Abstract
Polycomb group (PcG) proteins of the Polycomb repressive complex 1 (PRC1) are found to be diffusely distributed in nuclei of cells from various species. However they can also be localized in intensely fluorescent foci, whether imaged using GFP fusions to proteins of PRC1 complex, or by conventional immunofluorescence microscopy. Such foci are termed PcG bodies, and are believed to be situated in the nuclear intechromatin compartment. However, an ultrastructural description of the PcG body has not been reported to date. To establish the ultrastructure of PcG bodies in human U-2 OS cells stably expressing recombinant polycomb BMI1-GFP protein, we used correlative light electron microscopy (CLEM) implemented with high-pressure freezing, cryosubstitution and on-section labeling of BMI1 protein with immunogold. This approach allowed us to clearly identify fluorescent PcG bodies, not as distinct nuclear bodies, but as nuclear domains enriched in separated heterochromatin fascicles. Importantly, high-pressure freezing and cryosubstitution allowed for a high and clear-cut imunogold BMI1 labeling of heterochromatin structures throughout the nucleus. The density of immunogold labeled BMI1 in the heterochromatin fascicles corresponding to fluorescent "PcG bodies" did not differ from the density of labeling of heterochromatin fascicles outside of the "PcG bodies." Accordingly, an appearance of the fluorescent "PcG bodies" seems to reflect a local accumulation of the labeled heterochromatin structures in the investigated cells. The results of this study should allow to expand the knowledge about the biological relevance of the "PcG bodies" in human cells.
Introduction
Polycomb group (PcG) proteins are a set of conserved, essential regulatory factors, that through the assembly on key regulatory DNA elements, repress the transcription of their target genes.Citation1–Citation3 They are the best known for regulation of homeotic gene expression during embryogenesis.Citation4 However, PcG proteins also repress the genes whose products are implicated in cellular processes like cell cycle control, senescence, X-chromosome inactivation, cell fate decision and stem cell differentiation.Citation5–Citation8 PcG proteins execute their repressive function in two distinct multiprotein complexes: the Polycomb repressive complex 1 (PRC1) and the Polycomb repressive complex 2 (PRC2; in mammals also known as the Eed-Ezh2 complex).Citation9,Citation10 PRC2 is thought to be involved in the initiation of gene silencing by methylation of histone H3 at lysine 27 (H3K27),Citation11–Citation13 whereas PRC1 is implicated in stable maintenance of the repressed state of the genes.Citation14,Citation15 PRC1, consisting in human cells of HPC1–3, HPH1–3, BMI1, SCML and RING1–2 proteins,Citation16 are usually distributed diffusely in the nucleus. However, they are also found in intensely fluorescent foci, whether imaged using GFP fusion proteins or conventional immunofluorescence, with an accumulation of PcG proteins termed PcG bodies.Citation17–Citation21 Besides human, respectively mammalian cells, PcG bodies have been described in cells of a number of other species including Drosophila and Caenorhabditis elegans.Citation20,Citation22
The expression levels of PcG proteins vastly differ in human cell lines (reviewed in ref. Citation23). Reflecting the expression level, PcG bodies are conspicuous in human osteosarcoma U-2 OS cells where their relative sizes differ from 0.2 to 1.5 µm and their number varies between six and fourteen per nucleus.Citation18,Citation21 According to immuno-FISH data and karyotype analysis, these variations in expression also apparently result from karyotypic differences between cell lines.Citation24 In addition, nuclear positioning of PcG bodies is not completely random as the bodies appear to be preferentially associated with some loci on particular chromosomes.Citation21,Citation24 There are no observable co-localizations between PcG bodies and Cajal bodies, gemini of Cajal bodies and possibly also PML bodies.Citation21 PcG bodies thus appear to be unrelated to any other known nuclear body and are thought to represent a genuine structure within the nucleus.
The term nuclear body is used for a nuclear domain that is morphologically distinct from its surroundings when observed by transmission electron microscopy.Citation25,Citation26 Generally speaking, the cell nucleus has a compartmentalized structure made up of the chromatin domains and the interchromatin compartment (IC). Whereas the chromatin basically consists of euchromatin and heterochromatin, the IC is enriched in a number of various nuclear bodies.Citation25–Citation31 Nuclear bodies such as nucleoli, Cajal bodies, PML bodies and nuclear speckles are well characterized, even at the ultrastructural level.Citation26 In this respect, so far less well studied are the structures containing transcriptional regulators or RNAbinding proteins.Citation32 These structures are also termed orphan nuclear bodies. This group of bodies includes the clastosome, the cleavage body, the OPT domain, the SUMO body, the Sam68 body, and also the PcG body.Citation32 Even though the PcG bodies were described more than 10 years ago, no electron microscopy (EM) description of these bodies in human (mammalian) cells has been provided to date.
To identify, and to establish the fine structure of the PcG bodies in U-2 OS cell line expressing recombinant BMI1-GFP protein (U-2 OS BMI1-GFP cells), we used correlative light-electron microscopy (CLEM) to be able to assign fine structure corresponding to the distinct fluorescent foci of the PcG bodies. Surprisingly, we were able to identify the “PcG body” with the locally higher spatial accumulation of heterochromatin structures not bearing the characteristics of a nuclear body.
Results
All presented experiments were performed with the U-2 OS cell line since these cells contain a number of distinct PcG bodies, already studied in several reports in references Citation21, Citation24 and Citation33. U-2 OS cells stably expressing BMI1-GFP fusion proteinCitation33 furthermore allowed us to link the live cell imaging of the localization of BMI1-GFP with imaging, at the ultrastructural level, of immunolabeled ultrathin sections. It should be, however, mentioned that prior applying the CLEM technique, we tried to identify PcG bodies in non-transfected U-2 OS cells via post-embedding immunogold labeling procedures. Even though we were able to achieve a clean immunogold signal in heterochromatin (Sup. Fig. 1), we failed to identify distinct PcG bodies.
Correlation of live cell imaging and immunofluorescence.
With the transfected U-2 OS cells, we correlated BMI1-GFP fluorescent signals () with signals from the same cells which were immediately fixed and immunolabeled (). Within the resolution limit of light microscopy, we did not observe any major difference between fluorescence patterns generated by the two approaches (). At the same time, we compared patterns of BMI1 immunolabeling in both normal (non-transfected) U-2 OS cells and stably transfected U-2 OS BMI1-GFP cells. The two patterns were comparable (results not shown) and in agreement with their previous description (e.g., ref. Citation33). Except for an absence of an intranucleolar signal in most cells, the nuclear fluorescence consisted of the overall weaker nucleoplasmic signal together with several bright PcG foci/bodies of various size. The PcG bodies were often situated in a close proximity to nucleoli. Individual cells exhibited differences both concerning the number and size of PcG foci. These results from light microscopy were in agreement with the results of previous studies in references Citation21 and Citation33.
Correlation of “PcG bodies” fluorescence with DA/DAPI staining and DNA immunocytochemistry.
We wanted to know whether there is an increased density of DNA in the nuclear regions/domains that contain PcG bodies. Concerning DNA detection, there is a known cytogenetic problem due to the probe (e.g., DAPI, antibodies) penetration/epitope accessibility within compacted chromatin structures.
To overcome this problem, we first imaged GFP in fixed and permeabilized cells and then used the established counterstaining with DAPI in combination with distamycin A (DA/DAPI staining).Citation24,Citation34 A co-localization of the PcG bodies with an increased DAPI signal in the regions of the PcG bodies was observed (). Our results on the co-localization of PcG bodies with the DA/DAPI stained areas are in agreement with those of Voncken et al.Citation24
Alternatively, we first visualized PcG bodies in live U2-OS BMI-GFP cells, and after fixation, we permeabilized the cells with increasing concentrations of TritonX-100 up to 2% followed by immunocytochemistry using antibodies to GFP and DNA. The 2% detergent concentration allowed for a convenient co-localization of the PcG bodies with an increased DNA density in the regions of the PcG bodies ().
Taken together, we were able to show that there is an increased density of DNA in the nuclear domains that contain PcG bodies.
CLEM of high pressure frozen, cryosubstituted and on-section labeled cells.
To achieve structural preservation of the PcG bodies and high efficiency of the post-embedding immunocytochemistry we performed, together with CLEM, high-pressure freezing and cryosubstitution of transfected U-2 OS cells ( and ).
Using this approach, the ultrastructure of the cells was well preserved (, and Sup. Fig. 2). Thin sectioned cells exhibited well delineated electron-dense heterochromatin structures in the form of heterochromatin fascicles termed also largescale chromatin fibres.Citation35 In addition, they frequently exhibited nuclear envelope invaginations seen in their longitudinal or transverse sections ( and , black arrows and black arrowheads), this phenomena is typical for cultured, transformed cells.Citation36 The immunogold BMI1 label was specifically enriched within heterochromatin fascicles throughout the nucleus ( and ; see also Sup. Fig. 2) and appeared sometimes to line the heterochromatin borders (insert in ). In contrast, the label in the IC and nucleoli (see insert in ) was much lower, and was basically missing in the cytoplasm. With respect to the IC label, the gold particles were preferentially found in the vicinity of heterochromatin structures. The nucleolar label was usually found in intranucleolar heterochromatin clumps (insert in ); the nucleoli are known to exhibit such heterochromatin structures (reviewed in refs. Citation37 and Citation38).
Such a clear-cut immunogold signal thus does not necessitate any quantitative evaluation, and is of primary importance within the frame of this study. But it has to be mentioned that we were not able to achieve, in contrast to immunofluorescence light microscopy of fixed, non-embedded cells (), a convenient post-embedding on-section labeling with several antibodies to GFP. This illustrates a well known fact that the antibodies convenient for the fluorescence microscopy of fixed cells are frequently not useful for the detection of their respective targets in on-section labeling of resin embedded cells.
To identify the PcG bodies per se we correlated the snapshots (Z series) from live cell imaging with electron microscopy images. Because of the very small thickness of thin sections, we had to analyze serial sections in order to identify the EM area that fit well with the PcG body fluorescence signal (; for on-section immunofluorescence of the BMI1 signal see inserts in 3B and C). We never observed a nuclear region/domain with a locally increased immunogold label present outside of the heterochromatin structures. However, importantly, we found that there was a higher local accumulation of heterochromatin fascicles in the nuclear region/domain corresponding to the PcG body fluorescence signal (white and yellow arrows in and ; for a more detailed description of the analysis concerning the “PcG body” identification see Sup. Figs. 3–5) as compared to other nuclear domains seen in the thin sections. Accordingly, we cannot use the term “nuclear body” for the description of such a nuclear domain. Also, we cannot exactly draw a line delimiting a “PcG body” in thin sectioned nuclei as it is impossible to establish which part of a given heterochromatin fascicle still does, or does not, belong to the “PcG body” that is defined through its fluorescence signal. “PcG bodies” were frequenly associated with the nuclear envelope, together with its invaginations (black arrows and arrowheads in and ), and with nucleoli. In this respect, the vicinity of the nuclear envelope and the nucleoli are well known to be enriched in heterochromatin that is termed perinuclear and perinucleolar heterochromatin, respectively.
Importantly, the morphometric analysis did show that the number of gold particles per unit area of heterochromatin outside and inside the “PcG body” was about the same. The relative density of the immunolabeling of heterochromatin outside the “PcG body” compared to that inside of the “PcG body” was 1.05 ± 0.24.
Taken together, using this CLEM approach, we identified, and established the structure of “PcG bodies” at the ultrastructural level. Immunogold labeling provided high and clean labeling of nuclear heterochromatin structures, and the densities of gold particles confined to heterochromatin structures outside and inside the “PcG body” were comparable. However, the fine structure of “PcG bodies” did not correspond to the nuclear bodies as such,Citation25,Citation26 but to locally accumulated and BMI1 immunogold labeled heterochromatin structures. Generally speaking, nuclear regions/domains with higher 3-D () or 2-D (inserts in and C) BMI1 or GFP fluorescence intensity (including that of the “PcG bodies”) seen in the fluorescence microscope find thus their counterpart in the nuclear regions/domains with higher local accumulations of the thin sectioned (and BMI1 2-D immunogold labeled) heterochromatin fascicles seen in the EM (see also Sup. Figs. 3–5).
We ultrastructurally identified “PcG bodies” by additional CLEM approaches in which the chemical fixation of cells was performed. Since we consider only the above mentioned results as representing the convenient description of the “PcG body” (reviewed in refs. Citation39–Citation46), we document the two additional approaches in the Supplemental Material (Sup. Figs. 6 and 7). In summary, chromatin structures exhibit extraordinary sensitivity to enviromental factors, including the processing of cells for microscopy. While the resolution power of the fluorescence approaches we used does not allow for the detection of significant structural chromatin changes accompanying the processing of cells, such changes are put in evidence by electron microscopy.
Discussion
PcG bodies have been so far identified only by fluorescence microscopy as distinct accumulations of PcG proteins of the PRC1 (e.g., refs. Citation17 and Citation18). With a help of CLEM, we were here able to ultrastructurally identify the “PcG bodies” in thin sectioned U-2 OS cells expressing BMI1-GFP proteins. Further, we were also able to describe the well preserved structure of the “PcG body” together with its reference nuclear space. Importantly, the transfected cells and BMI1 commercial antibody used in this study were explored and characterized in several previous studies (e.g., in refs. Citation24, Citation33 and Citation47).
To establish the fine structure of the “PcG body” we used the HPF followed by the cryosubstitution, nowadays the method of choice for the best preservation of cellular ultrastructure, and the basis for an achieving of convenient on-section labeling (e.g., refs. Citation48–Citation50). This approach revealed that PcG fluorescence signals do not correspond to a nuclear body, but to a nuclear region/domain containing a local accumulation of the heterochromatin fascicles. This finding is supported by the immunofluorescence results documenting an increased density of DNA in the nuclear regions/domains that contain fluorescence “PcG bodies”.
The approach we used also allowed for a high and clear-cut immunogold BMI1 label of heterochromatin fascicles throughout the nucleus. Importantly, the densities of gold particles per unit area of the thin sectioned and 2-D labeled heterochromatin structures were comparable, whatever was the chosen heterochromatin area within the nucleus, either inside or outside of the “PcG body”. Accordingly, the appearance of the fluorescent “PcG bodies” should reflect a local accumulation of the BMI1 labeled heterochromatin in the investigated cells.
This being said we have to discuss two relevant matters. First, we provided in and , Supplemental Figures 1 and 2 only static snapshots of the cell at the moment of rapid freezing. But chromatin is highly dynamic, and specifically, BMI1 protein exhibits high mobility.Citation33 Most chromatin proteins have a high turnover on chromatin with a residence time on the order of seconds.Citation51,Citation52 This transient binding is a common property of chromatin-associated proteins, but the major fraction of each protein is bound to chromatin at steady state.Citation51,Citation52 The immunocytochemical result presented here, with most gold particles localized in heterochromatin structures (, and Sup. Fig. 1), describes exactly such a steady state. In other words, low BMI1 immunogold label found outside of heterochromatin structures seems to be in agreement with such a steady state concept.
Second, the ultrastructural immunogold results presented here in and (see also Sup. Fig. 1 in the Sup. Material) differ from those of the previous study.Citation53 In cultured transformed cells of human origin and in somatic rat liver cells, the authors mapped BMI1 protein, as well other PRC1 proteins, mainly outside of heterochromatin. They established that with respect to the BMI1 labeling density of heterochromatin, this density was 100 times and 10 times higher over the perichromatin region (region in the vicinity of heterochromatin) and over the IC, respectively.Citation53 The likely explanation could be that different cells and different antibodies were used in the two studies. In a reconciliation tone, even though the present immunogold label was mostly found in heterochromatin, it was sometimes lining the periphery of heterochromatin structures, and the low label in the IC was preferentially found in the vicinity of heterochromatin. But at the same time, the strength of the present study is based on the high and clear-cut immunogold labeling of heterochromatin structures throughout the whole nucleus.
Through the previous comment we touched the third matter. We were dealing here with heterochromatin structures since such electron-dense structures are morphologically well definable ( and ). How about euchromatin structures? The perichromatin region is considered to encompass transcriptionally active genes,Citation53 i.e., euchromatin structures. The aim of the present study was to establish the fine structure of the “PcG body”, and based just on the BMI1 immunogold labeling results, it is anyway impossible to speculate in whatever terms on euchromatin structures.
The current models of the “PcG body” arise mainly from the biochemical and fluorescence microscopy studies (reviewed in refs. Citation54 and Citation55). PcG bodies have been compared to transcription factories in these models.Citation54 This presumption raises from the fact that Polycomb mediated gene silencing involves chromosomekissing events, and that such events occur at PcG bodies. Similar kissing phenomena are thought to be induced also by the active or activation-prone genes and occur at the structures like transcription factories or splicing speckles.Citation54 Coming up from the expectation that the “PcG bodies” are typical nuclear bodies and the fact that the light microscopy does not allow to study the reference space in details, the “PcG bodies” have been, in such models, situated in the IC (reviewed in refs. Citation31 and Citation54). In the present study, the essence of “PcG bodies” was, at the EM level, associated with the locally accumulated and immunogold labeled heterochromatin structures, and not with a nuclear body situated in the IC. It should be also mentioned here that the progress in the field of PcG proteins has been initiated and is, with a great success, being carried out in the Drosophila model (reviewed in refs. Citation56–Citation58), and results obtained with this model organism can be to greater or lesser extend transposed to human cells.
The present results are related just to the transfected U-2 OS cell line. If such results are confirmed in other human cell lines, they will, in a straightforward way, help to expand the knowledge with respect to the biological relevance of the “PcG body” observed in human cells.
Materials and Methods
Cell culture.
U-2 OS cell line of human origin (human osteosarcoma cell line) and U-2 OS cell line stably expressing BMI1-GFP were cultured in Dulbeco's modified Eagle's medium (GIBCO) supplemented with 10% fetal bovine serum (GIBCO) and 1% penicillin/streptomycin sulfate (PAA Laboratories) under normal conditions.
Correlation of live cell imaging and immunofluorescence.
U-2 OS BMI1-GFP cells (kindly provided by Dr. Maarten van Lohuizen, Amsterdam) grown on the gridded Petri dish were imaged for PcG bodies using a confocal microscope Leica TCS SP5 with 40x/1.Citation25 NA oil immersion objective. After acquiring a Z-series of cells with a distinct, point-like GFP signal, the cells were fixed with 4% formaldehyde in 0.2 mM PIPES (pH 7.2) for 10 minutes, permeabilized with 0.3% TritonX-100 for 5 minutes and washed several times in PBS. Nonspecific sites were blocked with 5% normal goat serum (NGS; Sigma) in PBS. The cells were incubated with mouse anti-BMI1 (1:300, Clone F6, Upstate) and rabbit anti-GFP (1:300, Abcam) antibodies in 1% (w/v) BSA in PBS containing 0.5% Tween20 for 1 hour, then washed and incubated with secondary goat antimouse and goat anti-rabbit antibodies conjugated with TRITC or FITC (Jackson ImmunoResearch Laboratories) in PBS for 45 min. DNA was counterstained with DAPI (4′,6-diamidino-2-phenylindole, Sigma). Gridded Petri dishes were then mounted using a Polyvinyl alcohol mounting medium with DABCO (BioChemika, Fluka). Immunofluorescence images were taken with the Leica TCS SP5 confocal microscope. Non-transfected U-2 OS cells were processed for immunofluorescence in the same way as transfected cells.
Correlation of “PcG bodies” fluorescence with DA/DAPI staining and DNA immunocytochemistry.
For staining of the U-2 OS BMI1-GFP cells with DAPI (Sigma) in combination with distamycin A-HCl (Chemos), the cells were fixed with 4% formaldehyde in 0.2 mM PIPES (pH 7.2) for 10 min, permeabilized with 0.3% TritonX-100 for 5 min and washed several times in PBS. Then the cells were counterstained with DA/DAPI according to protocol of Schweizer and Ambros.Citation34 Briefly, the cells were incubated in 0.2 mg/ml distamycin A-HCl for 15 min, rinsed in McIlvaine's buffer (pH 7.0), counterstained with 0.2 µg/ml DAPI for 15 min and rinsed again.
Concerning the DNA detection, live cell images of U-2 OS BMI1-GFP cells were taken and correlated with the immunocytochemical images of GFP (anti-GFP antibody, Abcam) and DNA (anti-DNA antibody, Progen) taken after 2% formaldehyde in PBS (pH 7.2) fixation for 10 min and permeabilization with an increasing concentrations of TritonX-100 (from 0.3% up to 2% TritonX-100) for 5 min and several washes in PBS. In the immunocytochemical approach, the cells were incubated with diluted mouse monoclonal anti-DNA (1:30) and rabbit polyclonal anti-GFP (1:300) in 1% (w/v) BSA in PBS containing 0.5% TweenCitation20 for 2 h, washed and incubated with secondary goat anti-mouse and goat anti-rabbit antibodies conjugated with cy5 or TRITC (Jackson ImmunoResearch Laboratories) in PBS for 90 min. The results with the 2% concentration of TritonX-100 provided an evidence that there is an increased density of DNA in the nuclear regions/domains that contain PcG bodies.
In both approaches, the coverslips were then mounted using a Polyvinyl alcohol mounting medium with DABCO. The cells were imaged using a confocal microscope Leica TCS SP5 with 63x/1.4 NA oil immersion objective.
High-pressure freezing and freeze substitution.
U-2 OS cells were grown on 1.4 mm sapphire discs (Leica Microsystems) in Petri dishes. Only a single sapphire disc was placed in a given Petri dish. Samples were then dipped into cryofiller, 20% BSA (Sigma) in CO2 independent medium supplemented with L-glutamin (GIBCO, Invitrogen) and the addition of 10% FCS (GIBCO), transferred into the rapid loader under a stereomicroscope and frozen. For the high-pressure freezing (HPF) the Leica EM PACT2 with the rapid transfer system (Leica EM RTS) was used.
Frozen samples were then processed using a freeze substitution apparatus (Leica EM AFS2) equipped with an automatic processor (Leica EM FSP). As a freeze substitution medium, 0.1% uranyl acetate (10% stock solution in methanol) in aceton (EM grade, Polysciences) was used. Cells were freeze substituted at −90°C for 48 h. Thereafter, the temperature was raised to −50°C (5°C per h). The samples were then kept in the freeze substitution medium for 24 h. After freeze substitution, the cells were washed with acetone and gradually infiltrated with increasing concentrations of Lowicryl HM20 monostep (Electron Microscopy Sciences) in acetone (2 h with 25% HM20, 3 h with 50% HM20, 3 h with 75% HM20, 4 h with 100% HM20, 8 h with 100% HM20). During all steps of substitution, washing and infiltration the automatic processor agitated the samples using a syringe. Lowicryl used for low temperature embedding was bubbled with a stream of dry nitrogen to remove oxygen, which can interfere with the polymerization process. Polymerization was performed at −50°C for 26 h and followed by gradual warming to 20°C over a 14 h period (with a slope 5°C/h). Final hardening process of resin blocks was performed at 20°C for 24 h. The polymerization ran under a UV lamp (a component of the Leica EM FSP).
Correlative light-electron microscopy (CLEM).
U-2 OS cells expressing BMI1-GFP were grown on sapphire discs in Petri dishes. A sapphire disc was placed into a gold-coated live cell carrier (1.5 mm in diameter, 140 µm deep; Leica Microsystems) with the cells facing up. On top of this system, a molybdenum finder grid (1.48 mm; Leica Microsystems) was clamped. Such a sandwich was flipped over and transferred into a Fluorodish (World Precision Instruments, Inc.) with CO2-independent medium containing 10% FCS. After acquiring a Z series using the inverted Leica TCS SP5 confocal microscope using a long working distance, 63x/1.3 NA glycerol immersion objective, the sandwich was frozen. Frozen samples were processed in the freeze substitution apparatus with the mounted automatic processor according to the protocol described above.
Polymerized blocks were then removed from the plastic flow through rings (accessories for the AFS2, Leica). To release the carrier from the block, the residual resin was trimmed, and the blocks were dipped into liquid nitrogen and attached to a 40°C razor blade. Blocks prepared for CLEM experiments had the cells on the surface and the finder grid deeper. A pyramid was made to only leave the quadrant in which the cell of the interest was located as described by Verkade.Citation50 Serial ultrathin sections (70 nm) cut with Leica Ultracut S ultramicrotome were collected on nickel slot grids coated with formvar-carbon film. Ultrathin resin sections not further stained with heavy metal salts were then viewed with a FEI Tecnai G2 Sphera electron microscope.
Immunolabeling on resin sections.
With respect to on-section immunolabeling, the BMI1 monoclonal antibody was purchased from Upstate (Millipore), TRITC-conjugated and 15 nm gold-conjugated secondary antibodies from Aurion and 18 nm gold-conjugated antibody from Jackson ImmunoResearch. NGS pretreated sections were incubated for 1 h at room temperature with a primary antibody (diluted at 1:35) in PBS with 2% BSA and 0.5% Tween 20 (Sigma). After a second treatment with 5% NGS, 15 nm gold-conjugated secondary antibodies (goat anti-mouse) diluted in 1.2% BSA in PBS (1:3, Aurion or 1:10, Jackson ImmunoResearch) were reacted with sections for 45 minutes at room temperature. For immunofluorescence on resin sections with the TRITC-conjugated goat anti-mouse secondary antibody (diluted 1:100), the same protocol was used.
The quantitative evaluation of the density of the post-embedding immunogold labeling in heterochromatin was estimated by comparing the outside, with respect to the inside, of the “PcG bodies” in thin sectioned cells by counting the number of gold particles per unit area of heterochromatin. In the case of the “PcG body” domain, attention was paid that the evaluated heterochromatin area in thin section is a part of the “PcG body”, as judged from its immunofluorescence image. For the chosen heterochromatin domains outside of the “PcG body”, attention was paid to only include the heterochromatin area distant from any “PcG body” identified by fluorescence microscopy. The ratio of these two estimates gave us information about how much of the immunogold labeling was found per heterochromatin area outside the “PcG body” compared to heterochromatin inside the “PcG body”. Statistical measurements were obtained by averaging these ratios over six sectioned “PcG bodies”.
In all control immunocytochemical experiments, the primary antibodies were omitted resulting in negligible background signals.
Abbreviations
| 2-/3-D | = | two-/three-dimensional |
| BMI1 | = | B lymphoma Mo-MLV insertion region 1 homolog |
| BSA | = | bovine serum albumin |
| CLEM | = | correlative light-electron microscop |
| DAPI | = | 4′,6-diamidino-2-phenylindole |
| DA | = | distamycin A |
| EM | = | electron microscopy |
| FISH | = | fluorescence in situ hybridization |
| H3K27 | = | lysine 27 of histone H3 |
| HPC1–3 proteins | = | human Polycomb 1–3 proteins |
| HPF | = | high-pressure freezing |
| HPH1–3 protein | = | human polyhomeotic 1–3 protein |
| IC | = | interchromatin compartment |
| NGS | = | normal goat serum |
| OPT domain | = | Oct1/PTF/transcription domain |
| PBS | = | phosphate buffer saline |
| PcG proteins | = | Polycomb group proteins |
| PML bodies | = | promyelocytic leukemia nuclear bodies |
| PNC | = | perinucleolar compartment |
| PRC1(2) | = | Polycomb repressive complex 1(2) |
| RING1–2 proteins | = | ring finger 1–2 proteins |
| Sam68 protein | = | Src-associated in mitosis 68 kDa protein |
| SCML | = | sex comb on midleg-like protein |
| SUMO | = | small ubiquitin-like modifier |
| U-2 OS cell line | = | human osteosarcoma cell line |
| U-2 OS BMI1-GFP cells | = | U-2 OS cells expressing the recombinant BMI1-GFP protein |
Figures and Tables
Figure 1 Correlation of live cell imaging and fluorescence immunocytochemistry, and correlation of “PcG bodies” fluorescence with DA/DAPI staining and DNA immunofluorescence. (A1) U-2 OS BMI1-GFP cells were imaged in vivo in gridded Petri dishes. Live cell images are shown consecutively: localization of the cells in phase contrast, with the chosen cell of the interest being delineated in the rectangle (overview phase), differential interference contrast microscopy of the cell of interest (DIC), and Z-projection of fluorescence of GFP-tagged BMI1 protein in the same cell (in vivo GFP). (A2) Subsequently, the cells were aldehyde fixed, permeabilized and immunolabeled with antibodies. Maximum intensity projections of BMI1 (BMI1) and GFP (GFP) signals are shown. DNA was counterstained with DAPI (DAPI, middle confocal section). The in vivo fluorescence signal of the PcG bodies matches well both the BMI1 and GFP immunofluorescence signals. (B) Counterstaining of the fixed and permeabilized U-2 OS BMI1-GFP cells with DAPI in combination with distamycin A (DA/DAPI) to show the co-localization of increased DNA density with the PcG bodies identified with an anti-GFP antibody (GFP). The highest intensities of DA/DAPI fluorescence on maximum intensity projection co-localized with the fluorescence of the PcG bodies (white arrows). (C) Live cell imaging of the PcG bodies (in vivo GFP) was, after weak fixation accompanied by the use of 2% Triton X-100 treatment, correlated with the GFP (GFP) and DNA (DNA) immunocytochemistry images. The anti-DNA labeling revealed a high accumulation of DNA in the PcG bodies (white arrows).
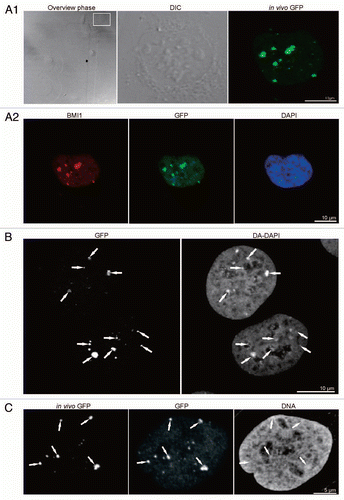
Figure 2 CLEM of high pressure frozen and cryosubstituted cells (the images of the same cell are also shown in ; however, encompasses different thin sections and documents images of a different “PcG body” in this same cell). U-2 OS BMI1-GFP cells grown on the sapphire discs were clamped in the live cell carrier with the finder grid and imaged face down. After live cell imaging the cells were high-pressure frozen, cryosubstituted and immunolabeled with anti-BMI1 antibody. In fluorescence and EM images in and , the white and yellow arrows point to a nuclear region/domain corresponding to the two GFP “PcG bodies”. Invaginations of the nuclear envelope are designated by black arrows and arrowheads. (A) An overview image (merge of fluorescence and phase contrast) to determine the quadrant of interest on the finder grid, (B) higher magnification of the quadrant with the cells of interest delineated in the rectangle. (B1) Phase contrast and (B2) maximum intensity projection of fluorescence of the cells from the rectangle. (C) The same two thin sectioned cells seen in the electron microscope. Some of the serial thin sections we also processed for on-section fluorescence immunocytochemistry (see inserts in and C as well as Sup. Figs. 3–5 in the Sup. Material). (D) Anti-BMI1 immunogold labeling on ultrathin sections (15 nm gold particles). The electron-dense heterochromatin structures (he) are specifically enriched in the BMI1 immunogold label while the cytoplasm (cy) rich in ribosomes is devoid of the label. The nucleolus (nu) in the insert of shows two BMI1 labeled intranucleolar heterochromatin clumps (white arrowheads). The white arrow points to a nuclear region/domain that correlates with (a section of) the “PcG body” fluorescence seen in , with the local accumulation of heterochromatin structures in this domain.
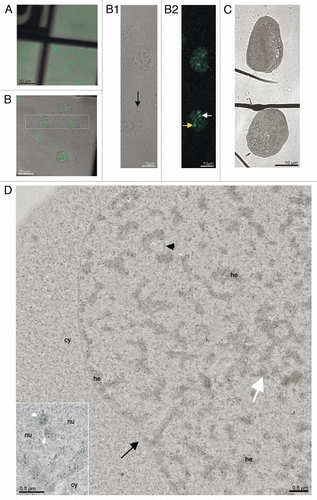
Figure 3 CLEM of a high pressure frozen, cryosubstituted and serially sectioned cell. (A) Anti-BMI1 immunogold labeling of heterochromatin structures (15 nm gold particles) of the same cell as in the , but a different thin section is shown in (A). The yellow arrow points to the nuclear region/domain that correlates with (the section of) the “PcG body” fluorescence seen in 2 with the local density of heterochromatin structures being enriched in this domain. The heterochromatin structures are specifically enriched in the BMI1 immunogold label. Gold particles are sometimes situated towards the periphery of the heterochromatin structures (insert). (B–E) Four consecutive serial sections from the same area as shown in (A). In (B and C), the thin sections were first used for on-section immunofluorescence mapping of the BMI1 protein (inserts; note that the intensity of the “PcG body” in is higher than that in ) to identify the position of the “PcG bodies”, and subsequently observed in the electron microscope (for a detailed analysis of the ultrastructural identification of the “PcG body”, see Sup. Figs. 3–5 in the Sup. Material). The two remaining serial thin sections (D and E) were on-section immunogold labeled for the BMI1 protein. Nucleolus (nu), cytoplasm (cy) and invaginagions of the nuclear envelope (black arrowheads) are designated in (A–E).
Additional material
Download Zip (17.6 MB)Acknowledgments
The authors would like to thank Dr. Maarten van Lohuizen for the U-2 OS cell line stably expressing BMI1-GFP protein and Dr. Paul Verkade for hosting J.S. in his laboratory and helpful consultations on CLEM. The authors thank Dr. Pavel Krizek for help with statistics of the immunogold labeling and Dr. Guy M. Hagen for careful reading of the manuscript. The results of this work were presented at the 50th Annual Meeting of the ASCB, Philadelphia, December 11–15, 2010. This work was supported by the Czech grants MSM0021620806, LC535 and AV0Z50110509.
References
- Tuckfield A, Clouston DR, Wilanowski TM, Zhao LL, Cunningham JM, Jane SM. Binding of the RING polycomb proteins to specific target genes in complex with the grainyhead-like family of developmental transcription factors. Mol Cell Biol 2002; 22:1936 - 1946
- Sawarkar R, Paro R. Interpretation of developmental signaling at chromatin: the Polycomb perspective. Dev Cell 2010; 19:651 - 661
- Hodgson JW, Brock HW. Are polycomb group bodies gene silencing factories?. Cell 2011; 144:170 - 171
- Liang Z, Biggin MD. Eve and ftz regulate a wide array of genes in blastoderm embryos: the selector homeoproteins directly or indirectly regulate most genes in Drosophila. Development 1998; 125:4471 - 4482
- Plath K, Talbot D, Hamer KM, Otte AP, Yang TP, Jaenisch R, et al. Developmentally regulated alterations in Polycomb repressive complex 1 proteins on the inactive X chromosome. J Cell Biol 2004; 167:1025 - 1035
- Sparmann A, van Lohuizen M. Polycomb silencers control cell fate, development and cancer. Nat Rev Cancer 2006; 6:846 - 856
- Villa R, Pasini D, Gutierrez A, Morey L, Occhionorelli M, Vire E, et al. Role of the polycomb repressive complex 2 in acute promyelocytic leukemia. Cancer Cell 2007; 11:513 - 525
- Boukarabila H, Saurin AJ, Batsche E, Mossadegh N, van Lohuizen M, Otte AP, et al. The PRC1 Polycomb group complex interacts with PLZF/RARA to mediate leukemic transformation. Genes Dev 2009; 23:1195 - 1206
- Martinez AM, Cavalli G. The role of polycomb group proteins in cell cycle regulation during development. Cell Cycle 2006; 5:1189 - 1197
- Enderle D, Beisel C, Stadler MB, Gerstung M, Athri P, Paro R. Polycomb preferentially targets stalled promoters of coding and noncoding transcripts. Genome Res 2011; http://dx.doi.org/10.1101/gr.114348.110
- Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, et al. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science 2002; 298:1039 - 1043
- Czermin B, Melfi R, McCabe D, Seitz V, Imhof A, Pirrotta V. Drosophila enhancer of Zeste/ESC complexes have a histone H3 methyltransferase activity that marks chromosomal Polycomb sites. Cell 2002; 111:185 - 196
- Trojer P, Reinberg D. Facultative heterochromatin: is there a distinctive molecular signature?. Mol Cell 2007; 28:1 - 13
- Lund AH, van Lohuizen M. Polycomb complexes and silencing mechanisms. Curr Opin Cell Biol 2004; 16:239 - 246
- Ringrose L, Paro R. Epigenetic regulation of cellular memory by the Polycomb and Trithorax group proteins. Annu Rev Genet 2004; 38:413 - 443
- Levine SS, King IF, Kingston RE. Division of labor in polycomb group repression. Trends Biochem Sci 2004; 29:478 - 485
- Gunster MJ, Satijn DP, Hamer KM, den Blaauwen JL, de Bruijn D, Alkema MJ, et al. Identification and characterization of interactions between the vertebrate polycomb-group protein BMI1 and human homologs of polyhomeotic. Mol Cell Biol 1997; 17:2326 - 2335
- Satijn DP, Gunster MJ, van der Vlag J, Hamer KM, Schul W, Alkema MJ, et al. RING1 is associated with the polycomb group protein complex and acts as a transcriptional repressor. Mol Cell Biol 1997; 17:4105 - 4113
- Schoorlemmer J, Marcos-Gutierrez C, Were F, Martinez R, Garcia E, Satijn DP, et al. Ring1A is a transcriptional repressor that interacts with the Polycomb-M33 protein and is expressed at rhombomere boundaries in the mouse hindbrain. EMBO J 1997; 16:5930 - 5942
- Buchenau P, Hodgson J, Strutt H, Arndt-Jovin DJ. The distribution of polycomb-group proteins during cell division and development in Drosophila embryos: impact on models for silencing. J Cell Biol 1998; 141:469 - 481
- Saurin AJ, Shiels C, Williamson J, Satijn DP, Otte AP, Sheer D, et al. The human polycomb group complex associates with pericentromeric heterochromatin to form a novel nuclear domain. J Cell Biol 1998; 142:887 - 898
- Zhang T, Sun Y, Tian E, Deng H, Zhang Y, Luo X, et al. RNA-binding proteins SOP-2 and SOR-1 form a novel PcG-like complex in C. elegans. Development 2006; 133:1023 - 1033
- Otte AP, Kwaks TH. Gene repression by Polycomb group protein complexes: a distinct complex for every occasion?. Curr Opin Genet Dev 2003; 13:448 - 454
- Voncken JW, Schweizer D, Aagaard L, Sattler L, Jantsch MF, van Lohuizen M. Chromatin-association of the Polycomb group protein BMI1 is cell cycle-regulated and correlates with its phosphorylation status. J Cell Sci 1999; 112:4627 - 4639
- Raska I. Nuclear ultrastructures associated with the RNA synthesis and processing. J Cell Biochem 1995; 59:11 - 26
- Matera AG, Izaguire-Sierra M, Praveen K, Rajendra TK. Nuclear bodies: random aggregates of sticky proteins or crucibles of macromolecular assembly?. Dev Cell 2009; 17:639 - 647
- Lamond AI, Earnshaw WC. Structure and function in the nucleus. Science 1998; 280:547 - 553
- Spector DL. Nuclear domains. J Cell Sci 2001; 114:2891 - 2893
- Gall JG. The centennial of the Cajal body. Nat Rev Mol Cell Biol 2003; 4:975 - 980
- Craig JM. Heterochromatin—many flavours, common themes. Bioessays 2005; 27:17 - 28
- Zhao R, Bodnar MS, Spector DL. Nuclear neighborhoods and gene expression. Curr Opin Genet Dev 2009; 19:172 - 179
- Carmo-Fonseca M, Berciano MT, Lafarga M. Orphan nuclear bodies. Cold Spring Harb Perspect Biol 2010; 2:703
- Hernandez-Munoz I, Taghavi P, Kuijl C, Neefjes J, van Lohuizen M. Association of BMI1 with polycomb bodies is dynamic and requires PRC2/EZH2 and the maintenance DNA methyltransferase DNMT1. Mol Cell Biol 2005; 25:11047 - 11058
- Schweizer D, Ambros PF. Gosden JR. Chromosome banding: stain combinations for specific regions. Methods in Molecular Biology. Chromosome Analysis Protocols 1994; 29:Totowa, New Jersey Humana Press 97 - 112
- Hu Y, Kireev I, Plutz M, Ashourian N, Belmont AS. Large-scale chromatin structure of inducible genes: transcription on a condensed, linear template. J Cell Biol 2009; 185:87 - 100
- Fricker M, Hollinshead M, White N, Vaux D. Interphase nuclei of many mammalian cell types contain deep, dynamic, tubular membrane-bound invaginations of the nuclear envelope. J Cell Biol 1997; 136:531 - 544
- Raska I, Rychter Z, Smetana K. Fibrillar centres and condensed nucleolar chromatin in resting and stimulated human lymphocytes. Z Mikrosk Anat Forsch 1983; 97:15 - 32
- Raska I, Armbruster BL, Frey JR, Smetana K. Analysis of ring-shaped nucleoli in serially sectioned human lymphocytes. Cell Tissue Res 1983; 234:707 - 711
- Raska I, Dundr M, Koberna K, Melcak I, Risueno MC, Torok I. Does the synthesis of ribosomal RNA take place within nucleolar fibrillar centers or dense fibrillar components?. J Struct Biol 1995; 114:1 - 22
- Raska I. Oldies but goldies: searching for Christmas trees within the nucleolar architecture. Trends Cell Biol 2003; 13:517 - 525
- Armbruster BL, Wunderli H, Turner BM, Raska I, Kellenberger E. Immunocytochemical localization of cytoskeletal proteins and histone 2B in isolated membrane-depleted nuclei, metaphase chromatin and whole Chinese hamster ovary cells. J Histochem Cytochem 1983; 31:1385 - 1393
- Dubochet J, Sartori Blanc N. The cell in absence of aggregation artifacts. Micron 2001; 32:91 - 99
- Hancock R. Internal organisation of the nucleus: assembly of compartments by macromolecular crowding and the nuclear matrix model. Biol Cell 2004; 96:595 - 601
- Richter K, Nessling M, Lichter P. Experimental evidence for the influence of molecular crowding on nuclear architecture. J Cell Sci 2007; 120:1673 - 1680
- Maeshima K, Hihara S, Eltsov M. Chromatin structure: does the 30 nm fibre exist in vivo?. Curr Opin Cell Biol 2010; 22:291 - 297
- Matsuda A, Shao L, Boulanger J, Kervrann C, Carlton PM, Kner P, et al. Condensed mitotic chromosome structure at nanometer resolution using PALM and EGFP-histones. PloS one 2010; 5:12768
- Alkema MJ, Bronk M, Verhoeven E, Otte A, van't Veer LJ, Berns A, et al. Identification of Bmi1-interacting proteins as constituents of a multimeric mammalian polycomb complex. Genes Dev 1997; 11:226 - 240
- McDonald KL, Morphew M, Verkade P, Muller-Reichert T. Recent advances in high-pressure freezing: equipment- and specimen-loading methods. Methods Mol Biol 2007; 369:143 - 173
- Muller-Reichert T, Srayko M, Hyman A, O'Toole ET, McDonald K. Correlative light and electron microscopy of early Caenorhabditis elegans embryos in mitosis. Methods Mol Biol 2007; 79:101 - 119
- Verkade P, Moving EM. The Rapid Transfer System as a new tool for correlative light and electron microscopy and high throughput for high-pressure freezing. J Microsc 2008; 230:317 - 328
- Misteli T. Protein dynamics: implications for nuclear architecture and gene expression. Science 2001; 291:843 - 847
- Phair RD, Scaffidi P, Elbi C, Vecerova J, Dey A, Ozato K, et al. Global nature of dynamic protein-chromatin interactions in vivo: three-dimensional genome scanning and dynamic interaction networks of chromatin proteins. Mol Cell Biol 2004; 24:6393 - 6402
- Cmarko D, Verschure PJ, Otte AP, van Driel R, Fakan S. Polycomb group gene silencing proteins are concentrated in the perichromatin compartment of the mammalian nucleus. J Cell Sci 2003; 116:335 - 343
- Cavalli G. Chromosome kissing. Curr Opin Genet Dev 2007; 17:443 - 450
- Sexton T, Umlauf D, Kurukuti S, Fraser P. The role of transcription factories in large-scale structure and dynamics of interphase chromatin. Semin Cell Dev Biol 2007; 18:691 - 697
- Platero JS, Hartnett T, Eissenberg JC. Functional analysis of the chromo domain of HP1. EMBO J 1995; 14:3977 - 3986
- Cavalli G, Paro R. The Drosophila Fab-7 chromosomal element conveys epigenetic inheritance during mitosis and meiosis. Cell 1998; 93:505 - 518
- Dellino GI, Schwartz YB, Farkas G, McCabe D, Elgin SC, Pirrotta V. Polycomb silencing blocks transcription initiation. Mol Cell 2004; 13:887 - 893