Abstract
Lmna yields two major protein products in somatic cells, lamin C and prelamin A. Mature lamin A is produced from prelamin A by four posttranslational processing steps-farnesylation of a carboxyl-terminal cysteine, release of the last three amino acids of the protein, methylation of the farnesylcysteine, and the endoproteolytic release of the carboxyl-terminal 15 amino acids of the protein (including the farnesylcysteine methyl ester). Although the posttranslational processing of prelamin A has been conserved in vertebrate evolution, its physiologic significance remains unclear. Here we review recent studies in which we investigated prelamin A processing with Lmna knock-in mice that produce exclusively prelamin A (LmnaPLAO), mature lamin A (LmnaLAO), or nonfarnesylated prelamin A (LmnanPLAO). We found that the synthesis of lamin C is dispensable in laboratory mice, that the direct production of mature lamin A (completely bypassing all prelamin A processing) causes no discernable pathology in mice, and that exclusive production of nonfarnesylated prelamin A leads to cardiomyopathy.
Introduction
The nuclear lamina is an intermediate filament meshwork that lies beneath the inner nuclear membrane.Citation1 Lamin A and lamin C, two key structural proteins of the nuclear lamina, are produced by alternative splicing of Lmna and are commonly known as A-type lamins.Citation2 Along with the B-type lamins, lamins A and C oligomerize to form a filamentous lattice that provides structural support for the nucleus.Citation1,Citation3 Both nuclear lamins also interact with a number of proteins in the heterochromatin and nuclear membrane.Citation4
Lmna knockout mice (Lmna−/−) clearly demonstrated the importance of A-type lamins in mammals.Citation5Lmna−/− mice appear normal at birth but grow slowly and die by 5–6 weeks of age, with histopathologic evidence of muscular dystrophy.Citation5 Heterozygous knockout mice (Lmna+/−) develop late-onset dilated cardiomyopathy and conduction system disease.Citation6 The absence of lamins A and C disrupts the structural integrity of the nuclear envelope, resulting in grossly misshapen cell nucleiCitation5 and defective nuclear mechanics.Citation7
The first 566 amino acids of lamins A and C are identical, but their carboxyl-terminal domains diverge.Citation2,Citation8–Citation10 Lamin C (572 amino acids) terminates with exon 10 sequences and contains six carboxyl-terminal amino acids not found in prelamin A. Prelamin A (664 amino acids) contains 98 additional amino acid sequences encoded by exons 11 and 12 not found in lamin C, and terminates with a carboxyl-terminal CaaX motif. Prelamin A is converted to mature lamin A by four enzymatic post-translational processing steps—farnesylation of a cysteine located four amino acids from the carboxyl terminus of the protein (carried out by protein farnesyltransferase), endoproteolytic cleavage of the last three amino acids of the protein (a redundant activity of RCE1 and ZMPSTE24), carboxyl methylation of the newly exposed farnesylcysteine (by ICMT), and endoproteolytic release of the last 15 amino acids of the protein, including the farnesylcysteine methyl ester (by ZMPSTE24).Citation11–Citation19 The final cleavage step releases mature lamin A, which contains 646 amino acids.
Disruption of prelamin A processing has been linked to progeroid disorders in humans and mice. Hutchinson-Gilford progeria syndrome (HGPS) is caused by heterozygosity for LMNA point mutations that alter mRNA splicing and results in a 50-amino acid internal deletion in prelamin A.Citation20 This deletion does not affect prelamin A's CaaX motif but removes the site for the final cleavage by ZMPSTE24, preventing normal lamin A biogenesis and causing an accumulation of a shortened prelamin A (often called progerin) that retains a carboxyl-terminal farnesylcysteine methyl ester.Citation21,Citation22
In humans, a deficiency in ZMPSTE24 causes a perinatal-lethal progeroid syndrome, restrictive dermopathy (RD). Without ZMPSTE24, the final processing step that releases mature lamin A cannot occur, resulting in an accumulation of a farnesylated and methylated prelamin A. Unlike progerin, the prelamin A in RD does not contain the internal deletion but retains the carboxyl-terminal 15-amino acid segment that is normally clipped off in the formation of mature lamin A.
The accumulation of farnesylated progerin (in HGPS) and noncleaved farnesylated prelamin A (in RD) disrupts the structural integrity of the nuclear lamina. HGPS fibroblasts (both mouse and human) as well as ZMPSTE24-deficient fibroblasts (both human and mouse) contain dramatically misshapen cell nuclei, with a high frequency of blebs and folds.Citation18–Citation20,Citation23
Prenylation and Progeria
The fact that farnesylated progerin in HGPS and the farnesylated prelamin A in the setting of ZMPSTE24 deficiency cause misshapen nuclei and severe disease phenotypes is well established, but the mechanism by which they elicit disease is less clear. Several studies have suggested that the retention of the carboxyl-terminal farnesylcysteine methyl ester is a key factor in disease pathogenesis. The frequency of misshapen nuclei in HGPS and RD fibroblasts can be reduced with a protein farnesyltransferase inhibitor (FTI).24-28 Also, the progeria-like disease phenotypes in HGPS knock-in mice (LmnaHG/+ mice, expressing farnesylated progerin) and Zmpste24-deficient mice (expressing noncleaved farnesylated prelamin A) could be ameliorated with an FTI.Citation29–Citation32 However, recent studies by Yang et al.Citation22 have suggested that the persistence of the farnesylcysteine may not be the entire story. They showed that knock-in mice expressing a nonfarnesylated version of progerin (LmnanHG/+) still developed progeria, although the severity of the disease was somewhat less than in the mice producing farnesylated progerin.Citation22 The latter finding implied that the farnesyl lipid may not be the sole factor in disease pathogenesis, and that the 50-amino acid deletion may also be relevant.
The discovery of significant disease in the LmnanHG/+ mice led Davies et al.Citation33 to hypothesize that the retention of the last 15 amino acids of prelamin A in ZMPSTE24-deficiency, and not merely the terminal farnesyl cysteine methyl ester, might be important in the pathogenesis of RD. To test this idea, they generated a Lmna knock-in allele that exclusively produced nonfarnesylated prelamin A. At the same time, a knock-in allele that produced exclusively wild-type prelamin A was generated.Citation33 The “nonfarnesylated prelamin A-only” allele (LmnanPLAO) and the “prelamin A-only” allele (LmnaPLAO) were identical except that the cysteine in the CaaX motif of the LmnanPLAO allele was changed to serine, thereby abolishing protein prenylation. Introns 10 and 11 were removed from both alleles, abolishing lamin C synthesis. Western blots of tissue extracts and embryonic fibroblasts from homozygous “nonfarnesylated prelamin A-only” (LmnanPLAO/nPLAO) mice showed that the LmnanPLAO allele produced exclusively prelamin A; subsequent metabolic labeling studies showed that this prelamin A was not farnesylated. Western blots of tissue extracts from LmnaPLAO/PLAO mice revealed the production of only mature lamin A (generated by the normal posttranslational processing of wild-type prelamin A).Citation33
Davies and colleagues reasoned that if the disease phenotypes in RD were truly elicited by the retention of the last 15 amino acids of prelamin A (rather than the farnesyl lipid), then the phenotypes of LmnanPLAO/nPLAO mice would be expected to resemble those of Zmpste24−/− mice. This was not the case. Unlike Zmpste24−/− mice, which gain weight slowly, develop micrognathia and osteolytic lesions in the bones, and die prematurelyCitation16,Citation18,Citation19 (phenotypes shared by humans with prelamin A-related progeroid syndromesCitation20,Citation34,Citation35), LmnanPLAO/nPLAO mice have no progeria phenotypes of any kind; they maintain virtually normal body weights and lack osteolytic lesions.Citation33 These findings suggest that—at least in the setting of full-length prelamin A—the farnesyl lipid is critical for the emergence of disease phenotypes.
Cardiomyopathy in LmnanPLAO/nPLAO Mice
Although the LmnanPLAO/nPLAO mice did not exhibit progeroid-like phenotypes, they were not phenotypically normal. Male LmnanPLAO/nPLAO mice died prematurely, with a median survival of 38.5 weeks. Female LmnanPLAO/nPLAO mice lived slightly longer but still had a markedly reduced median survival of 49.5 weeks.Citation33 Echocardiography revealed that LmnanPLAO/nPLAO mice had a dilated cardiomyopathy, with a reduced left ventricular ejection fraction and an increased end-systolic left ventricular chamber size. As is typical for cardiomyopathy,Citation36 the expression of myosin heavy chain alpha (Myh6) was reduced in the hearts of LmnanPLAO/nPLAO mice, and the expression of myosin heavy chain beta (Myh7) was increased.Citation33 LmnaPLAO/PLAO mice displayed no signs of cardiomyopathy and exhibited normal survival.
The finding of cardiomyopathy in LmnanPLAO/nPLAO mice was not entirely unexpected, given that LMNA missense mutations in humans have been linked to dilated cardiomyopathy and muscular dystrophy.Citation37–Citation39 When some of these disease-associated mutations were introduced into mouse Lmna, they have resulted in cardiomyopathy and premature death.Citation40,Citation41
There are two potential explanations for the cardiomyopathy in LmnanPLAO/nPLAO mice. One possibility is that nonfarnesylated prelamin A is a toxic molecule that leads to cardiac dysfunction. A toxic effect is quite plausible, especially in light of the observation that progeria disease phenotypes are positively correlated with the amount of farnesylated prelamin A. When the levels of farnesylated prelamin A in mice are reduced by one-half in Zmpste24−/− mice (by introducing a Lmna knockout allele or a lamin C-only allele), disease phenotypes are completely eliminated.Citation18,Citation42 Conversely, increasing farnesylated prelamin A levels in Zmpste24−/− mice (by introducing the LmnaPLAO allele) causes more severe disease.Citation33 The second potential explanation for cardiomyopathy in LmnanPLAO/nPLAO mice is that the nonfarnesylated prelamin A is simply a less effective lamin than wild-type lamin A. In other words, nonfarnesylated prelamin A is a poor substitute for lamin A (or lamin C) and is functionally less active—rather than being toxic. Heterozygosity for Lmna deficiency in mice causes cardiomyopathy in mice;Citation6 thus, it is quite plausible that a functionally hypoactive lamin could underlie the cardiomyopathy in LmnanPLAO/nPLAO mice.
To differentiate between these two possibilities (i.e., a toxic lamin vs. a hypoactive lamin), LmnanPLAO/+ mice were intercrossed with Lmna+/− mice to generate LmnanPLAO/− mice. LmnanPLAO/− mice produce exclusively nonfarnesylated prelamin A, but only half as much as LmnanPLAO/nPLAO mice. We reasoned that if nonfarnesylated prelamin A were toxic, LmnanPLAO/− mice would be protected from cardiomyopathy. On the other hand, if the cardiomyopathy in LmnanPLAO/nPLAO mice was due to a functionally hypoactive lamin, we would expect LmnanPLAO/− mice to have more severe disease. The outcome of this experiment was unequivocal; LmnanPLAO/− mice had greatly shortened survival (median survival, 14.6 weeks) compared with LmnanPLAO/nPLAO mice (median survival, 38.5 weeks) (). These genetic studies suggest that the cardiomyopathy in LmnanPLAO/nPLAO is caused by a functionally hypoactive lamin, not a toxic lamin.
The examination of a Lmna knock-in allele yielding the farnesylated form of progerin (LmnaHG) led to the opposite conclusion. In that case, the mutant lamin was clearly toxic. Mice carrying a single copy of the mutant allele and one copy of the knockout allele (LmnaHG/−) survived longer than mice carrying two copies of the mutant allele (LmnaHG/HG).Citation43 Thus, we suggest that lamin A mutations can be divided into two categories—those yielding a toxic lamin (e.g., farnesylated progerin in HGPS) and those yielding a functionally hypoactive lamin (e.g., nonfarnesylated prelamin A in cardiomyopathy).
Purpose of Prelamin A Processing
Several studies have suggested that prelamin A's hydrophobic lipid anchor is important for the targeting of the protein to the inner nuclear membrane.Citation15,Citation44–Citation46 If this were the case in vivo, one could reasonably propose that nonfarnesylated prelamin A is a poorly functioning lamin protein because it cannot reach the nuclear rim. However, we found scant evidence for defective targeting of nonfarnesylated prelamin A. In embryonic fibroblasts, the amount of nonfarnesylated prelamin A at the nuclear rim was somewhat less than the amount of mature lamin A. However, the targeting of nonfarnesylated prelamin A to the nuclear rim in tissues of LmnanPLAO/nPLAO mice was perfectly normal, identical to that of mature lamin A in LmnaPLAO/PLAO tissues.Citation33 Thus, the incorporation of prelamin A into the nuclear lamina does not appear to be dependent on protein farnesylation. These findings are consistent with several other recent reports. First, lamin C (which is never farnesylated) reaches the nuclear rim in the absence of lamin A.Citation42 Also, Lee et al.Citation47 showed that the nuclear rim localization of nonfarnesylated prelamin A in protein farnesyltransferase-deficient keratinocytes is indistinguishable from that of mature lamin A in wild-type keratinocytes.
To further explore the purpose of the multistep posttranslational processing pathway for prelamin A, Coffinier et al.Citation48 generated a knock-in Lmna allele (LmnaLAO) that yields mature lamin A directly, bypassing prelamin A synthesis and processing. These “mature lamin A-only” mice were generated by introducing a stop codon into Lmna immediately after the codon for the last amino acid of mature lamin A.Citation48 LmnaLAO/LAO mice produced only mature lamin A (and no prelamin A or lamin C), and the levels of mature lamin A in these mice were identical to those in LmnaPLAO/PLAO mice (where all of the mature lamin A is derived from prelamin A). We fully expected that the absence of prelamin A processing in LmnaLAO/LAO mice would be accompanied by significant pathology, particularly in view of the conservation of the prelamin A processing pathway in vertebrate evolution.Citation45,Citation49–Citation51 To our surprise, this was not the case. LmnaLAO/LAO mice had normal body weights, normal survival, and lacked discernable disease phenotypes—even in mice >18 months of age.Citation48 And quite remarkably, the mature lamin A in LmnaLAO/LAO tissues was normally positioned at the nuclear rim, indistinguishable from the lamin A in the tissues of wild-type or LmnaPLAO/PLAO mice. The only abnormality that we identified was in LmnaLAO/LAO fibroblasts; those fibroblasts had more nuclear shape deformities than wild-type fibroblasts, and a greater percentage of the mature lamin A in LmnaLAO/LAO fibroblasts seemed to be located in the nucleoplasm (as opposed to the nuclear rim).Citation48
Given that the prelamin A processing pathway is evolutionarily conserved, we believe that it is overwhelmingly likely that the pathway serves some important function. At this point, however, current evidence suggests that the prelamin A processing pathway is largely dispensable, at least in laboratory mice. It is entirely possible that a more subtle phenotype in the LmnaLAO/LAO (or LmnaLCO/LCO; see below) mice may not have been detectable in our initial studies, and that future studies very well may uncover a unique functional role for prelamin A processing in mammals.
Lamin A or Lamin C: Why Two Isoforms?
Both LmnaLAO/LAO and LmnaPLAO/PLAO mice appeared to be phenotypically normal, despite a complete absence of lamin C.Citation33,Citation48 Similarly, Fong et al.Citation42 found that knock-in mice that produced exclusively lamin C and no lamin A (LmnaLCO/LCO) were phenotypically normal. In contrast, mice lacking both lamin A and lamin C succumb to muscular dystrophy.Citation5 These observations strongly imply that lamin A and lamin C are crucial in mammals, but that they play largely redundant roles and that either isoform is sufficient to prevent disease. Once again, however, we believe that much remains to be learned, and that sooner or later, distinct functional properties for the two A-type lamins will emerge.
Concluding Remarks
Since the discovery of protein isoprenylation and the recognition that prelamin A is prenylated, prevailing dogma has held that the carboxyl-terminal posttranslational modifications must be critically important. Also, because lamin A biogenesis involved not only protein isoprenylation but three other processing steps, lamin A has generally been considered essential for the formation of the nuclear lamina. While miscellaneous studies in transfected cells have tended to support these views,Citation15,Citation44–Citation46 no one had actually tested this dogma in vivo. With the development of an array of knock-in mouse models (), this situation has started to change. The study of several of these mice has already shown that lamin A and lamin C are functionally redundant in laboratory mice.Citation33,Citation42,Citation48 Knock-in mouse experiments have also shown that exclusive production of nonfarnesylated prelamin A leads to cardiomyopathy, but not progeria.Citation33 Finally, we have learned that the direct synthesis of mature lamin A, bypassing prelamin A synthesis and processing, has no discernible adverse effects on either the targeting of lamin A to the nuclear envelope in vivo or mouse vitality.Citation48
While the absence of prelamin A processing appears to be inconsequential, defects in prelamin A processing wreak havoc. Defects that block lamin A biogenesis and lead to an accumulation of farnesylated prelamin A cause severe progeroid syndromes. More work is required to understand the importance of the farnesyl lipid in disease pathogenesis. FTI treatment studies in HGPS mice have shown that blocking protein farnesylation has salutary effects; however, genetic studies with a “nonfarnesylated progerin mouse” have raised questions about whether protein prenylation is the only important factor in disease pathogenesis. Additional studies in mice, along with the ongoing clinical trials of FTI therapy in humans with HGPS,Citation52 should help to clarify this issue.
Abbreviations
| HGPS | = | hutchinson-gilford progeria syndrome |
| RD | = | restrictive dermopathy |
| FTI | = | farnesyltransferase inhibitor |
Figures and Tables
Figure 1 Survival curves for Lmna+/+, LmnanPLAO/+ and LmnanPLAO/− mice. The survival time for male LmnanPLAO/− mice was shorter than for Lmna+/+ or LmnanPLAO/+ mice (n = 4 for each group) (p = 0.002). Previous studies showed that male LmnanPLAO/nPLAO mice had an average survival time of 38.5 weeks (a milder phenotype than LmnanPLAO/− mice),Citation33 suggesting that nonfarnesylated prelamin A is a poorly functioning lamin.
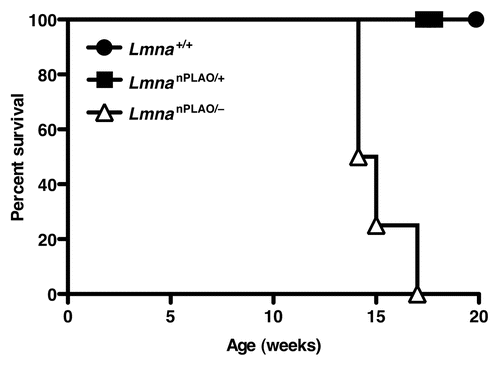
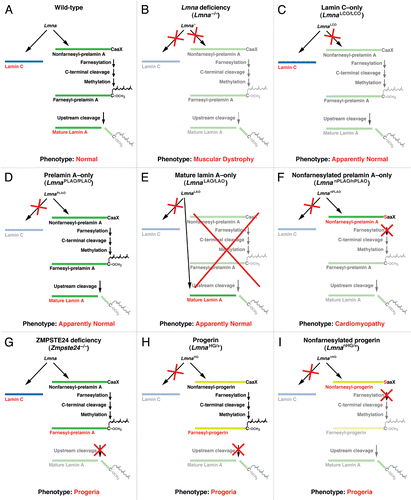
Figure 2 The effects of disrupting A-type lamin synthesis and processing in laboratory mice. (A) Wild-type mice produce both lamin C and prelamin A. Prelamin A then undergoes four sequential posttranslational processing steps to generate mature lamin A. (B) Lmna−/− mice produce neither lamin A nor lamin C. These mice have muscular dystrophy and die at 5–6 weeks of age.Citation5 (C) LmnaLCO/LCO mice produce only lamin C. These mice appear to be phenotypically normal.Citation42 (D) LmnaPLAO/PLAO mice do not produce lamin C. The prelamin A produced by these mice is efficiently converted to mature lamin A. These mice also appear to be normal.Citation33 (E) LmnaLAO/LAO mice produce mature lamin A directly, bypassing prelamin A synthesis and processing. These mice make no lamin C. Like LmnaLCO/LCO and LmnaPLAO/PLAO mice, LmnaLAO/LAO mice appear to be normal.Citation48 (F) LmnanPLAO/nPLAO produce exclusively nonfarnesylated prelamin A. Because of a cysteine-to-serine substitution in prelamin A's CaaX motif, the prelamin A cannot be farnesylated. These mice develop dilated cardiomyopathy and have reduced survival.Citation33 (G) Zmpste24-deficient mice produce both lamin C and prelamin A, but the final endoproteolytic step does not occur, resulting in the accumulation of farnesylated prelamin A. These mice display multiple progeria-like disease phenotypes.Citation16,Citation19 (H) In addition to making normal lamin C and prelamin A from one allele, LmnaHG/+ mice make a prelamin A with a 50-amino acid internal deletion (from the LmnaHG allele). This deletion prevents the final, ZMPSTE24-mediated, endoproteolytic step from occurring and leads to the accumulation of a farnesylated and truncated form of prelamin A called progerin. Despite the presence of normal forms of lamin A and lamin C, these mice develop progeria-like disease phenotypes.Citation28,Citation29 (I) LmnanHG/+ mice are identical to LmnaHG/+ mice except that the truncated prelamin A generated from the LmnanHG allele cannot be farnesylated (due to a cysteine-to-serine substitution in the CaaX motif), leading to the accumulation of nonfarnesylated progerin. These mice also have progeria, though it is somewhat less severe than that found in LmnaHG/+ mice.Citation22
Acknowledgements
This work was supported by the National Institutes of Health Grants (HL76839, HL86683, HL089781, GM66152, AG035626); a March of Dimes Grant (6-FY2007-1012); the Ellison Medical Foundation Senior Scholar Program; a postdoctoral fellowship award from the American Heart Association, western States Affiliate; and a Scientist Development Grant from the American Heart Association (0835489N).
Extra View to: Davies BS, Barnes RH 2nd, Tu Y, Ren S, Andres DA, Spielmann HP, et al. An accumulation of non-farnesylated prelamin A causes cardiomyopathy but not progeria. Hum Mol Genet 2010; 19:2682 - 2694; PMID: 20421363; http://dx.doi.org/10.1093/hmg/ddq158
References
- Muchir A, Worman HJ. The nuclear envelope and human disease. Physiology (Bethesda) 2004; 19:309 - 314
- Lin F, Worman HJ. Structural organization of the human gene encoding nuclear lamin A and nuclear lamin C. J Biol Chem 1993; 268:16321 - 16326
- Hutchison CJ, Worman HJ. A-type lamins: guardians of the soma?. Nat Cell Biol 2004; 6:1062 - 1067
- Wilson KL. The nuclear envelope, muscular dystrophy and gene expression. Trends Cell Biol 2000; 10:125 - 129
- Sullivan T, Escalante-Alcalde D, Bhatt H, Anver M, Bhat N, Nagashima K, et al. Loss of A-type lamin expression compromises nuclear envelope integrity leading to muscular dystrophy. J Cell Biol 1999; 147:913 - 920
- Wolf CM, Wang L, Alcalai R, Pizard A, Burgon PG, Ahmad F, et al. Lamin A/C haploinsufficiency causes dilated cardiomyopathy and apoptosis-triggered cardiac conduction system disease. J Mol Cell Cardiol 2008; 44:293 - 303
- Lammerding J, Schulze PC, Takahashi T, Kozlov S, Sullivan T, Kamm RD, et al. Lamin A/C deficiency causes defective nuclear mechanics and mechanotransduction. J Clin Invest 2004; 113:370 - 378
- Burke B, Stewart CL. Life at the edge: the nuclear envelope and human disease. Nat Rev Mol Cell Biol 2002; 3:575 - 585
- Fisher DZ, Chaudhary N, Blobel G. cDNA sequencing of nuclear lamins A and C reveals primary and secondary structural homology to intermediate filament proteins. Proc Natl Acad Sci USA 1986; 83:6450 - 6454
- Mounkes LC, Burke B, Stewart CL. The A-Type lamins: nuclear structural proteins as a focus for muscular dystrophy and cardiovascular diseases. Trends Cardiovasc Med 2001; 11:280 - 285
- Casey PJ. Biochemistry of protein prenylation. J Lipid Res 1992; 33:1731 - 1740
- Casey PJ, Seabra MC. Protein prenyltransferases. J Biol Chem 1996; 271:5289 - 5292
- Clarke S. Protein isoprenylation and methylation at carboxyl-terminal cysteine residues. Annu Rev Biochem 1992; 61:355 - 386
- Davies BS, Fong LG, Yang SH, Coffinier C, Young SG. The posttranslational processing of prelamin A and disease. Annu Rev Genomics Hum Genet 2009; 10:153 - 174
- Sinensky M, Fantle K, Trujillo M, McLain T, Kupfer A, Dalton M. The processing pathway of prelamin A. J Cell Sci 1994; 107:61 - 67
- Bergo MO, Gavino B, Ross J, Schmidt WK, Hong C, Kendall LV, et al. Zmpste24 deficiency in mice causes spontaneous bone fractures, muscle weakness and a prelamin A processing defect. Proc Natl Acad Sci USA 2002; 99:13049 - 13054
- Corrigan DP, Kuszczak D, Rusinol AE, Thewke DP, Hrycyna CA, Michaelis S, et al. Prelamin A endoproteolytic processing in vitro by recombinant Zmpste24. Biochem J 2005; 387:129 - 138
- Fong LG, Ng JK, Meta M, CotÇ N, Yang SH, Stewart CL, et al. Heterozygosity for Lmna deficiency eliminates the progeria-like phenotypes in Zmpste24-deficient mice. Proc Natl Acad Sci USA 2004; 101:18111 - 18116
- Pendas AM, Zhou Z, Cadinanos J, Freije JMP, Wang J, Hultenby K, et al. Defective prelamin A processing and muscular and adipocyte alterations in Zmpste24 metalloproteinase-deficient mice. Nat Genet 2002; 31:94 - 99
- Eriksson M, Brown WT, Gordon LB, Glynn MW, Singer J, Scott L, et al. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature 2003; 423:293 - 298
- Dechat T, Shimi T, Adam SA, Rusinol AE, Andres DA, Spielmann HP, et al. Alterations in mitosis and cell cycle progression caused by a mutant lamin A known to accelerate human aging. Proc Natl Acad Sci USA 2007; 104:4955 - 4960
- Yang SH, Andres DA, Spielmann HP, Young SG, Fong LG. Progerin elicits disease phenotypes of progeria in mice whether or not it is farnesylated. J Clin Invest 2008; 118:3291 - 3300
- Goldman RD, Shumaker DK, Erdos MR, Eriksson M, Goldman AE, Gordon LB, et al. Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci USA 2004; 101:8963 - 8968
- Capell BC, Erdos MR, Madigan JP, Fiordalisi JJ, Varga R, Conneely KN, et al. Inhibiting farnesylation of progerin prevents the characteristic nuclear blebbing of Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci USA 2005; 102:12879 - 12884
- Glynn MW, Glover TW. Incomplete processing of mutant lamin A in Hutchinson-Gilford progeria leads to nuclear abnormalities, which are reversed by farnesyltransferase inhibition. Hum Mol Genet 2005; 14:2959 - 2969
- Mallampalli MP, Huyer G, Bendale P, Gelb MH, Michaelis S. Inhibiting farnesylation reverses the nuclear morphology defect in a HeLa cell model for Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci USA 2005; 102:14416 - 14421
- Toth JI, Yang SH, Qiao X, Beigneux AP, Gelb MH, Moulson CL, et al. Blocking protein farnesyltransferase improves nuclear shape in fibroblasts from humans with progeroid syndromes. Proc Natl Acad Sci USA 2005; 102:12873 - 12878
- Yang SH, Bergo MO, Toth JI, Qiao X, Hu Y, Sandoval S, et al. Blocking protein farnesyltransferase improves nuclear blebbing in mouse fibroblasts with a targeted Hutchinson-Gilford progeria syndrome mutation. Proc Natl Acad Sci USA 2005; 102:10291 - 10296
- Yang SH, Meta M, Qiao X, Frost D, Bauch J, Coffinier C, et al. A farnesyltransferase inhibitor improves disease phenotypes in mice with a Hutchinson-Gilford progeria syndrome mutation. J Clin Invest 2006; 116:2115 - 2121
- Yang SH, Chang SY, Andres DA, Spielmann HP, Young SG, Fong LG. Assessing the efficacy of protein farnesyltransferase inhibitors in mouse models of progeria. J Lipid Res 2010; 51:400 - 405
- Yang SH, Qiao X, Fong LG, Young SG. Treatment with a farnesyltransferase inhibitor improves survival in mice with a Hutchinson-Gilford progeria syndrome mutation. Biochim Biophys Acta 2008; 1781:36 - 39
- Fong LG, Frost D, Meta M, Qiao X, Yang SH, Coffinier C, et al. A protein farnesyltransferase inhibitor ameliorates disease in a mouse model of progeria. Science 2006; 311:1621 - 1623
- Davies BS, Barnes RH 2nd, Tu Y, Ren S, Andres DA, et al. An accumulation of non-farnesylated prelamin A causes cardiomyopathy but not progeria. Hum Mol Genet 2010; 19:2682 - 2694
- DeBusk FL. The Hutchinson-Gilford progeria syndrome. Report of 4 cases and review of the literature. J Pediatr 1972; 80:697 - 724
- Fernandez-Palazzi F, McLaren AT, Slowie DF. Report on a case of Hutchinson-Gilford progeria, with special reference to orthopedic problems. Eur J Pediatr Surg 1992; 2:378 - 382
- Lowes BD, Minobe W, Abraham WT, Rizeq MN, Bohlmeyer TJ, Quaife RA, et al. Changes in gene expression in the intact human heart. Downregulation of alphamyosin heavy chain in hypertrophied, failing ventricular myocardium. J Clin Invest 1997; 100:2315 - 2324
- Bonne G, Barletta MRD, Varnous S, Becane HM, Hammouda EH, Merlini L, et al. Mutations in the gene encoding lamin A/C cause autosomal dominant Emery-Dreifuss muscular dystrophy. Nat Genet 1999; 21:285 - 288
- Muchir A, Bonne G, van der Kooi AJ, van Meegen M, Baas F, Bolhuis PA, et al. Identification of mutations in the gene encoding lamins A/C in autosomal dominant limb girdle muscular dystrophy with atrioventricular conduction disturbances (LGMD1B). Hum Mol Genet 2000; 9:1453 - 1459
- Vytopil M, Benedetti S, Ricci E, Galluzzi G, Dello Russo A, Merlini L, et al. Mutation analysis of the lamin A/C gene (LMNA) among patients with different cardiomuscular phenotypes. J Med Genet 2003; 40:132
- Arimura T, Helbling-Leclerc A, Massart C, Varnous S, Niel F, Lacene E, et al. Mouse model carrying H222P-Lmna mutation develops muscular dystrophy and dilated cardiomyopathy similar to human striated muscle laminopathies. Hum Mol Genet 2005; 14:155 - 169
- Mounkes LC, Kozlov SV, Rottman JN, Stewart CL. Expression of an LMNA-N195K variant of A-type lamins results in cardiac conduction defects and death in mice. Hum Mol Genet 2005; 14:2167 - 2180
- Fong LG, Ng JK, Lammerding J, Vickers TA, Meta M, CotÇ N, et al. Prelamin A and lamin A appear to be dispensable in the nuclear lamina. J Clin Invest 2006; 116:743 - 752
- Yang SH, Qiao X, Farber E, Chang SY, Fong LG, Young SG. Eliminating the synthesis of mature lamin A reduces disease phenotypes in mice carrying a Hutchinson-Gilford Progeria Syndrome allele. J Biol Chem 2008; 283:7094 - 7099
- Beck LA, Hosick TJ, Sinensky M. Incorporation of a product of mevalonic acid metabolism into proteins of Chinese hamster ovary cell nuclei. J Cell Biol 1988; 107:1307 - 1316
- Lutz RJ, Trujillo MA, Denham KS, Wenger L, Sinensky M. Nucleoplasmic localization of prelamin A: implications for prenylation-dependent lamin A assembly into the nuclear lamina. Proc Natl Acad Sci USA 1992; 89:3000 - 3004
- Hennekes H, Nigg EA. The role of isoprenylation in membrane attachment of nuclear lamins. A single point mutation prevents proteolytic cleavage of the lamin A precursor and confers membrane binding properties. J Cell Sci 1994; 107:1019 - 1029
- Lee R, Chang SY, Trinh H, Tu Y, White AC, Davies BS, et al. Genetic studies on the functional relevance of the protein prenyltransferases in skin keratinocytes. Hum Mol Genet 2010; 19:1603 - 1617
- Coffinier C, Jung HJ, Li Z, Nobumori C, Yun UJ, Farber EA, et al. Direct synthesis of lamin A, bypassing prelamin a processing, causes misshapen nuclei in fibroblasts but no detectable pathology in mice. J Biol Chem 2010; 285:20818 - 20826
- Beck LA, Hosick TJ, Sinensky M. Isoprenylation is required for the processing of the lamin A precursor. J Cell Biol 1990; 110:1489 - 1499
- Vorburger K, Kitten GT, Nigg EA. Modification of nuclear lamin proteins by a mevalonic acid derivative occurs in reticulocyte lysates and requires the cysteine residue of the C-terminal CXXM motif. EMBO J 1989; 8:4007 - 4013
- Weber K, Plessmann U, Traub P. Maturation of nuclear lamin A involves a specific carboxy-terminal trimming, which removes the polyisoprenylation site from the precursor; implications for the structure of the nuclear lamina. FEBS Lett 1989; 257:411 - 414
- Kieran MW, Gordon L, Kleinman M. New approaches to progeria. Pediatrics 2007; 120:834 - 841