Abstract
Centromeres are important structural constituents of chromosomes that ensure proper chromosome segregation during mitosis by providing defined sites for kinetochore attachment. In higher eukaryotes, centromeres have no specific DNA sequence and thus, they are rather determined through epigenetic mechanisms. A fundamental process in centromere establishment is the incorporation of the histone variant CENP-A into centromeric chromatin, which provides a binding platform for the other centromeric proteins. The Mis18 complex, and, in particular, its member M18BP1 was shown to be essential for both incorporation and maintenance of CENP-A.
Here we show that M18BP1 displays a cell cycle-regulated association with centromeric chromatin in mouse embryonic stem cells. M18BP1 is highly enriched at centromeric regions from late anaphase through to G1 phase. An interaction screen against 16 core centromeric proteins revealed a novel interaction of M18BP1 with CENP-C. We mapped the interaction domain in M18BP1 to a central region containing a conserved SANT domain and in CENP-C to the C-terminus. Knock-down of CENP-C leads to reduced M18BP1 association and lower CENP-A levels at centromeres, suggesting that CENP-C works as an important factor for centromeric M18BP1 recruitment and thus for maintaining centromeric CENP-A.
Introduction
Centromeres are sites for kinetochore attachment during mitosis. In order to prevent chromosome segregation defects, cells have to ensure that each chromosome has one functional centromere. Centromeres have no fixed DNA sequence that can be recognized by specific binding proteins, therefore it is assumed that epigenetic mechanisms ensure maintenance of the centromeric structure. The histone H3 variant CENP-A is a central component of centromeric chromatin. CENP-A aids in recruiting numerous proteins that build the constitutive centromere-associated network (CCAN),Citation1-Citation3 an essential step in establishing a proper kinetochore structure.Citation4 Two proteins directly bind CENP-A and have the potential to bridge centromeric chromatin with kinetochore components. The first protein, CENP-C, recognizes the C-terminal region of CENP-A through an internal region.Citation5 The C-terminus of CENP-C mediates its dimerization,Citation6,Citation7 the extreme N-terminus interacts with the Mis12 complex, which, in turn, bridges to outer kinetochore components.Citation8 The second protein directly recognizing CENP-A is CENP-N,Citation9 which also interacts with other centromeric components. Notably, disruption of either CENP-C or CENP-N leads to reduced levels of CENP-A at centromeres, suggesting that both proteins have additional functions in establishment or maintenance of centromere identity.Citation5,Citation9
The incorporation of CENP-A into the centromere is a strictly cell cycle-regulated process. During replication of centromeres, CENP-A is equally distributed onto the daughter strands, diluting the amount per centromere to 50%. To preserve centromere function, CENP-A needs to be subsequently replenished. Expression levels of CENP-A peak in G2 phase, though incorporation into the centromere only occurs in late mitosis and early G1 phase.Citation10-Citation13 The histone chaperone that mediates incorporation of CENP-A is the Holliday junction-recognizing protein (HJURP).Citation14,Citation15 HJURP can incorporate CENP-A only in domains that show a signature of actively transcribed chromatin.Citation16 Therefore, centromeric chromatin needs to be prepared (licensed), by currently unknown mechanisms. Mis18α, Mis18β and M18BP1 which form the Mis18 complex in humans have been suggested to play an important role in this licensing mechanism.Citation17-Citation19 Disruption of Mis18 complex components leads to failure in CENP-A incorporation,Citation17,Citation19 which could be explained by lack of HJURP recruitment to centromeres.Citation20,Citation21 Neither of the Mis18 complex proteins directly interact with CENP-A,Citation9 therefore, an important question within the understanding of CENP-A establishment is how this complex is specifically targeted to centromeric chromatin.
Here we show that M18BP1 is a cell cycle-regulated component of centromeric chromatin. By screening 16 CCAN proteins we identify CENP-C as a novel interaction partner of M18BP1. We mapped the interaction domain to a central region of M18BP1 encompassing the conserved SANT domain. CENP-C facilitates the recruitment of M18BP1 to centromeric chromatin during specific stages of the cell cycle, as RNAi depletion of CENP-C leads to reduced levels of centromeric M18BP1. In summary, our work identifies CENP-C as an important centromere component that recruits M18BP1 to centromeric chromatin.
Results
M18BP1 is a centromere-associated protein in mouse ES (mES) cells
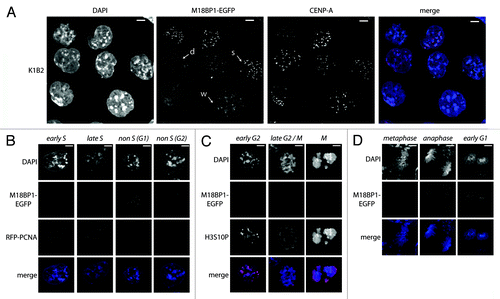
In HeLa cells, prominent centromeric association of Mis 18 complex members was observed from late mitosis (ana/telophase) until the end of G1 phase.Citation17 In order to determine the localization of M18BP1 in mouse cells we generated M18BP1 knock-in mES cells (K1B2) by introducing an EGFP tag into the endogenous M18BP1 locus (Fig. S1A). The K1B2 cells express M18BP1 at near endogenous levels (Fig. S1B), suggesting that the transcriptional regulation of M18BP1 is not impaired by the alterations to this locus. We determined the localization of M18BP1-EGFP in these cells by comparing the EGFP signal with CENP-A staining to ask whether M18BP1 localizes to the centromeres. Three classes of staining patterns could be observed (): (1) diffuse nuclear; (2) weak centromeric; and (3) strong centromeric foci. We then asked which cell cycle stage would correspond to the strong centromeric association of M18BP1. K1B2 cells show the typical cell cycle profile of mES cells, that is, the majority of cells are in S phase with an additional high percentage of cells in G2/M phase. In contrast to other frequently analyzed cell types, such as HeLa cells and mouse fibroblasts, the population of G1 cells is comparably low in mES cells. We visualized the different cell cycle stages in K1B2 cells using specific markers. First we expressed RFP-tagged PCNA in K1B2 cells, which is an indicator of different S phase stages.Citation22 All K1B2 cells which showed defined PCNA dots, indicative of ongoing replication showed weak or diffuse M18BP1-EGFP signals (). A significant number of cells outside S phase showed comparatively stronger signals, suggesting that centromeric M18BP1 association is low throughout S phase () and is only enriched in G1 or G2/M phase. We therefore tested whether M18BP1 begins to be enriched after S phase, in G2/M. In order to visualize G2/M cells, we performed immunofluorescence staining for H3S10 phosphorylation.Citation23 All K1B2 cells that were positive for H3S10P showed only weak centromeric M18BP1 signals (, early G2), leading to the conclusion that the highly enriched M18BP1 signals appear in G1 phase cells. Interestingly, in G2 phase cells, M18BP1 does not seem to associate with all centromeres as we detect numerous CENP-A spots without M18BP1 enrichment (Fig. S2). Late G2 and prometaphase cells did not show significant centromeric signals for M18BP1 (, late G2/M). Finally, we tested at which step after mitosis M18BP1 starts being localized to centromeric regions by examining distinct mitotic stages in K1B2 cells. Notably, we find that metaphase cells still display very low M18BP1 signals, though as soon as cells enter anaphase/telophase, M18BP1 is highly enriched at centromeres (). In those cells centromeric CENP-A signals are still relatively low as deposition of new CENP-A only occurs at later stages in the cell cycle.Citation12 In summary, our localization analysis demonstrates that in mES cells, M18BP1 is not constitutively enriched at centromeric chromatin but rather associates with centromeres from anaphase continuing to G1 phase. This is the time when CENP-A incorporation takes place.
Figure 1. M18BP1 associates with centromeres in a cell-cycle dependent manner. (A) Localization of endogenously tagged M18BP1-EGFP in the K1B2 mES cell line. K1B2 cells were stained for CENP-A and confocal stacks were recorded. Maximum intensity projections are shown. M18BP1-EGFP showed different patterns: strong enrichment at centromeres (s), weak enrichment (w), no enrichment/diffuse nuclear (d). Scale bars are 20 µm. (B) M18BP1 distribution during S phase. K1B2 cells were transfected with a RFP-PCNA expression construct to detect cells in different S phase stages. M18BP1-EGFP showed intermediate to low centromeric enrichment throughout S phase. Cells which are not in S phase fall into two different staining patterns: M18BP1 is highly enriched at centromeres (presumably G1) and cells with low/no centromeric M18BP1 signals (presumably G2). (C) M18BP1 distribution in G2/M phase. K1B2 cells were stained with H3S10P antibodies to visualize different stages of G2 and M phase. Starting from early G2 phase (weak H3S10P signal) to M phase (strong H3S10P signal) M18BP1 appeared to be largely absent from centromeres. (D) M18BP1 localization in different mitotic stages. In metaphase cells, M18BP1 is absent from centromeres, however, starting from late anaphase, M18BP1 showed strong signals at centromeric regions. Scale bars in (B–D) are 5 µm.
Centromere interaction screen for M18BP1
M18BP1 is known to be important for preparing centromeric chromatin for CENP-A incorporation. However, still very little is known about how M18BP1 actually recognizes centromeric chromatin. We pursued the idea that M18BP1 might be recruited through interaction with components of the CCAN network. To test this hypothesis we performed an F3H interaction screen of M18BP1 with proteins of the CCAN network. The F3H interaction assay utilizes a BHK cell line with a lac operator repeat array stably integrated into its genome (). This cell line was transfected with an expression vector encoding the lac repressor (lacI) which directly binds to the lac operator sequence fused with a GFP binding protein (GBP). These cells further expressed the EGFP tagged bait protein (M18BP1-EGFP) and individual RFP/mCherry tagged prey proteins (CCAN proteins). M18BP1-EGFP is bound by the lacI-GBP fusion protein at the lac operator arrays and can be detected at the well-discernible nuclear lacO focus. Prey protein interaction with M18BP1 is identified by localization to this nuclear focus (). The red/green signal intensity ratio provides a measure for the strength of the tested interaction.
Figure 2. F3H interaction screen for M18BP1 interaction partners. (A) Scheme depicting the F3H screening strategy. Cells containing a lac operator array were transfected with plasmids expressing a lac repressor-GBP fusion protein, M18BP1-EGFP and mCherry/RFP-CCAN proteins. The lac repressor binds to the lac operator array and through the GBP recruits M18BP1-EGFP. CCAN proteins interacting with M18BP1 are consequently enriched at the lac operator array. (B) Representative examples for M18BP1 interacting (CENP-C) and non-interacting (CENP-A and CENP-W) proteins are shown. Scale bar is 5µm. (C) Summary of interaction tests between M18BP1 and CCAN proteins. Interactions were tested with the F3H assay using M18BP1-EGFP and 16 RFP or mCherry fusions with CCAN proteins. From all 16 tested CCAN proteins, only CENP-C showed a clear interaction with M18BP1.
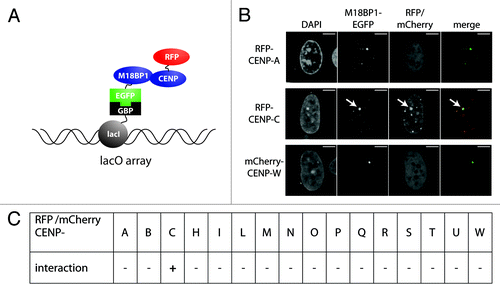
We tested M18BP1 for interaction with 16 proteins of the CCAN network using the F3H assay. In agreement with previous analyses we did not detect a direct interaction with CENP-A (). Interestingly, we found a strong interaction with another protein of the inner centromere, CENP-C. None of the other CCAN proteins that we tested showed significant interaction with M18BP1 ().
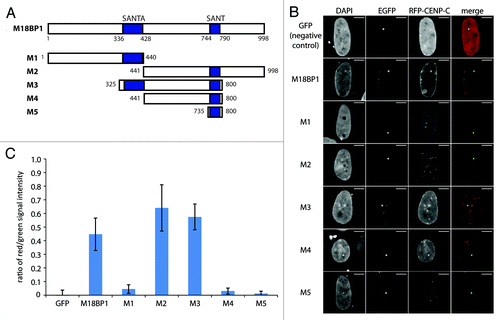
M18BP1 harbors two evolutionarily conserved domains, the SANT domain in the C-terminal part of the protein and the SANTA domain which is toward the N-terminus. To test which region of M18BP1 participates in the interaction with CENP-C we tested a panel of M18BP1 truncated proteins (). The N-terminus of M18BP1 (M1, aa1–440) showed no interaction with CENP-C, but the C-terminus (M2, aa441–998) clearly interacted (). The central region (M3, aa325–800) and a truncated protein lacking the SANTA domain (M4, aa441–800) displayed clear interactions with CENP-C. In order to then test whether the SANT domain is sufficient for the CENP-C interaction, we assessed a truncated protein harboring only the SANT domain (M5, aa735–800). This failed to interact with CENP-C, suggesting that additional parts of M18BP1 participate in this interaction. We scrutinized these observations by quantifying the interactions in several hundred cells per construct through measurement of the ratio between red and green intensity values at the M18BP1-EGFP foci. Although, by confocal imaging we can detect red/green colocalization of M18BP1-M4 and CENP-C in 73% of the cells (), the average red/green signal ratio is relatively low (). This, however, can be explained by relatively low expression levels of RFP-CENP-C in the combination with M18BP1-M4. In summary, our F3H data show that CENP-C interacts with a central region of M18BP1 comprising the SANT domain.
Figure 3. Mapping of the M18BP1-CENP-C interaction domains by F3H. (A) Scheme of M18BP1 truncations used in the interaction tests. (B) Representative F3H images of the negative control (GFP only) and EGFP tagged M18BP1 truncations tested with RFP-CENP-C. Overlap of red and green signals at the nuclear lacO focus indicates interaction. (C) Quantification of the F3H M18BP1-CENP-C interaction data. Intensities of red and green signals at the nuclear lacO focus were measured in several hundred cells each. The ratio between red and green signals was determined to measure the strength of the tested interactions.
M18BP1 directly interacts with CENP-C
We then aimed to further define the M18BP1-CENP-C interaction. First, we wanted to analyze whether M18BP1 co-localizes with CENP-C. To do this, we transfected K1B2 cells with a plasmid expressing RFP-tagged CENP-C and performed confocal imaging. We found many cells showing a clear overlap between M18BP1-EGFP and RFP-CENP-C signals. However, there was also a large percentage of cells with prominent CENP-C signals, with no M18BP1 co-localization (). These data suggest that the interaction between these two proteins is highly regulated in vivo.
Figure 4. M18BP1 and CENP-C interact in vitro. (A) Co-localization of RFP-CENP-C and M18BP1-EGFP. K1B2 cells were transfected with a RFP-CENP-C expression construct. Maximum intensity projections of two representative staining patterns are shown. Scale bar is 5µm. (B) Co-immunoprecipitation of CENP-C and M18BP1. HEK293FT cells were transfected with expression plasmids for EGFP-CENP-C and myc-M18BP1. Nuclear extracts from these cells were incubated with agarose beads (control) and GFP-Trap affinity beads to enrich for EGFP-CENP-C and interacting bound proteins. Protein gel blot analysis shows the nuclear extract (Inp), proteins bound to agarose beads (mock) and proteins that were enriched with GFP-Trap agarose beads (IP). An empty lane is indicated by “-”. EGFP-CENP-C and myc-M18BP1 were detected using antibodies against GFP and myc, respectively. (C) Interaction tests between M18BP1 and CENP-C truncations. The scheme shows the domain structure of mouse CENP-C and the truncation constructs that were used in this assay. Recombinant GST-tagged M18BP1 truncations (M1-M5) were incubated with in vitro translated myc-CENP-C truncation proteins and bound to GST beads. The bound CENP-C protein truncations were detected using myc antibody. Only the C-terminal CENP-C fragment showed clear interaction with M18BP1. The M18BP1 fragments M1-M5 are depicted in .
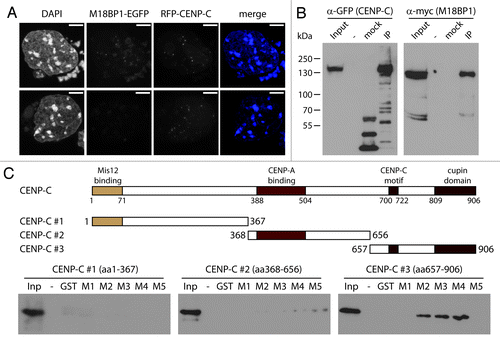
In order to test whether CENP-C and M18BP1 can interact in vivo, we performed co-immunoprecipitation experiments. We transfected HEK293 cells with expression plasmids for CENP-C-EGFP and myc-M18BP1, prepared nuclear extract and purified CENP-C-EGFP using GFP trap affinity beads. In the bound material we could clearly detect co-purification of the EGFP-tagged CENP-C and the myc-tagged M18BP1 (). We then wanted to further map the interaction domains between M18BP1 and CENP-C using in vitro binding assays. CENP-C has several conserved domains which have already been implicated in different biochemical interactions and in vivo functions, such as connection to the outer kinetochore proteins, CENP-A binding and CENP-C dimerization (, schematic). We generated in vitro translated proteins of three CENP-C truncations and tested their interaction with recombinant GST tagged M18BP1 truncations (M1-M5). In these assays we could only detect significant interaction of M18BP1 with CENP-C #3, containing the CENP-C motif and the cupin domain (). These data provide an extension of the F3H analysis; confirming that the large central region of M18BP1 is required for CENP-C binding.
CENP-C is required for the recruitment of M18BP1 to centromeres
Our data show that M18BP1 interacts with CENP-C in vitro and in vivo. CENP-C itself binds to centromeres through direct interaction with CENP-A. We therefore hypothesized that CENP-C facilitates the recruitment of M18BP1 to centromeric chromatin. In order to test this hypothesis we performed CENP-C knock-down experiments in K1B2 cells in which we could easily assess the localization of endogenously expressed M18BP1-EGFP. We prepared pLKO-based lentiviral vectors with three independent shRNA oligos against CENP-C and one control oligo containing an unrelated sequence (Table S1). CENP-C knock-down cells were analyzed by qPCR five days post infection to determine the knock-down efficiency of the individual oligos. Importantly, all three knock-down oligos resulted in effective downregulation of CENP-C mRNA () and protein (Fig. S3), with shCENP-C #3 showing the strongest knock-down. Crucially, the expression level of M18BP1 was unchanged. CENP-C knock-down did not lead to significant changes in the cell cycle profile of K1B2 cells, however, we did notice an increase in the number of cells with sub-G1 DNA content (Fig. S4) as well as reduced cell numbers at day five after knock-down (Fig. S5), indicating elevated cell death upon CENP-C knock-down.
Figure 5. CENP-C knock-down leads to impaired centromeric recruitment of M18BP1. (A) RT-qPCR for CENP-C and M18BP1 five days after knock-down. Expression levels in the control knock-down cell line (shControl) and the three CENP-C knock-down cell lines (shCENP-C #1-#3) were normalized to the geometric mean of GAPDH and Actin. (B) Representative maximum intensity projections of confocal stacks of control and CENP-C knock-down cells that were stained for CENP-A. Arrows point to example cells for the three classes of M18BP1 signals: strong (s), weak (w) and no enrichment/diffuse nuclear (d). Scale bars are 10µm. (C) Quantification of the M18BP1 signals in control vs. CENP-C knock-down cell lines. M18BP1 and CENP-A staining patterns were classified in several hundred cells. The bar graph depicts the percentages of each class.
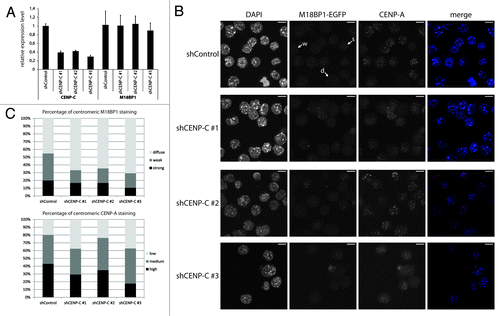
To test whether CENP-C affects the localization of M18BP1 we investigated M18BP1-EGFP and CENP-A patterns in the CENP-C knock-down cells. In control knock-down cells, M18BP1-EGFP shows the typical distribution of different centromere enrichment levels: strong, weak and diffuse nuclear (, arrows). We observed that the number of cells with centromeric association of M18BP1 was reduced in all three knock-down cell lines (). In order to quantify this phenotype we determined the distribution of M18BP1 staining patterns in control and CENP-C knock-down cells. Importantly, in all knock-down cell lines the percentage of cells with “weak” M18BP1-EGFP enrichment at centromeres was reduced, whereas M18BP1-EGFP “diffuse” cells were increased (). In knock-down shCENP-C #3 we even detected reduced numbers of M18BP1 ‘strong’ cells, suggesting that more efficient CENP-C knock-down more severely impairs centromeric M18BP1 recruitment. We then asked whether the reduced centromeric M18BP1 recruitment corresponds to specific cell cycle stages by co-staining of control and CENP-C knock-down cells with specific cell cycle markers (Fig. S6). In shControl cells we could reproduce the results of our initial cell cycle analysis in K1B2 cells: G1 cells showed strong centromeric signals, G2 cells showed weak signals. Importantly, upon CENP-C knock-down we detected G1 and G2 phase cells which had clearly lost centromeric M18BP1 (Fig. S6), indicating that the role of CENP-C in ensuring centromeric M18BP1 localization is not restricted to a particular cell cycle stage.
M18BP1 was proposed to ‘prime’ centromeres for deposition of CENP-A.Citation17,Citation19 In order to investigate whether reduced centromeric M18BP1 recruitment would also lead to less efficient CENP-A incorporation, we divided CENP-A staining patterns into low, medium and high and determined the percentage of cells showing these patterns in control and CENP-C knock-down cells. In particular knock-down shCENP-C #3 which had the strongest effect on centromeric M18BP1 recruitment lead to significantly reduced CENP-A levels ().
In summary our data demonstrate that the interaction between CENP-C and M18BP1 is an important recruitment mechanism for M18BP1 to centromeric chromatin, which appears necessary for the correct deposition of CENP-A.
Discussion
The deposition of CENP-A into centromeric chromatin is essential to ensure proper segregation of chromosomes. The Mis18 complex member M18BP1 was shown as an essential factor to prepare centromeric chromatin for CENP-A deposition and to ensure its maintenance.Citation17,Citation19,Citation24 Our data constitute the first analysis of endogenously expressed M18BP1 in mES cells. We report that M18BP1 associates with centromeric chromatin during distinct cell cycle stages. M18BP1 shows highest abundance at centromeres from anaphase through to late G1 phase. These data are in agreement with observations in human cells, where M18BP1 also associates with centromeres starting from late telophase through to G1 phaseCitation17 and suggest that M18BP1-mediated processes might be evolutionarily conserved in higher mammals.
We have furthered the understanding of how M18BP1 is recruited to centromeres through identification of a novel interaction between the C-terminus of CENP-C with a central region in M18BP1, which contains a SANT domain. The SANT domain is highly conserved and found in many chromatin-associated proteins, but very little is known about its potential functions. It has been implicated in the mediation of protein-protein interactions and binding to histone modifications.Citation25 We do not detect a direct interaction between the isolated M18BP1 SANT domain and CENP-C, however, it is possible that this domain is only functional in a larger protein context. More detailed experiments are necessary to further understand the functional roles of this domain in M18BP1. Our interaction data are consistent with a very recent study which appeared during the preparation of this manuscript.Citation26 Moree et al. found that X. laevis xM18BP1 isoforms interact with xCENP-C, and they could also show that human M18BP1 interacts with human CENP-C. In their study the interaction domain with xM18BP1 was mapped to the C-terminus of xCENP-C containing the CENP-C motif and the cupin domain. Mouse CENP-C (906aa) is much smaller than xCENP-C (1400aa), however, the major domains, such as CENP-A binding domain, CENP-C motif and cupin domain are conserved. We could show that in the mouse the interaction with M18BP1 is also mediated through a C-terminal fragment of CENP-C containing the CENP-C motif and the cupin domain, and thus the M18BP1 interaction site in CENP-C seems to be evolutionarily conserved.
Our data moreover demonstrate an important function for CENP-C in mediating the centromeric recruitment of M18BP1. When CENP-C protein levels are reduced to around 40–50% (shCENP-C #1 and #2) we found reduced numbers of “weak” centromeric M18BP1 cells. The numbers of “strong” centromeric M18BP1 cells, a staining pattern which we found characteristic for G1 phase cells, seemed to be unaltered. More severely reduced CENP-C levels (shCENP-C #3) resulted in lower numbers of cells with “weak” and “strong” centromeric M18BP1. Our cell cycle marker analysis revealed that the role of CENP-C in mediating centromeric M18BP1 recruitment is not restricted to selective cell cycle stages. However, we cannot exclude the possibility that CENP-C has additional functions that indirectly regulate the centromeric association of M18BP1 during distinct cell cycle phases. In this context it is interesting to note that when we transiently express CENP-C in K1B2 cells, we find a high percentage of cells in which CENP-C is abundantly associated with centromeres, but M18BP1 does not co-localize. It is therefore plausible to assume that further regulatory mechanisms exist, e.g., post-translational modifications, which influence the in vivo interaction between these two proteins. Both M18BP1 and CENP-C can be phosphorylated and sumoylated at multiple sites.Citation27-Citation30 It will be challenging to understand how these modifications are regulated and how they influence interactions between the different centromeric proteins in a cell cycle-dependent manner.
The centromeric recruitment of M18BP1 appears important for correct deposition of CENP-A. In particular strong depletion of CENP-C with knock-down oligo shCENP-C #3 leads to reduced levels of centromeric CENP-A. These data are consistent with Moree et al., which demonstrate in the Xenopus system that upon xCENP-C depletion, centromeric deposition of new CENP-A is impaired.Citation26 The failure to correctly establish CENP-A might be due to loss of centromeric M18BP1 at critical cell cycle stages. In human cells, CENP-A deposition is mediated by HJURP during G1 phase. Loss of M18BP1 leads to reduced HJURP association with centromeres and consequently to reduced deposition of newly synthesized CENP-A.Citation14,Citation15 In our mES cell system strong CENP-C knock-down results in cells which lose M18BP1 during G1 phase when CENP-A deposition normally occurs. We therefore postulate that those cells will also have problems in correctly establishing centromeric CENP-A patterns. We do not detect a large number of cells which have lost CENP-A upon CENP-C knock-down. We think that this could be explained by the high cell lethality of the CENP-C knock-down. Therefore, at the current stage of analysis, we cannot distinguish whether critically low CENP-A levels would induce apoptosis in ES cells, or whether CENP-C has additional functions that could be critical for survival of ES cells. Also, the functions of M18BP1 need to be investigated in more detail to understand how the centromeric recruitment of this molecule drives the subsequent deposition of CENP-A during G1 phase and whether M18BP1 features additional roles during other cell cycle stages when its centromeric recruitment is much lower but still detectable.
Materials and Methods
M18BP1 knock-in cell line
The M18BP1-EGFP targeting constructs were obtained using the recombineering cloning technique described previously.Citation31 To generate retrieval and mini-targeting vectors, PCR fragments were amplified from the BAC clone RP23–396P24 (Children's Hospital Oakland Research Institute). For the retrieval plasmid, PCR fragments were cloned into the pL253 plasmid using NotI, HindIII and SpeI. A genomic region of 7 kb, spanning the last exons of M18BP1, was retrieved from the BAC clone using recombineering in EL350 bacteria. The mini-targeting plasmid was constructed by generating PCR fragments flanking the M18BP1 stop codon. These PCR fragments were cloned together with the floxed Neomycin selection cassette from pL452 (EcoRI-BamHI fragment) into pBluescript IISK+ using NotI, EcoRI, BamHI and SalI. In a subsequent cloning step, the EGFP tag was inserted with EcoRI. For the final targeting vector, the 7 kb region was mini-targeted by recombineering with the NotI-SalI fragment containing the EGFP and floxed Neomycin selection cassette from the mini-targeting plasmid.
To generate M18BP1 knock-in cells, the NotI-linearized targeting vector was electroporated into feeder-independent wild type mES cells. Cells were selected in 180 µg/ml G418 (PAA) and 2 µM Ganciclovir (Invivogen). Single colonies were picked and screened by nested PCR to obtain the final mES cell clone (K1B2). Primers used for cloning and PCR screening are listed in Table S1.
Cell culture and transfections
BHK cells containing a lac operator repeat arrayCitation32 were cultured in DMEM medium with 10% FCS and seeded on coverslips in 6-well plates for microscopy. After attachment cells were co-transfected with expression vectors for the indicated fluorescent fusion proteins and a LacI-GBP fusionCitation33,Citation34 using polyethylenimine (Sigma). After about 16 h cells were fixed with 3.7% formaldehyde in PBS for 10 min, washed with PBST (PBS with 0.02% Tween), stained with DAPI and mounted in Vectashield medium (Vector Laboratories).
Mouse ES cells were cultivated on gelatinized plates in High Glucose DMEM with L-Glutamine and sodium pyruvate, complemented with 15% FCS, β-mercaptoethanol, non essential amino acids (PAA), penicillin/streptomycin (PAA) and LIF in a 37°C incubator at 5% CO2. For transfection with Lipofectamine 2000 (Invitrogen), mES cells were seeded on matrigel (BD Biosciences) coated coverslips.
HEK 293FT cells (Invitrogen) were cultivated on gelatinizes plates in High Glucose DMEM with L-Glutamine and sodium pyruvate (PAA) complemented with 10% FCS, β-mercaptoethanol, non essential amino acids (PAA) and penicillin/streptomycin (PAA) in a 37°C incubator at 5% CO2. The cells were transiently transfected one day after seeding using standard calcium phosphate transfection.
Microscopy
F3H samples were analyzed with a confocal fluorescence microscope (TCS SP5, Leica) equipped with a 63 × /1.4 numerical aperture Plan-Apochromat oil immersion objective as described.Citation34 DAPI, EGFP and mCherry/RFP were excited by 405 nm diode laser, 488 nm argon laser and 561 nm diode-pumped solid-state laser, respectively. Images were recorded with a frame size of 512 × 512 pixels.
K1B2 cells were imaged using a Leica TCS SP5 confocal laser scanning microscope with a HCX PL APO CS 63x/1.3 NA glycerol immersion objective. Sequential excitation at 405 nm, 488 nm, 543 nm and 633 nm was provided by diode, argon and helium-neon gas lasers, respectively. Emission detection ranges of the photomultipliers were adjusted to avoid crosstalk between the channels. Maximum intensity projections of the confocal sections were generated using ImageJ software.
Intensity ratio measurement
Images were acquired with an IN Cell Analyzer 2000 (GE Healthcare) using a 40 × air objective and analyzed with the IN Cell Analyzer 1000 Workstation 3.7 (GE Healthcare). Green and red fluorescence intensities at the lac spots were quantified. After background subtraction, intensity ratios of red (prey) to green (bait) were calculated and plotted using Excel software (Microsoft).
Lentiviral knockdown and infection of K1B2 cells
Lentiviral shRNA sequences (Table S2) were selected from the TRC libraryCitation35 or designed using the TRC shRNA designer (http://www.broadinstitute.org/rnai/public/).
For lentiviral knock-downs one non-targeting shRNA and three shRNAs targeting mouse CENP-C mRNA (NCBI RefSeq NM_007683.3) were cloned into the lentiviral knock-down vector pLKOmod1Citation36 with MluI/XmaI.
For restricting lentiviral transduction to mouse cells, we replaced the commonly used VSVg protein during viral packaging with the ecotropic envelope protein of Moloney Murine Leukemia Virus. The new packaging vector pLP-ecoenv was generated by removing the VSVg sequence of pLP-VSVg (Invitrogen) by EcoRI digest, followed by T4 polymerase filling of the remaining vector and ligation of a EcoRI/NotI cut and T4 polymerase filled PCR product of the M-MLV ecotropic envelope sequence
(Primers: eco env fw 5′-CGAATTCGCCGCCACCATGGCGCGTTCAACGCTCTCAAAA-3′; eco env rw 5′-TACGCGGCCGCTATGGCTCGTACTCTAT-3′).
Lentiviral production was performed by seeding 4 million HEK293FT cells (Invitrogen) one day before transfection in gelatinized 10 cm dishes. On the following day, cells were transiently cotransfected with 8 µg psPAX2, 8 µg pLP-ecoenv and 8 µg of the respective pLKOmod1 vector using standard calcium phosphate transfection. Conditioned medium containing recombinant lentiviruses was harvested 48 h post transfection, aliquoted, snap frozen and stored at -80°C until further use.
K1B2 cells were transduced by seeding 3 × 106 cells onto gelatinized 15 cm dishes containing mES cell medium supplemented with 4 µg/ml Polybrene (Sigma) and up to 20% conditioned virus medium. After 24 h the medium was replaced. 48 h post transduction, cells with stable integration of the pLKOmod1 vector were selected in mES cell medium containing 1.4–2 µg/ml puromycin (PAA) and then maintained in this selection medium until analysis.
Knock-down efficiency was determined at day five post infection by qRT-PCR and protein gel blotting. The following antibodies were used: CENP-C (Abcam ab50974), Suv4–20h2 (Hahn et al., in preparation).
RT-qPCR for monitoring M18BP1 and CENP-C expression levels
RNA of control and CENP-C knock-down cells was harvested at day 5 after transduction using RNeasy (Qiagen). 1.25 µg RNA was used for cDNA synthesis using Superscript III Kit (Invitrogen) and random hexameric primers (NEB). QPCR reactions were performed in technical triplicates using a Roche Light Cycler 480 with FAST SYBR® Master Mix (Applied Biosystems), and gene-specific primers (Table S3). Ct-values were normalized to the geometric mean of Actin and GAPDH for each individual cDNA and fold changes where calculated by the 2-ΔΔCt-method.Citation37
Immunofluorescence
Immunofluorescence analyses were performed as describedCitation38 using the following antibodies: CENP-A (C51A7, Cell Signaling Technology), H3S10P (06–570, Upstate) and Alexa 647 (A31573, Molecular Probes).
Plasmids
Encoding sequences of CENPs were amplified by PCR (Expand high fidelityPLUS PCR System, Roche, Penzberg, Germany). As forward primers we used 5′-GGGGACAAGTTTGTACAAAAAAGCAGGCTTCGAAAACCTGATTTTCAGGGCGCCACC-3′as flanking regions followed by 20–26 bases of coding regions starting with 5-ATGG-3′and as reverse primers we used 5′-GGGGACCACTTTGTACAAGAAAGCTGGGT-3′as flanking regions followed by 20–26 bases of coding sequences without stop codon. CENP encoding PCR fragments were transferred into vector pDONR221 by BP recombination reaction (Invitrogen, Carlsbad, CA, USA). After verification by DNA sequencing (MWG Biotech, Ebersberg, München, Germany), genes were transferred by LR recombination reactions to various modified pEGFP-C and pmCh-C (BD Biosciences, Clontech, Palo Alto, CA, USA) based destination vectors. The resulting expression vectors encode CENPs fused to the C-termini of EGFP and mCherry with SGTSLYKKAGFENLYFQGAT as linker sequence and TQLSCTKW added to the C-terminal ends of the FP-CENPs fusions. Complete sequences are provided upon request. Correct full length expression of fusion constructs was confirmed by protein gel blots.
Full-length open reading frames of mouse M18BP1, the M18BP1 truncations and the mouse CENP-C truncations were PCR amplified from mouse cDNA derived from mES cells and cloned into the pDONR/Zeo GATEWAY entry vector (Invitrogen) using Gateway BP Clonase II enzyme mix (Invitrogen). PCR primers are listed in Table S4. Entry clones were recombined into target vectors pEGFP-N1-GW, pCMVmyc-GW and pGEX6P1-GWCitation39 using LR Clonase II enzyme mix (Invitrogen).
In vitro binding assays
Recombinant M18BP1 protein truncations (C1 aa1–440, C2 aa441–998, C3 aa325–800, C4 aa441–800, C5 aa735–800) were expressed as GST tagged versions in E. coli and purified on Glutathione-S-Sepharose (GE Healthcare). In vitro translation of CENP-C protein truncations (aa1–367, aa368–656, aa657–906) was performed using TnT® Quick Coupled Transcription/Translation System (Promega). 10µl of the in vitro translated myc-tagged CENP-C and 5µg M18BP1 GST-fusion protein coupled to Glutathione-S-Sepharose were incubated in IP buffer (50 mM Tris pH7.5, 150 mM NaCl, 1 mM EDTA, 0.1% NP40, 20% glycerol and proteinase inhibitor cocktail (Roche)) overnight at 4°C on a rotating wheel. The beads were washed four times with IP buffer containing 1 M NaCl and resuspended in 50 μl SDS loading buffer (Roth). Bound proteins were separated on SDS polyacrylamidgels and detected by immunoblotting using α-myc antibody (9E10).
Co-immunoprecipitation in HEK293FT cells
HEK293FT cells (Invitrogen) were co-transfected with plasmids expressing EGFP-CENP-C and myc-M18BP1. Isolated nuclei were resuspended in high salt IP buffer (50 mM Tris pH 7.5, 500 mM NaCl, 1 mM EDTA, 0.1% NP40, 20% glycerol) with 4 strokes through a 19.5G syringe needle. After incubation on ice for 30 min the solution was sonicated 3x10” at an amplitude of 30 in a Branson sonifier. The nuclear extract was diluted to a final concentration of 150 mM NaCl with no salt IP buffer and precipitates were removed by centrifugation. The extract was incubated overnight at 4°C on a rotating wheel with GFP-Trap beads (ChromoTek) and agarose beads. The beads were washed five times with IP buffer containing 300 mM NaCl and afterwards resuspended in SDS loading buffer (Roth). Proteins were separated on SDS-polyacrylamidgels and analyzed by protein gel blotting using α-myc (9E10) and α-GFP (Roche # 11814460001) antibodies.
Additional material
Download Zip (8.2 MB)Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We would like to thank S. F. Lichtenthaler and P.-H. Kuhn for generously providing the pLKOmod1 vector. We thank N.G. Copeland for providing us with reagents for the bacterial recombineering system. We further thank A. Schmid and F. Büddefeld for help with cloning and C. Foster for critical reading of the manuscript.
G.S. and H.L. were supported by grants from DFG (TR-SFB) and BMBF (Episys). G.S. received additional support from Weigand’sche Stiftung. H.L. was further supported by the BioImaging Network and the Nanosystems Initiative Munich (NIM). SD wants to thank the DFG (SPP1128, SPP1395) and the TAB (2007 FE 9011) for support.
References
- Foltz DR, Jansen LE, Black BE, Bailey AO, Yates JR 3rd, Cleveland DW. The human CENP-A centromeric nucleosome-associated complex. Nat Cell Biol 2006; 8:458 - 69; http://dx.doi.org/10.1038/ncb1397; PMID: 16622419
- Obuse C, Yang H, Nozaki N, Goto S, Okazaki T, Yoda K. Proteomics analysis of the centromere complex from HeLa interphase cells: UV-damaged DNA binding protein 1 (DDB-1) is a component of the CEN-complex, while BMI-1 is transiently co-localized with the centromeric region in interphase. Genes Cells 2004; 9:105 - 20; http://dx.doi.org/10.1111/j.1365-2443.2004.00705.x; PMID: 15009096
- Okada M, Okawa K, Isobe T, Fukagawa T. CENP-H-containing complex facilitates centromere deposition of CENP-A in cooperation with FACT and CHD1. Mol Biol Cell 2009; 20:3986 - 95; http://dx.doi.org/10.1091/mbc.E09-01-0065; PMID: 19625449
- Guse A, Carroll CW, Moree B, Fuller CJ, Straight AF. In vitro centromere and kinetochore assembly on defined chromatin templates. Nature 2011; 477:354 - 8; http://dx.doi.org/10.1038/nature10379; PMID: 21874020
- Carroll CW, Milks KJ, Straight AF. Dual recognition of CENP-A nucleosomes is required for centromere assembly. J Cell Biol 2010; 189:1143 - 55; http://dx.doi.org/10.1083/jcb.201001013; PMID: 20566683
- Cohen RL, Espelin CW, De Wulf P, Sorger PK, Harrison SC, Simons KT. Structural and functional dissection of Mif2p, a conserved DNA-binding kinetochore protein. Mol Biol Cell 2008; 19:4480 - 91; http://dx.doi.org/10.1091/mbc.E08-03-0297; PMID: 18701705
- Sugimoto K, Kuriyama K, Shibata A, Himeno M. Characterization of internal DNA-binding and C-terminal dimerization domains of human centromere/kinetochore autoantigen CENP-C in vitro: role of DNA-binding and self-associating activities in kinetochore organization. Chromosome Res 1997; 5:132 - 41; http://dx.doi.org/10.1023/A:1018422325569; PMID: 9146917
- Screpanti E, De Antoni A, Alushin GM, Petrovic A, Melis T, Nogales E, et al. Direct binding of Cenp-C to the Mis12 complex joins the inner and outer kinetochore. Curr Biol 2011; 21:391 - 8; http://dx.doi.org/10.1016/j.cub.2010.12.039; PMID: 21353556
- Carroll CW, Silva MC, Godek KM, Jansen LE, Straight AF. Centromere assembly requires the direct recognition of CENP-A nucleosomes by CENP-N. Nat Cell Biol 2009; 11:896 - 902; http://dx.doi.org/10.1038/ncb1899; PMID: 19543270
- Bernad R, Sanchez P, Rivera T, Rodriguez-Corsino M, Boyarchuk E, Vassias I, et al. Xenopus HJURP and condensin II are required for CENP-A assembly. J Cell Biol 2011; 192:569 - 82; http://dx.doi.org/10.1083/jcb.201005136; PMID: 21321101
- Hemmerich P, Weidtkamp-Peters S, Hoischen C, Schmiedeberg L, Erliandri I, Diekmann S. Dynamics of inner kinetochore assembly and maintenance in living cells. J Cell Biol 2008; 180:1101 - 14; http://dx.doi.org/10.1083/jcb.200710052; PMID: 18347072
- Jansen LE, Black BE, Foltz DR, Cleveland DW. Propagation of centromeric chromatin requires exit from mitosis. J Cell Biol 2007; 176:795 - 805; http://dx.doi.org/10.1083/jcb.200701066; PMID: 17339380
- Schuh M, Lehner CF, Heidmann S. Incorporation of Drosophila CID/CENP-A and CENP-C into centromeres during early embryonic anaphase. Curr Biol 2007; 17:237 - 43; http://dx.doi.org/10.1016/j.cub.2006.11.051; PMID: 17222555
- Dunleavy EM, Roche D, Tagami H, Lacoste N, Ray-Gallet D, Nakamura Y, et al. HJURP is a cell-cycle-dependent maintenance and deposition factor of CENP-A at centromeres. Cell 2009; 137:485 - 97; http://dx.doi.org/10.1016/j.cell.2009.02.040; PMID: 19410545
- Foltz DR, Jansen LE, Bailey AO, Yates JR 3rd, Bassett EA, Wood S, et al. Centromere-specific assembly of CENP-a nucleosomes is mediated by HJURP. Cell 2009; 137:472 - 84; http://dx.doi.org/10.1016/j.cell.2009.02.039; PMID: 19410544
- Bergmann JH, Rodriguez MG, Martins NM, Kimura H, Kelly DA, Masumoto H, et al. Epigenetic engineering shows H3K4me2 is required for HJURP targeting and CENP-A assembly on a synthetic human kinetochore. EMBO J 2011; 30:328 - 40; http://dx.doi.org/10.1038/emboj.2010.329; PMID: 21157429
- Fujita Y, Hayashi T, Kiyomitsu T, Toyoda Y, Kokubu A, Obuse C, et al. Priming of centromere for CENP-A recruitment by human hMis18alpha, hMis18beta, and M18BP1. Dev Cell 2007; 12:17 - 30; http://dx.doi.org/10.1016/j.devcel.2006.11.002; PMID: 17199038
- Hayashi T, Fujita Y, Iwasaki O, Adachi Y, Takahashi K, Yanagida M. Mis16 and Mis18 are required for CENP-A loading and histone deacetylation at centromeres. Cell 2004; 118:715 - 29; http://dx.doi.org/10.1016/j.cell.2004.09.002; PMID: 15369671
- Maddox PS, Hyndman F, Monen J, Oegema K, Desai A. Functional genomics identifies a Myb domain-containing protein family required for assembly of CENP-A chromatin. J Cell Biol 2007; 176:757 - 63; http://dx.doi.org/10.1083/jcb.200701065; PMID: 17339379
- Pidoux AL, Choi ES, Abbott JK, Liu X, Kagansky A, Castillo AG, et al. Fission yeast Scm3: A CENP-A receptor required for integrity of subkinetochore chromatin. Mol Cell 2009; 33:299 - 311; http://dx.doi.org/10.1016/j.molcel.2009.01.019; PMID: 19217404
- Williams JS, Hayashi T, Yanagida M, Russell P. Fission yeast Scm3 mediates stable assembly of Cnp1/CENP-A into centromeric chromatin. Mol Cell 2009; 33:287 - 98; http://dx.doi.org/10.1016/j.molcel.2009.01.017; PMID: 19217403
- Leonhardt H, Rahn HP, Weinzierl P, Sporbert A, Cremer T, Zink D, et al. Dynamics of DNA replication factories in living cells. J Cell Biol 2000; 149:271 - 80; http://dx.doi.org/10.1083/jcb.149.2.271; PMID: 10769021
- Hsu JY, Sun ZW, Li X, Reuben M, Tatchell K, Bishop DK, et al. Mitotic phosphorylation of histone H3 is governed by Ipl1/aurora kinase and Glc7/PP1 phosphatase in budding yeast and nematodes. Cell 2000; 102:279 - 91; http://dx.doi.org/10.1016/S0092-8674(00)00034-9; PMID: 10975519
- Lagana A, Dorn JF, De Rop V, Ladouceur AM, Maddox AS, Maddox PS. A small GTPase molecular switch regulates epigenetic centromere maintenance by stabilizing newly incorporated CENP-A. Nat Cell Biol 2010; 12:1186 - 93; http://dx.doi.org/10.1038/ncb2129; PMID: 21102442
- Boyer LA, Latek RR, Peterson CL. The SANT domain: a unique histone-tail-binding module?. Nat Rev Mol Cell Biol 2004; 5:158 - 63; http://dx.doi.org/10.1038/nrm1314; PMID: 15040448
- Moree B, Meyer CB, Fuller CJ, Straight AF. CENP-C recruits M18BP1 to centromeres to promote CENP-A chromatin assembly. J Cell Biol 2011; 194:855 - 71; http://dx.doi.org/10.1083/jcb.201106079; PMID: 21911481
- Chung TL, Hsiao HH, Yeh YY, Shia HL, Chen YL, Liang PH, et al. In vitro modification of human centromere protein CENP-C fragments by small ubiquitin-like modifier (SUMO) protein: definitive identification of the modification sites by tandem mass spectrometry analysis of the isopeptides. J Biol Chem 2004; 279:39653 - 62; http://dx.doi.org/10.1074/jbc.M405637200; PMID: 15272016
- Mayya V, Lundgren DH, Hwang SI, Rezaul K, Wu L, Eng JK, et al. Quantitative phosphoproteomic analysis of T cell receptor signaling reveals system-wide modulation of protein-protein interactions. Sci Signal 2009; 2:ra46; http://dx.doi.org/10.1126/scisignal.2000007; PMID: 19690332
- Nousiainen M, Sillje HH, Sauer G, Nigg EA, Korner R. Phosphoproteome analysis of the human mitotic spindle. Proc Natl Acad Sci USA 2006; 103:5391 - 6; http://dx.doi.org/10.1073/pnas.0507066103; PMID: 16565220
- Olsen JV, Blagoev B, Gnad F, Macek B, Kumar C, Mortensen P, et al. Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 2006; 127:635 - 48; http://dx.doi.org/10.1016/j.cell.2006.09.026; PMID: 17081983
- Liu P, Jenkins NA, Copeland NG. A highly efficient recombineering-based method for generating conditional knockout mutations. Genome Res 2003; 13:476 - 84; http://dx.doi.org/10.1101/gr.749203; PMID: 12618378
- Tsukamoto T, Hashiguchi N, Janicki SM, Tumbar T, Belmont AS, Spector DL. Visualization of gene activity in living cells. Nat Cell Biol 2000; 2:871 - 8; http://dx.doi.org/10.1038/35046510; PMID: 11146650
- Rothbauer U, Zolghadr K, Tillib S, Nowak D, Schermelleh L, Gahl A, et al. Targeting and tracing antigens in live cells with fluorescent nanobodies. Nat Methods 2006; 3:887 - 9; http://dx.doi.org/10.1038/nmeth953; PMID: 17060912
- Zolghadr K, Mortusewicz O, Rothbauer U, Kleinhans R, Goehler H, Wanker EE, et al. A fluorescent two-hybrid assay for direct visualization of protein interactions in living cells. Mol Cell Proteomics 2008; 7:2279 - 87; http://dx.doi.org/10.1074/mcp.M700548-MCP200; PMID: 18622019
- Moffat J, Grueneberg DA, Yang X, Kim SY, Kloepfer AM, Hinkle G, et al. A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell 2006; 124:1283 - 98; http://dx.doi.org/10.1016/j.cell.2006.01.040; PMID: 16564017
- Kuhn PH, Wang H, Dislich B, Colombo A, Zeitschel U, Ellwart JW, et al. ADAM10 is the physiologically relevant, constitutive alpha-secretase of the amyloid precursor protein in primary neurons. EMBO J 2010; 29:3020 - 32; http://dx.doi.org/10.1038/emboj.2010.167; PMID: 20676056
- Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, et al. 2002; Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol 2002; 3: H0034; http://dx.doi.org/10.1186/gb-2002-3-7-research0034; PMID: 12184808
- Lehnertz B, Ueda Y, Derijck AA, Braunschweig U, Perez-Burgos L, Kubicek S, et al. Suv39h-mediated histone H3 lysine 9 methylation directs DNA methylation to major satellite repeats at pericentric heterochromatin. Curr Biol 2003; 13:1192 - 200; http://dx.doi.org/10.1016/S0960-9822(03)00432-9; PMID: 12867029
- Schotta G, Sengupta R, Kubicek S, Malin S, Kauer M, Callen E, et al. A chromatin-wide transition to H4K20 monomethylation impairs genome integrity and programmed DNA rearrangements in the mouse. Genes Dev 2008; 22:2048 - 61; http://dx.doi.org/10.1101/gad.476008; PMID: 18676810