ub-dependent recruitment of RNF168 to sites of damage and its subsequent activity is required for the localization of RAP80 at damaged sites. This in turn is necessary for focus formation by BRCA1. On the other hand, focus formation by 53BP1 is mediated by the binding of its tTudor domains to methylated histones. Ubiquitination of histones and/or other proteins at sites of damage might expose these modified histones and facilitate the recruitment of 53BP1 to sites of damage.
Abbreviations
| 53BP1 | = | p53 binding protein 1 |
| APC/C | = | anaphase-promoting complex/cyclosome |
| AT | = | ataxia telangiectasia |
| ATLD | = | AT-like disease |
| BARD1 | = | BRCA1-associated ring domain protein 1 |
| BRCT | = | BRCA1 C-terminal |
| CDK | = | cyclin dependent kinase |
| CK2 | = | casein kinase 2 |
| DDR | = | DNA damage response |
| DSB | = | double-strand break |
| FHA | = | forkhead-associated |
| HR | = | homologous recombination |
| IP | = | immunoprecipitation |
| IR | = | ionizing radiation |
| MRN | = | Mre11/Rad50/NBS1 complex |
| MDC1 | = | mediator of DNA damage checkpoint 1 |
| MIU | = | motif interacting with Ub |
| NBS | = | Nijmegen breakage syndrome |
| NHEJ | = | non-homologous end joining |
| PIKK | = | phospho-inositide-3-kinase-related protein kinase |
| siRNA | = | small interfering RNA |
| tBRCT | = | tandem BRCT |
| Ub | = | ubiquitin |
| UIM | = | Ub-interacting motif |
| UV | = | ultraviolet |
Figures and Tables
Figure 1 (See previous page). A model for focus formation at a double-strand break. After a DSB is induced (A), the MRN complex quickly relocalizes to the break site, bringing ATM along through an interaction with a C-terminal motif in NBS1 (B). ATM phosphorylates adjacent histone H2AX molecules, tagging nearby chromatin with γ-H2AX (C). MDC1 binds γ-H2AX through its tBRCT domain and recruits additional ATM molecules, both directly via its FHA domain, as well as indirectly via an interaction with NBS1 (D). This allows ATM to phosphorylate more distant histone H2AX molecules, thus spreading the initial signal and amplifying it. MDC1 also recruits the E3 Ub ligase RNF8, which attaches Ub molecules onto histones H2A and H2AX (D). RNF168 binds ubiquitinated histones H2A/X and catalyzes the formation of poly-Ub chains on these histones (E). These events are required for the subsequent arrival of important DDR proteins such as 53BP1 and the BRCA1-Abraxas-RAP80 complex. The events described in (D and E) occur on both sides of the break but only one side of the break is depicted for the sake of simplicity.
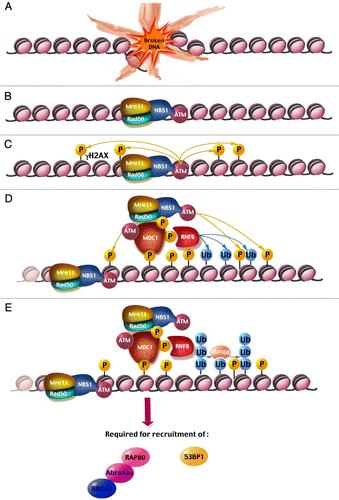
Figure 2 A schematic representation of human MDC1. All features are drawn to scale. Above each feature appears its name with relevant post-translational modifications and below are proteins that interact with it. The fourth TQXF repeat (depicted darker) is not as conserved as the first three. Also, the first three and last three PST repeats (light orange) show weak homology to the central 13 repeats (dark orange). See main text for more details.
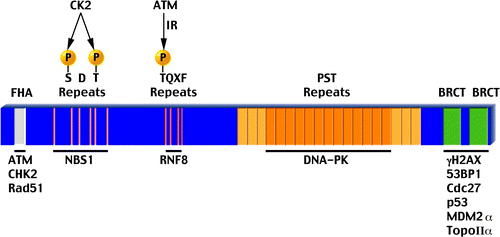
Table 1 A summary of all currently known MDC1 interactions
Acknowledgements
We thank members of our laboratories, Yosef Shiloh and Assaf Friedler for critical input and discussions and to Zehava Cohen for graphic support. Our research regarding MDC1 was supported by the Israel Science Foundation, Association for International Cancer Research and the Abisch-Frenkel Foundation.
References
- Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem 2004; 73:39 - 85
- Harper JW, Elledge SJ. The DNA damage response: Ten years after. Molecular Cell 2007; 28:739 - 745
- Bartek J, Lukas J. DNA damage checkpoints: from initiation to recovery or adaptation. Curr Opin Cell Biol 2007; 19:238 - 245
- Petrini J, Stracker T. The cellular response to DNA double-strand breaks: defining the sensors and mediators. Trends Cell Biol 2003; 13:458 - 462
- Fernandez-Capetillo O, Lee A, Nussenzweig M, Nussenzweig A. H2AX: the histone guardian of the genome. DNA Repair 2004; 3:959 - 967
- Soutoglou E, Misteli T. Activation of the cellular DNA damage response in the absence of DNA lesions. Science 2008; 320:1507 - 1510
- Huen MSY, Grant R, Manke I, Minn K, Yu XC, Yaffe MB, et al. RNF8 transduces the DNA-damage signal via histone ubiquitylation and checkpoint protein assembly. Cell 2007; 131:901 - 914
- Mailand N, Bekker-Jensen S, Faustrup H, Melander F, Bartek J, Lukas C, et al. RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins. Cell 2007; 131:887 - 900
- Kolas NK, Chapman JR, Nakada S, Ylanko J, Chahwan R, Sweeney FD, et al. Orchestration of the DNA-damage response by the RNF8 ubiquitin ligase. Science 2007; 318:1637 - 1640
- Wang B, Elledge SJ. Ubc13/Rnf8 ubiquitin ligases control foci formation of the Rap80/Abraxas/Brca1/Brcc36 complex in response to DNA damage. Proc Natl Acad Sci USA 2007; 104:20759 - 20763
- Goldberg M, Stucki M, Falck J, D'Amours D, Rahman D, Pappin D, et al. MDC1 is required for the intra-S-phase DNA damage checkpoint. Nature 2003; 421:952 - 956
- Lou Z, Minter-Dykhouse K, Wu X, Chen J. MDC1 is coupled to activated CHK2 in mammalian DNA damage response pathways. Nature 2003; 421:957 - 961
- Mochan TA, Venere M, DiTullio RA, Halazonetis TD. 53BP1 and NFBD1/MDC1-Nbs1 function in parallel interacting pathways activating ataxia-telangiectasia mutated (ATM) in response to DNA damage. Cancer Research 2003; 63:8586 - 8591
- Stewart G, Wang B, Bignell C, Taylor A, Elledge S. MDC1 is a mediator of the mammalian DNA damage checkpoint. Nature 2003; 421:961 - 966
- Xu XZ, Stern DF. NFBD1/MDC1 regulates ionizing radiation-induced focus formation by DNA checkpoint signaling and repair factors. Faseb J 2003; 17:1842 - 1848
- Lou Z, Minter-Dykhouse K, Franco S, Gostissa M, Rivera M, Celeste A, et al. MDC1 maintains genomic stability by participating in the amplification of ATM-dependent DNA damage signals. Mol Cell 2006; 21:187 - 200
- Peng A, Chen P. NFBD1, like 53BP1, is an early and redundant transducer mediating Chk2 phosphorylation in response to DNA damage. J Biol Chem 2003; 278:8873 - 8876
- Dimitrova N, de Lange T. MDC1 accelerates nonhomologous end-joining of dysfunctional telomeres. Genes Dev 2006; 20:3238 - 3243
- Zhang J, Ma Z, Treszezamsky A, Powell S. MDC1 interacts with Rad51 and facilitates homologous recombination. Nat Struct Mol Biol 2005; 12:902 - 909
- Xie AY, Hartlerode A, Stucki M, Odate S, Puget N, Kwok A, et al. Distinct roles of chromatin-associated proteins MDC1 and 53BP1 in mammalian double-strand break repair. Mol Cell 2007; 28:1045 - 1057
- Luo KT, Yuan J, Chen JJ, Lou ZK. Topoisomerase II alpha controls the decatenation checkpoint. Nature Cell Biol 2009; 11:196 - 204
- Townsend K, Mason H, Blackford AN, Miller ES, Chapman JR, Sedgwick GG, et al. Mediator of DNA Damage Checkpoint 1 (MDC1) Regulates Mitotic Progression. J Biol Chem 2009; 284:33939 - 33948
- Lou Z, Chen B, Asaithamby A, Minter-Dykhouse K, Chen D, Chen J. MDC1 regulates DNA-PK autophosphorylation in response to DNA damage. J Biol Chem 2004; 279:46359 - 46362
- Lukas C, Falck J, Bartkova J, Bartek J, Lukas J. Distinct spatiotemporal dynamics of mammalian checkpoint regulators induced by DNA damage. Nat Cell Biol 2003; 5:212 - 255
- Bekker-Jensen S, Lukas C, Kitagawa R, Melander F, Kastan M, Bartek J, et al. Spatial organization of the mammalian genome surveillance machinery in response to DNA strand breaks. J Cell Biol 2006; 173:195 - 206
- van den Bosch M, Bree RT, Lowndes NF. The MRN complex: coordinating and mediating the response to broken chromosomes. EMBO Reports 2003; 4:844 - 849
- D'Amours D, Jackson SP. The Mre11 complex: At the crossroads of dna repair and checkpoint signalling. Nat Rev Mol Cell Biol 2002; 3:317 - 327
- Uziel T, Lerenthal Y, Moyal L, Andegeko Y, Mittelman L, Shiloh Y. Requirement of the MRN complex for ATM activation by DNA damage. EMBO J 2003; 22:5612 - 5621
- Falck J, Coates J, Jackson SP. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature 2005; 434:605 - 611
- Rogakou EP, Boon C, Redon C, Bonner WM. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J Cell Biol 1999; 146:905 - 915
- Burma S, Chen BP, Murphy M, Kurimasa A, Chen DJ. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J Biol Chem 2001; 276:42462 - 42467
- Stiff T, O'Driscoll M, Rief N, Iwabuchi K, Lobrich M, Jeggo PA. ATM and DNA-PK function redundantly to phosphorylate H2AX after exposure to ionizing radiation. Cancer Res 2004; 64:2390 - 2396
- Ward IM, Chen JJ. Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. J Biol Chem 2001; 276:47759 - 47762
- Celeste A, Difilippantonio S, Difilippantonio M, Fernandez-Capetillo O, Pilch D, Sedelnikova O, et al. H2AX haploinsufficiency modifies genomic stability and tumor susceptibility. Cell 2003; 114:371 - 383
- Bassing C, Suh H, Ferguson D, Chua K, Manis J, Eckersdorff M, et al. Histone H2AX: A dosage-dependent suppressor of oncogenic translocations and tumors. Cell 2003; 114:359 - 370
- Celeste A, Petersen S, Romanienko P, Fernandez-Capetillo O, Chen H, Sedelnikova O, et al. Genomic instability in mice lacking histone H2AX. Science 2002; 296:922 - 927
- Yamauchi M, Oka Y, Yamamoto M, Niimura K, Uchida M, Kodama S, et al. Growth of persistent foci of DNA damage checkpoint factors is essential for amplification of G1 checkpoint signaling. DNA Repair 2008; 7:405 - 417
- Celeste A, Fernandez-Capetillo O, Kruhlak M, Pilch D, Staudt D, Lee A, et al. Histone H2AX phosphorylation is dispensable for the initial recognition of DNA breaks. Nat Cell Biol 2003; 5:651 - 675
- Fernandez-Capetillo O, Chen HT, Celeste A, Ward I, Romanienko PJ, Morales JC, et al. DNA damage-induced G(2)-M checkpoint activation by histone H2AX and 53BP1. Nat Cell Biol 2002; 4:993 - 997
- Stucki M, Clapperton J, Mohammad D, Yaffe M, Smerdon S, Jackson S. MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell 2005; 123:1213 - 1226
- Rodriguez M, Yu XC, Chen JJ, Songyang Z. Phosphopeptide binding specificities of BRCA1 COOH-terminal (BRCT) domains. J Biol Chem 2003; 278:52914 - 52918
- Lee M, Edwards R, Thede G, Glover J. Structure of the BRCT repeat domain of MDC1 and its specificity for the free COOH-terminal end of the gamma-H2AX histone tail. J Biol Chem 2005; 280:32053 - 32056
- Chowdhury D, Keogh MC, Ishii H, Peterson CL, Buratowski S, Lieberman J. gamma-H2AX dephosphorylation by protein phosphatase 2A facilitates DNA double-strand break repair. Mol Cell 2005; 20:801 - 809
- Nakada S, Chen GI, Gingras AC, Durocher D. PP4 is a gamma H2AX phosphatase required for recovery from the DNA damage checkpoint. EMBO Rep 2008; 9:1019 - 1026
- Bassing C, Chua K, Sekiguchi J, Suh H, Whitlow S, Fleming J, et al. Increased ionizing radiation sensitivity and genomic instability in the absence of histone H2AX. Proc Natl Acad Sci USA 2002; 99:8173 - 8178
- Xiao A, Li HT, Shechter D, Ahn SH, Fabrizio LA, Erdjument-Bromage H, et al. WSTF regulates the H2A.X DNA damage response via a novel tyrosine kinase activity. Nature 2009; 457:57
- Cook PJ, Ju BG, Telese F, Wang XT, Glass CK, Rosenfeld MG. Tyrosine dephosphorylation of H2AX modulates apoptosis and survival decisions. Nature 2009; 458:553 - 591
- Shiloh Y. The ATM-mediated DNA-damage response: taking shape. Trends Biochem Sci 2006; 31:402 - 410
- Lavin MF, Kozlov S. ATM activation and DNA damage response. Cell Cycle 2007; 6:931 - 942
- Lee JH, Paull TT. Activation and regulation of ATM kinase activity in response to DNA double-strand breaks. Oncogene 2007; 26:7741 - 7748
- Kozlov SV, Graham ME, Peng C, Chen P, Robinson PJ, Lavin MF. Involvement of novel autophosphorylation sites in ATM activation. EMBO J 2006; 25:3504 - 3514
- Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 2003; 421:499 - 506
- Sun YL, Jiang XF, Chen SJ, Fernandes N, Price BD. A role for the Tip60 histone acetyltransferase in the acetylation and activation of ATM. Proc Natl Acad Sci USA 2005; 102:13182 - 13187
- Sun YL, Xu Y, Roy K, Price BD. DNA damage-induced acetylation of lysine 3016 of ATM activates ATM kinase activity. Mol Cell Biol 2007; 27:8502 - 8509
- Pellegrini M, Celeste A, Difilippantonio S, Guo R, Wang WD, Feigenbaum L, et al. Autophosphorylation at serine 1987 is dispensable for murine Atm activation in vivo. Nature 2006; 443:222 - 225
- Daniel JA, Pellegrini M, Lee JH, Paull TT, Feigenbaum L, Nussenzweig A. Multiple autophosphorylation sites are dispensable for murine ATM activation in vivo. J Cell Biol 2008; 183:777 - 783
- Andegeko Y, Moyal L, Mittelman L, Tsarfaty I, Shiloh Y, Rotman G. Nuclear retention of ATM at sites of DNA double strand breaks. J Biol Chem 2001; 276:38224 - 38230
- So S, Davis AJ, Chen DJ. Autophosphorylation at serine 1981 stabilizes ATM at DNA damage sites. J Cell Biol 2009; 7:977 - 990
- Durocher D, Jackson SP. The FHA domain. FEBS Lett 2002; 513:58 - 66
- Lukas C, Melander F, Stucki M, Falck J, Bekker-Jensen S, Goldberg M, et al. Mdc1 couples DNA double-strand break recognition by Nbs1 with its H2AX-dependent chromatin retention. EMBO J 2004; 23:2674 - 2683
- Wu LM, Luo KT, Lou ZK, Chen JJ. MDC1 regulates intra-S-phase checkpoint by targeting NBS1 to DNA double-strand breaks. Proc Natl Acad Sci USA 2008; 105:11200 - 11205
- Spycher C, Miller E, Townsend K, Pavic L, Morrice N, Janscak P, et al. Constitutive phosphorylation of MDC1 physically links the MRE11-RAD50-NBS1 complex to damaged chromatin. J Cell Biol 2008; 181:227 - 240
- Melander F, Bekker-Jensen S, Falck J, Bartek J, Mailand N, Lukas J. Phosphorylation of SDT repeats in the MDC1 N terminus triggers retention of NBS1 at the DNA damage-modified chromatin. J Cell Biol 2008; 181:213 - 226
- Chapman JR, Jackson SP. Phospho-dependent interactions between NBS1 and MDC1 mediate chromatin retention of the MRN complex at sites of DNA damage. EMBO Rep 2008; 9:795 - 801
- Lloyd J, Chapman JR, Clapperton JA, Haire LF, Hartsuiker E, Li JJ, et al. A Supramodular FHA/BRCT-Repeat Architecture Mediates Nbs1 Adaptor Function in Response to DNA Damage. Cell 2009; 139:100 - 111
- Williams RS, Dodson GE, Limbo O, Yamada Y, Williams JS, Guenther G, et al. Nbs1 Flexibly Tethers Ctp1 and Mre11-Rad50 to Coordinate DNA Double-Strand Break Processing and Repair. Cell 2009; 139:87 - 99
- Botuyan MV, Lee J, Ward IM, Kim JE, Thompson JR, Chen JJ, et al. Structural basis for the methylation state-specific recognition of histone H4-K20 by 53BP1 and Crb2 in DNA repair. Cell 2006; 127:1361 - 1373
- Charier G, Couprie J, Alpha-Bazin B, Meyer V, Quemeneur E, Guerois R, et al. The tudor tandem of 53BP1: A new structural motif involved in DNA and RG-rich peptide binding. Structure 2004; 12:1551 - 1562
- Huyen Y, Zgheib O, DiTullio RA, Gorgoulis VG, Zacharatos P, Petty TJ, et al. Methylated lysine 79 of histone H3 targets 53BP1 to DNA double-strand breaks. Nature 2004; 432:406 - 411
- Iwabuchi K, Basu BP, Kysela B, Kurihara T, Shibata M, Guan DY, et al. Potential role for 53BP1 in DNA end-joining repair through direct interaction with DNA. J Biol Chem 2003; 278:36487 - 36495
- Zgheib O, Pataky K, Brugger J, Halazonetis TD. An Oligomerized 53BP1 Tudor Domain Suffices for Recognition of DNA Double-Strand Breaks. Mol Cell Biol 2009; 29:1050 - 1058
- Eliezer Y, Argaman L, Rhie A, Doherty AJ, Goldberg M. The Direct Interaction between 53BP1 and MDC1 Is Required for the Recruitment of 53BP1 to Sites of Damage. J Biol Chem 2009; 284:426 - 435
- Coster G, Hayouka Z, Argaman L, Strauss C, Friedler A, Brandeis M, et al. The DNA damage response mediator MDC1 directly interacts with the anaphase-promoting complex/cyclosome. J Biol Chem 2007; 282:32053 - 32064
- Meek K, Gupta S, Ramsden DA, Lees-Miller SP. The DNA-dependent protein kinase: the director at the end. Immunol Rev 2004; 200:132 - 141
- Chan DW, Chen BPC, Prithivirajsingh S, Kurimasa A, Story MD, Qin J, et al. Autophosphorylation of the DNA-dependent protein kinase catalytic subunit is required for rejoining of DNA double-strand breaks. Genes Dev 2002; 16:2333 - 2338
- Dimitrova N, Chen YCM, Spector DL, de Lange T. 53BP1 promotes non-homologous end joining of telomeres by increasing chromatin mobility. Nature 2008; 456:524 - 551
- Difilippantonio S, Gapud E, Wong N, Huang CY, Mahowald G, Chen HT, et al. 53BP1 facilitates long-range DNA end-joining during V(D)J recombination. Nature 2008; 456:529 - 557
- Bartek J, Lukas J. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell 2003; 3:421 - 429
- Nakanishi M, Ozaki T, Yamamoto H, Hanamoto T, Kikuchi H, Furuya K, et al. NFBD1/MDC1 associates with p53 and regulates its function at the crossroad between cell survival and death in response to DNA damage. J Biol Chem 2007; 282:22993 - 23004
- Inoue K, Nakanjishi M, Kikuchi H, Yamamoto H, Todo S, Nakagawara A, et al. NFBD1/MDC1 stabilizes oncogenic MDM2 to contribute to cell fate determination in response to DNA damage. Biochem Biophys Res Commun 2008; 371:829 - 833
- Lakin ND, Jackson SP. Regulation of p53 in response to DNA damage. Oncogene 1999; 18:7644 - 7655
- Harris SL, Levine AJ. The p53 pathway: positive and negative feedback loops. Oncogene 2005; 24:2899 - 2908
- Brooks CL, Gu W. p53 ubiquitination: Mdm2 and beyond. Mol Cell 2006; 21:307 - 315
- Peters JM. The anaphase promoting complex/cyclosome: a machine designed to destroy. Nat Rev Mol Cell Biol 2006; 7:644 - 656
- Savitsky K, Bar-Shira A, Gilad S, Rotman G, Ziv Y, Vanagaite L, et al. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science 1995; 268:1749 - 1753
- Stewart GS, Maser RS, Stankovic T, Bressan DA, Kaplan MI, Jaspers NG, et al. The DNA double-strand break repair gene hMRE11 is mutated in individuals with an ataxia-telangiectasia-like disorder. Cell 1999; 99:577 - 587
- Matsuura S, Tauchi H, Nakamura A, Kondo N, Sakamoto S, Endo S, et al. Positional cloning of the gene for Nijmegen breakage syndrome. Nat Genet 1998; 19:179 - 181
- Varon R, Vissinga C, Platzer M, Cerosaletti KM, Chrzanowska KH, Saar K, et al. Nibrin, a novel DNA double-strand break repair protein, is mutated in Nijmegen breakage syndrome. Cell 1998; 93:467 - 476
- Miki Y, Swensen J, Shattuck-Eidens D, Futreal PA, Harshman K, Tavtigian S, et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 1994; 266:66 - 71
- Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature 2009; 461:1071 - 1078
- Hofmann K, Bucher P. The FHA domain: a putative nuclear signalling domain found in protein kinases and transcription factors. Trends Biochem Sci 1995; 20:347 - 349
- Li S, Ting NS, Zheng L, Chen PL, Ziv Y, Shiloh Y, et al. Functional link of BRCA1 and ataxia telangiectasia gene product in DNA damage response. Nature 2000; 406:210 - 215
- Yaffe MB, Smerdon SJ. The use of in vitro peptide-library screens in the analysis of phosphoserine/threonine-binding domain structure and function. Annu Rev Biophys Biomol Struct 2004; 33:225 - 244
- Durocher D, Jackson SP. The FHA domain. FEBS Lett 2002; 513:58 - 66
- Koonin EV, Altschul SF, Bork P. BRCA1 protein products… Functional motifs. Nat Genet 1996; 13:266 - 268
- Castilla LH, Couch FJ, Erdos MR, Hoskins KF, Calzone K, Garber JE, et al. Mutations in the BRCA1 gene in families with early-onset breast and ovarian cancer. Nat Genet 1994; 8:387 - 391
- Huyton T, Bates PA, Zhang X, Sternberg MJ, Freemont PS. The BRCA1 C-terminal domain: structure and function. Mutat Res 2000; 460:319 - 332
- Yu X, Chini CC, He M, Mer G, Chen J. The BRCT domain is a phospho-protein binding domain. Science 2003; 302:639 - 642
- Manke IA, Lowery DM, Nguyen A, Yaffe MB. BRCT repeats as phosphopeptide-binding modules involved in protein targeting. Science 2003; 302:636 - 639
- Rodriguez M, Yu X, Chen J, Songyang Z. Phosphopeptide binding specificities of BRCA1 COOH-terminal (BRCT) domains. J Biol Chem 2003; 278:52914 - 52918
- Stucki M, Clapperton JA, Mohammad D, Yaffe MB, Smerdon SJ, Jackson SP. MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell 2005; 123:1213 - 1226
- Stewart GS. Solving the RIDDLE of 53BP1 recruitment to sites of damage. Cell Cycle 2009; 8:1532 - 1538
- Panier S, Durocher D. Regulatory ubiquitylation in response to DNA double-strand breaks. DNA Repair (Amst) 2009; 8:436 - 443
- van Attikum H, Gasser SM. Crosstalk between histone modifications during the DNA damage response. Trends Cell Biol 2009; 19:207 - 217
- Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem 1998; 67:425 - 479
- Kim I, Rao H. What's Ub chain linkage got to do with it?. Sci STKE 2006; 2006:18
- Hurley JH, Lee S, Prag G. Ubiquitin-binding domains. Biochem J 2006; 399:361 - 372
- Sobhian B, Shao G, Lilli DR, Culhane AC, Moreau LA, Xia B, et al. RAP80 targets BRCA1 to specific ubiquitin structures at DNA damage sites. Science 2007; 316:1198 - 1202
- Kim H, Chen J, Yu X. Ubiquitin-binding protein RAP80 mediates BRCA1-dependent DNA damage response. Science 2007; 316:1202 - 1205
- Wang B, Matsuoka S, Ballif BA, Zhang D, Smogorzewska A, Gygi SP, et al. Abraxas and RAP80 form a BRCA1 protein complex required for the DNA damage response. Science 2007; 316:1194 - 1198
- Xu B, Kim S, Kastan MB. Involvement of Brca1 in S-phase and G(2)-phase checkpoints after ionizing irradiation. Mol Cell Biol 2001; 21:3445 - 3450
- Gudmundsdottir K, Ashworth A. The roles of BRCA1 and BRCA2 and associated proteins in the maintenance of genomic stability. Oncogene 2006; 25:5864 - 5874
- Nishikawa H, Ooka S, Sato K, Arima K, Okamoto J, Klevit RE, et al. Mass spectrometric and mutational analyses reveal Lys-6-linked polyubiquitin chains catalyzed by BRCA1-BARD1 ubiquitin ligase. J Biol Chem 2004; 279:3916 - 3924
- Wu-Baer F, Lagrazon K, Yuan W, Baer R. The BRCA1/BARD1 heterodimer assembles polyubiquitin chains through an unconventional linkage involving lysine residue K6 of ubiquitin. J Biol Chem 2003; 278:34743 - 34746
- Zhao GY, Sonoda E, Barber LJ, Oka H, Murakawa Y, Yamada K, et al. A critical role for the ubiquitin-conjugating enzyme Ubc13 in initiating homologous recombination. Mol Cell 2007; 25:663 - 675
- Wang B, Elledge SJ. Ubc13/Rnf8 ubiquitin ligases control foci formation of the Rap80/Abraxas/Brca1/Brcc36 complex in response to DNA damage. Proc Natl Acad Sci USA 2007;
- Huen MS, Grant R, Manke I, Minn K, Yu X, Yaffe MB, et al. RNF8 Transduces the DNA-Damage Signal via Histone Ubiquitylation and Checkpoint Protein Assembly. Cell 2007; 131:901 - 914
- Doil C, Mailand N, Bekker-Jensen S, Menard P, Larsen DH, Pepperkok R, et al. RNF168 binds and amplifies ubiquitin conjugates on damaged chromosomes to allow accumulation of repair proteins. Cell 2009; 136:435 - 446