Abstract
Up-regulated expression of lamin A has been implicated in increased cell invasiveness and mortality in colorectal cancer. Here we use quantitative proteomics to investigate lamin A regulated changes in the cytoskeleton that might underpin increased cell motility. Using siRNA knockdown of lamin A in a model cell line (SW480/lamA) we confirm that the presence of lamin A promotes cell motility. Using an enhanced technique to prepare cytoskeleton fractions in combination with 2D DiGE we were able to accurately and reproducibly detect changes in the representation of protein species within the cytoskeleton as low as 20%. In total 64 protein spots displayed either increased or decreased representation within the cytoskeleton of SW480/lamA cells compared to controls. Of these the identities of 29 spots were determined by mass spectrometry. A majority were multiple forms of three classes of proteins, including components of the actin and IF cytoskeletons, protein chaperones and translation initiation and elongation factors. In particular our data reveal that the representation of tissue transglutaminase 2, which is known to modify elements of the cytoskeleton and is associated with cancer progression, was highly over-represented in the cytoskeleton fraction of SW480/lamA cells. Overall, our data are consistent with changed protein cross-linking and folding that favours the formation of dynamic actin filaments over stress fibres accounting for the altered cell motility properties in SW480/lamA cells.
Introduction
Lamins are type V intermediate filament proteins that are strongly implicated in human diseases. Lamins fall into two sub-types, the B-type lamins, which are housekeeping proteins and the A-type lamins, which have roles in differentiation and cellular fitness.Citation1 Lamins are multifunctional proteins that are often aberrantly expressed or localised in tumours. The nature of lamin function in cancer is still unclear. There is no overall pattern of A-type lamin expression in cancers and frequently inconsistent patterns are observed between cancer subtypes. Many studies show lamin A/C to be downregulated in tumour cells,Citation2–Citation4 but others show positive or upregulated expression.Citation2,Citation5,Citation6 Sometimes expression levels can vary dramatically even within cancer subtypes; for example, in colorectal and basal cell carcinomas, A-type lamin expression can be positive,Citation4,Citation5,Citation7 reducedCitation3,Citation4 or negativeCitation3,Citation4,Citation7 in tumour tissue. Finally, lamin A/C has been shown to aberrantly localize to the cytoplasm in lung carcinomas, colon adenomas and adenocarcinomas, pancreatic and gastric cancers.Citation2,Citation3,Citation7
The link between cancer prognosis and A-type lamin expression is also complex. Some studies point to a lack of lamin A/C expression as a sign of poor prognosis. CpG island promoter hypermethylation, which is predicted to silence LMNA leading to loss of A-type lamin expression in nodal diffuse large B-cell lymphoma, has been correlated with a decrease in overall survival.Citation8 Moreover, a recent study has revealed that patients with gastric carcinoma cells containing downregulated A-type lamin expression have poorer prognosis compared with those expressing A-type lamins.Citation9 In contrast, in colorectal cancer (CRC), Willis et al. discovered that patients with CRC tumours expressing A-type lamins were almost twice as likely to die from the cancer compared with clinicopathologically identical patients with tumours showing negative expression of A-type lamins.Citation7 A-type lamins are therefore potential biomarkers of poor prognosis in CRC.
Willis et al. found that expression of lamin A in colon carcinoma promotes increased cell motility.Citation7 In wounding assays, wound closure was significantly faster in CRC cells transfected with GFP-lamin A compared with control cells transfected with GFP alone. GFP-lamin A expression was shown to control a pathway in which upregulated expression of T plastin, an actin bundling protein, led to downregulated expression of the cell adhesion molecule E-cadherin, resulting in increased cell motility. Loss of E-cadherin and expression of plastins are often hallmarks of tumours, correlating with invasive and metastatic behaviour.Citation10,Citation11
The findings of Willis et al. may reveal a function of lamin A as a regulator of a pathway involving actin dynamics, cell adhesion and cell motility. It has been recently discovered that the cytoskeleton is connected to the nucleus through LINC (linker of nucleoskeleton and cytoskeleton) complexes, formed through binding of KASH (Klarsicht/ANC-1/Syne homology) domain proteins such as nesprins to SUN (Sad1 and UNC-84 homology) domain proteins.Citation12 The nucleoplasmic regions of human SUN domain proteins bind to A-type laminsCitation13,Citation14 and nesprins have been shown to bind to actin microfilaments,Citation15 plectinCitation16 and emerin.Citation17,Citation18 This provides a pathway by which signals can travel between the outside of the cell and the nucleus. Lamin A expression may therefore control the reorganization of the cytoskeleton through its association with LINC complexes, causing alterations in cell migration.
In this study we have used quantitative proteomics to begin to understand how expression of A-type lamins in CRC cell lines might influence cytoskeleton organisation. We show that expression of A-type lamins is correlated with changes in the actin and IF cytoskeleton that is accompanied by reduced association of protein chaperones with the cytoskeleton and may be underpinned by altered protein cross-linking.
Results
We have previously shown that expression of GFP-lamin A in SW480 CRC cells increases the expression of endogenous lamin A and also increases cell motility.Citation7 To confirm this finding we performed immunoblotting experiments on SW480/lamA and SW480/cntl cells lines and found that the expression of endogenous lamin A was 40% higher in the presence of GFP-lamin A and that the combined representation of lamin A and GFP lamin A was four fold higher in SW480/lamA cells when compared to SW480/cntl (). Next we performed scratch wound assays on both cell lines and found that the rate of wound closure was >20% faster in SW480/lamA cells compared to SW480/cntl ( and E). To confirm that these motility changes were a direct consequence of increased expression of lamin A forms, we used siRNA to downregulate lamin A and GFP-lamin A expression and then used scratch wound assays to measure rates of cell motility. Using siRNA specific for lamin A we found that endogenous lamin A was almost undetectable 96 h after transfection whereas GFP lamin A was downregulated 120 h after transfection. At this time point both cell lines maintained viability as judged by trypan blue exclusion (not shown) and by the normal morphology of cells as seen in time-lapse video microscopy (Fig. S1A and B). Consistent with this finding, rates of wound closure were only slightly slower in SW480/lamA cells treated with siRNA specific to lamin A compared to cells treated with scrambled siRNA 72–96 h after transfection, but were >40% slower 96–120 h after transfection ( and H). Taken together, these data confirm that upregulated expression of lamin A in SW480 cells increases rates of cell motility.
Previously, we found that increased cell motility in SW480/lamA cells was associated with increased expression of the actin bundling protein T-plastin.Citation7 We wanted to extend this finding by investigating whether increased expression of lamin A caused more widespread changes in the cytoskeleton. To do this we first used an optimised extraction protocol to isolate cytoskeleton preparations.Citation19 Initially, we used immunoblotting to compare the solubility properties of the major elements of the cytoskeleton and representative membrane proteins in SW480/lamA and SW480/cntl cells. We found that a greater fraction of actin was solubilised by detergent and nuclease treatment in SW480/cntl cells compared to SW480/lamA ( and B), but the solubility properties of α-tubulin and keratin 18 were very similar in each cell line (). Overall, in both cell lines, a-tubulin and keratin 18 were mainly retained in the insoluble cytoskeleton fraction (P4), whereas only a minority of actin was retained in this fraction. Actin displayed increased solubility in SW480/ctl cells (compared to SW480/lamA) throughout the extraction procedure, perhaps reflecting not only altered distribution between F and G actin populations but also within the F actin population, altered distributions between stress fibres and dynamic actin filaments. As expected, vinculin was solubilised following detergent extraction ( and H).
We adopted a proteomic approach to compare the protein complement of the final cytoskeleton fraction obtained from SW480/lamA cells with the same fraction obtained from SW480/cntl cells. First, we determined the reproducibility of the method by using 2D gel electrophoresis (2D GE) to compare the proteins in each cytoskeleton fraction from six replicate experiments. Overall, the representation of proteins in the cytoskeleton fractions was very similar in both cell lines. In addition, the cytoskeleton fractions appeared highly reproducible between replicate experiments (). For the final proteomic analysis, we decided to discard replicate 2 ( and D), because the fractions were prepared from cells that had not reached the same cell density as those used in the other five replicate experiments.
Next we labelled each fraction with Cy-5 dye, whilst a pooled internal standard was labelled with Cy-3. The efficiency of dye labelling was confirmed by resolving 4 µg of each sample on 12% SDS PAGE (Fig. S2). One replicate from SW480/lamA cells appeared to be poorly labelled (SW480/lamA lane 1). Nonetheless all samples were resolved on large format gels using a pH range of 4–7 in the first dimension and 12% SDS-PAGE in the second dimension. Gels were scanned using a Typhoon Variable Mode Imager and the images were processed using Progenesis Samespots. In the first instance gel images were aligned in automatic mode and checked manually. By this process we confirmed that one of the replicates was anomalous and this replicate was eliminated from final analysis. Of the remaining four replicates, an ANOVA test was used to identify spots with a p value of <0.05 and a power of >0.7 for spots with a >1.2 fold change in representation between cytoskeleton fractions obtained from SW480/lamA and SW480/cntl cells (). Using this approach a list of 64 spots showed changed representation within the cytoskeleton between each cell line. Of these spots, 29 were chosen for mass spectrometry analysis based upon the likelihood that they contained sufficient protein for accurate identification.
To identify the proteins represented by the 29 selected spots, 2-D GE preparative gels were run using 500 µg of protein for each gel. The spots were picked robotically and subjected to trypsin digestion prior to MALDI ToF-ToF analysis. Mass spectra were matched to theoretical trypsin digests of proteins from the NCBInr database using MASCOT software at a mass accuracy of 50 ppm. Proteins with a MOWSE probability score of 82 or higher were considered as significant (). Seven of the proteins migrated as a train of two or three spots with similar Mr but differing pI values. One protein (β-actin) was identified in a train of seven spots. Thus the 29 spots picked for protein identification, in fact represented modified forms of a total of thirteen proteins. Of these, eight proteins were over-represented in the cytoskeleton fraction obtained from SW480/lamA cells whilst five proteins were under-represented ( and and ). The spots found in trains of differing mobility all had very similar fold changes. The range of fold changes was between 1.2 to 3.5 and all of these fold changes were completely reproducible between replicate samples. The majority of proteins displayed fold changes within the 20–100% range, which is rather typical for disease states. Only two proteins, Histone cluster 2H4b and transglutaminase 2 displayed significantly higher fold representation in SW480/lama cells, which is exceptional and may reflect a controlling influence on other state changes. A majority of the proteins selected fell into three classes of proteins. Four of the proteins that were over represented in the cytoskeleton fraction of SW480/lamA cells were either cytoskeleton proteins or proteins known to modify the cytoskeleton (), whilst three of the proteins under-represented in the same cytoskeleton fraction were protein chaperones (). Five of the remaining proteins were translation initiation and elongation factors and of these three were over-represented and two were under-represented ( and ). Only histone cluster 2, H4b, which was over-represented in the SW480/lamA cytoskeleton, did not fall into a common category of proteins.
Discussion
In this investigation, we have used 2D DiGE technology as a first attempt to understand modifications in the cytoskeleton associated with lamin A mediated cell motility changes in a cancer cell line. Whilst a number of investigations have previously used a quantitative proteomics approach to investigate cell motility changes in cancer cells to our knowledge this is the first study to focus on a cytoskeleton fraction. On 2-D DiGE, we found that 64 spots displayed completely reproducible changes in their representation within the cytoskeleton of SW480/lamA cells (compared to controls) but less than half of these spots contained sufficient material for significant protein identification. We further found that of the spots that could be identified accurately most represented multiple isoforms of a much smaller subset of thirteen proteins. If the same were true of the 35 spots that were not subjected to mass spectrometry then perhaps as few as thirty proteins are needed to modify the cytoskeleton in order to promote increased cell motility. Recent work has shown that a combination of SILAC technology and affinity isolation significantly improves the sensitivity of detection and identification of protein changesCitation20 and in future work we will adopt this method to indentify the remaining proteins in our list.
Of the thirteen proteins identified as having changed representation in the cytoskeleton fraction, twelve fell into three distinct categories. β-actin,Citation21,Citation22 α4-actininCitation23–Citation25 and vimentinCitation26,Citation27 are all components of the cytoskeleton that have previously been implicated in cancer progression, cell migration and invasiveness. Tissue transglutaminase 2 (TG2) is a Ca2+ regulated transamidase that cross-links proteins via the formation of ε-(-γ-glutamyl) lysine isopeptide bonds. TG2 is thought to play a role in cytoskeleton organisation, since β-actin, α-actinin, tubulin, cofilin and vimentin are all TG2 substrates.Citation28–Citation31
Three protein chaperones, B23, Hsp60 and mortalin (heat shock 70 kDa protein 9) were all under-represented in the cytoskeleton fraction of SW480/lamA cells. All three proteins have been implicated in cancer progressionCitation32–Citation34 and all three bind to cytoskeleton proteins and influence cytoskeleton dynamics and organisation.Citation35–Citation37 In particular, changes in B23 expression alters the balance between stress fibre formation and other more dynamic forms of F actin.Citation37 Interestingly, Hsp60 and members of the Hsp70 family are also substrates for TG2.Citation29
Five proteins that had altered representation levels in the SW480/lamA cytoskeleton were translation initiation and elongation proteins. Three of those proteins EF-1γ, EF-2α and EF5α all have increased representation in the cytoskeleton, whilst two other proteins EF-1δ and TUFM both have decreased representation in the cytoskeleton. EF-1γ and EF-2α are both substrates of TG2.Citation29 Altered levels of expression of many of these translation proteins have been observed in gastrointestinal cancersCitation38–Citation40 and these changes have been correlated with tumour aggressiveness.Citation41,Citation42 As yet no clear role in carcinogenesis has been ascribed to any of these proteins. The final protein that was over-represented in the cytoskeleton fraction of SW480/lamA cells is histone cluster 2H4b. This protein has only recently been identified and has no assigned function.Citation43
Based upon our initial findings we propose a hypothetical model that coherently links the changed representation of cytoskeleton proteins to the altered cell motility rates observed in SW480/lamA cells. Our data suggest that there is a 50%–60% increase in the amount of F actin in these cells accompanied by a 40% increase in polymerised vimentin and a 20% enrichment of α4-actinin. Increases in the ratio of F:G actin has previously been implicated in a transition to a metastatic step in colon adenocarcinoma cell lines.Citation21 Similarly, enrichment of α-actinin at the leading edge of CRC has been shown to underpin increased cell motility.Citation23 Finally, expression of vimentin in CRC is also associated with poor prognosis and invasiveness.Citation27,Citation44 Our data suggest that these changes in the cytoskeleton could be achieved by modification and cross-linking of all three proteins to other cytoskeleton proteins by TG2, which is the most highly enriched protein in the cytoskeleton of SW480/lamA cells (320% enrichment). It is interesting from this point of view that all three proteins are represented as multiple spots in 2D DiGE and future studies will seek to understand whether these multiple spots represents differential post translational modifications of these proteins. The depletion of chaperonin proteins from the cytoskeleton might also be a result of altered regulation of the cross-linking of these proteins to the cytoskeleton by TG2. The reduced association, particularly of B23, within the cytoskeleton might for example alter the balance between stress fibre formation and other forms of filamentous actin, which is a hallmark of invasive cells.Citation45,Citation46
The altered association of translation initiation and elongation factors within the cytoskeleton might reflect an increasingly accepted concept, which is that the cytoskeleton and nuclear lamina form a trans-cellular network that physically links the interior of the nucleus via the cytoskeleton to the extracellular matrix, thus allowing hardwired signalling between the extracellular environment, the cytoplasm and the nucleoplasm. This hardwired signalling could link changes in cytoskeleton dynamics with altered protein expression either at the level of transcription or translation. Future studies will seek to test this hypothesis.
Materials and Methods
Cell culture.
The human pre-metastatic colon adenocarcinoma cell line SW480 was obtained from the European Collection of Cell Cultures. Cells were stably transfected with DNA constructs encoding EGFP-lamin A (SW480/lamA) or EGFP (SW480/cntl).Citation7 Cells were grown in Leibovitz-15 medium (Invitrogen) supplemented with 2 mM L-Glutamine, 10% FBS, 100 units/ml penicillin and 100 µg/ml streptomycin. Cells were maintained in a humidified environment at 37°C, without CO2.
siRNA transfection.
To knockdown lamin A, Silencer® Select Custom Designed siRNA (Ambion) ID#s238117 (si-lamin A) was used, which was specific for lamin A but not lamin C (sense: 5′-UCA UCU AUC UCA AUC CUA Att-3′, antisense: 5′-UUA GGA UUG AGA UAG AUG Aga-3′). Silencer® Select Negative Control #1 siRNA (si-control) was used as a negative control. Cells were seeded at a density of 4.0 × 105 cells per well 24 hours before transfection. Cell media was changed immediately preceding transfection, and L-15 media containing FBS but no antibiotics was added. Cells were treated with a transfection mixture containing 200 µl serum-free L-15 medium, 10 µl Oligofectamine reagent (Invitrogen) and 10 µl si-RNA (20 µM). Media was changed after 24 hours. Cells were processed for protein extraction or wounding assays between 24 and 120 hours post-transfection.
Preparation of whole cell extracts.
Cells were harvested at 70–100% confluency. Cell pellets were washed with ice-cold PBS before incubation in 500 µl hypotonic buffer (10 mM Tris-HCl pH 7.4, 10 mM KCl, 3 mM MgCl2, 0.1% (v/v) Triton X-100) plus 2 µl DNase (5 units/µl) containing protease inhibitor cocktail and 40 mM NEM on ice for 10 minutes. 500 µl 2x sample buffer (125 mM Tris-HCl pH 6.8, 2% (v/v) sodium dodecyl sulphate (SDS), 2 mM dithiothreitol (DTT), 20% (v/v) glycerol, 5% (v/v) β-mercaptoethanol and 0.25% (w/v) bromophenol blue) was then added to each sample and tubes were heated to 95°C for 3 minutes then centrifuged for 1 minute at 13,000 g.
One-dimensional SDS-PAGE and immunoblotting.
1D SDS-PAGE was performed according to Laemmli (1970).Citation47 Proteins separated on gels were electrophoretically transferred onto nitrocellulose membranes (Protran®, Scleicher and Schuell). Membranes were blocked with 4% skimmed milk powder (w/v) in blot rinse buffer for 16 h at 4°C with constant agitation. Primary antibodies used were anti-lamin A/C, JoL2 [1:200], anti-α-tubulin [1:1,000] (Sigma), anti-keratin 18 [1:1,000] (Oncogene) and anti-vinculin (VIN-11-5) [1:500] (Sigma). Anti-actin clone AC-40 (Sigma) was used as a loading control [1:1,000]. Donkey anti-mouse secondary antibody conjugated to HRP (Jackson Immuno-Research Laboratories) was used at 1:2,000 dilution. Immunological detection of proteins was performed according to standard protocols (Willis). Differences in protein expression were quantified using densitometry. Developed X-ray films were scanned in a Fujifilm Intelligent Dark Box II (Fujifilm Medical Systems) directed by Fujifilm Image Reader LAS-1000 Pro Ver. 2.11 software and intensities were quantified using Image J (http://imagej.nih.gov/ij/).
Scratch wound assay.
SW480/lamA and SW480/cntl cells were seeded at 7.5 × 105 cells per well in a 12 well plate (Grenier Bio-one). Once cells reached 100% confluency, wounds were made using a 10 µl disposable pipette tip. Identical wound locations were visualized using a live cell imaging phase contrast microscope (Zeiss) at x10 magnification every 15 minutes for 24 hours. The width of the wound at the start and end of the experiment was measured six times for each wound at 100 µm intervals using Axiovision Rel. 4.8 (Zeiss). The mean distance the cells moved in 24 hours was calculated and standard errors were calculated from the biological replicates. A paired student t-test was used to test for statistical significance.
In further experiments, SW480/lamA cells were seeded at 4.0 × 105 cells per well in a 6 well plate and transfected with si-lamin A or si-cntl after 24 hours. The wounding assay was started 96 hours post-transfection, when the cells were 100% confluent.
Biochemical fractionation.
SW480/lamA and SW480/cntl cells were extracted to prepare cytoskeleton fractions using a modification of the protocol described by Dyer et al. using icecold buffers and freshly added protease inhibitor cocktail. After each extraction step, soluble (S1–4) and insoluble (P1–4) fractions were separated by centrifugation at 1,200 g for 5 minutes at 4°C and the supernatants and one of the pellets were snap frozen.
Briefly, cell pellets were resuspended in CSK buffer (10 mM Pipes pH 6.8, 10 mM KCl, 300 mM sucrose, 3 mM MgCl2, 1 mM EGTA pH 8.0, 40 mM NEM), split into four aliquots and centrifuged. The remaining insoluble fractions were then incubated in CSK buffer supplemented with 0.5% (v/v) Triton X-100 (CSK/T buffer) for 5 minutes on ice before centrifugation. Next, the remaining insoluble fractions were resuspended in digestion (Dig) buffer (10 mM Pipes pH 6.8, 50 mM NaCl, 300 mM sucrose, 3 mM MgCl2, 1 mM EGTA pH 8.0, 40 mM NEM) and centrifuged before incubation of the pellets in 500 units/ml DNase1 in digestion buffer for 20 minutes at room temperature followed by centrifugation. In order to remove digested material, the remaining insoluble fraction was incubated in extraction (Ext) buffer (10 mM Pipes pH 6.8, 250 mM ammonium sulphate, 300 mM sucrose, 3 mM MgCl2, 1 mM EGTA pH 8.0, 40 mM NEM) for 5 minutes on ice and centrifuged.
To prepare fractions for immunoblotting, pellets were incubated with 86 µl ice-cold hypotonic buffer (10 mM Tris-HCl pH 7.4, 10 mM KCl, 3 mM MgCl2, 0.1% (v/v) Triton X-100, 40 mM NEM) plus 2 µl DNase (5 units/µl) and 2 µl protease inhibitor cocktail for 10 min on ice. Each P1 was homogenised with a Dounce homogeniser to extract the proteins using 10 gentle strokes of the pestle. 95 µl 2x sample buffer was then added to each pellet and 50 µl 5x sample buffer was added to each supernatant. All samples were then heated for 3 min at 95°C and centrifuged for 30 seconds at 13,000 g.
To prepare the cytoskeletal fraction (P4) for 2D gel electrophoresis, each P4 was solubilised in 86 µl lysis buffer (9 M urea, 2 M thiourea, 4% (w/v) CHAPS) plus 40 mM NEM, 2 µl protease inhibitor cocktail and 2 µl DNase (5 units/µl).
Mini-format 2D SDS-PAGE.
7 cm linear pH 4–7 IPG strips were re-swelled overnight with 125 µl rehydration solution (100 µg protein solubilised in 9 M urea, 2 M thiourea, 4% (w/v) CHAPS, 1% (w/v) DTT, 2% (v/v) IPG buffer pH 4–7 (GE Healthcare), 0.002% (w/v) bromophenol blue). IEF was performed on a Multiphor II Electrophoresis System (Amersham Biosciences) at 50 µA/strip with a power of 5 W for a total of 8,010 Vh. Strips were then equilibrated for 15 mins in equilibration buffer (6 M urea, 30% (v/v) glycerol, 2% (w/v) SDS, 50 mM Tris HCl pH 8.8, 0.002% (w/v) bromophenol blue) supplemented with 1% (w/v) DTT followed by another 15 min incubation with equilibration buffer containing 4.8% (w/v) iodoacetamide. The IPG strips were placed on top of 12% resolving gels and SDS-PAGE was performed as described above. Gels were fixed, stained with Coomassie Brilliant Blue overnight and destained using standard procedures. Proteins were visualised using a Fujifilm system as described above.
2D DIGE: acetone precipitation and determination of protein concentration.
Protein was extracted by acetone precipitation overnight at −20°C before centrifugation at 15,000 g for 10 minutes at 4°C. The supernatants were discarded and pellets were washed again with 80% (v/v) acetone before centrifugation at 15,000 g for 10 minutes at 4°C, removal of the supernatant and air drying. The pellets were resuspended in 500 µl 30 mM Tris-HCl pH 8.8, vortexed for 1 hour and centrifuged at 15,000 g for 10 minutes at 4°C. To measure protein concentration, a modified Bradford assay was used in reference 88, with BSA as a standard. 2 µl of protein samples and standards and 10 µl 0.1 M HCl were mixed with 25% Protein Assay Dye Reagent Concentrate (BioRad) in a total volume of 1 ml. Samples were vortexed and left to stand for 15 min at room temperature. Absorbance at 595 nm was measured using a spectrophotometer (Thistle Scientific).
2D DIGE: fluorescent labelling with CyDyes.
CyDye DIGE Fluor minimal dyes (GE Healthcare) that had been reconstituted with DMF (≤0.005% H2O, ≤99.8% pure) were used at a concentration of 0.04 mM. A two dye DiGE designCitation49 was used for comparison where each sample was labelled with Cy-5 against a pooled standard labelled with Cy-£ run together on the same gel. For each sample, 1 µl of Cy-5 dye was added to 50 µg protein. And the pooled standard was prepared containing 50 µg protein from each sample and 10 µl Cy-3 dye. Samples were incubated on ice in the dark for exactly 30 minutes before the reaction was quenched by addition of 1 µl of 10 mM lysine to each sample except the pooled sample, to which 10 µl of 10 mM lysine was added. Samples were then incubated on ice for 10 minutes.
10 µg of each Cy-5-labelled sample was mixed with 10 µg of Cy-3-labelled pooled internal standard, 80% (v/v) acetone and 5 µl 1.5 M Tris pH 8.8 to make a total volume of 0.5 ml. Samples were incubated for an hour at room temperature then centrifuged for 10 minutes at 14,000 g. The supernatant was removed and the pellets were air dried for 3 minutes. Samples were resuspended in lysis buffer supplemented with 1% (w/v) DTT and 2% (v/v) ampholytes (pH 4–7) in a final volume of 70 µl and vortexed for two hours.
2D DIGE: IEF and SDS-PAGE.
70 µl samples containing 20 µg total protein were loaded using anodic cups onto re-swelled 24 cm linear pH 4–7 IPG strips. IEF was performed on an IPGphor system (Amersham Biosciences) for a total of 70 kVh. Strips were then equilibrated as described above. Second dimension SDS-PAGE was performed using an Ettan™ DALTtwelve system (Amersham Biosciences). Equilibrated strips were loaded on top of 12% large format polyacrylamide gels and electrophoresis was carried out at 5 W per gel for 30 minutes followed by 17 W per gel for 4 hours at 25°C.
2D DIGE gel imaging.
Gels were imaged using a Typhoon Variable Mode Imager (GE Healthcare/Amersham Biosciences) immediately after SDS-PAGE. Cy-3 images were scanned using a 532 nm laser and a 580 nm BP 30 emission filter. Cy-5 images were scanned using a 633 nm laser and a 670 nm BP 30 emission filter. Final images were acquired at 100 µm (pixel size) resolution and an appropriate photomultiplier tube voltage was chosen to avoid pixel saturation.
2D DIGE analysis.
Gel images were processed using Progenesis Samespots (Nonlinear Dynamics) software for spot detection and alignment first in automatic mode and then checked manually. Sot values were calculated automatically by the software and an Anova test was performed, and spots changing across all replicates and those with a p-value of < 0.05 and a power of >0.7 were chosen for analysis by mass spectrometry.
Spot excision and in-gel tryptic digestion.
Protein spots were picked from preparative gels containing 500 µg protein stained with SYPRO™ Ruby Protein Stain and imaged using a Typhoon Variable Mode Imager (GE Healthcare/Amersham Biosciences). Trypic digestion of proteins was performed on a ProGest Workstation (Genomic Solutions Ltd.,) using a ProGest robot according to the long trypsin digestion protocol. Protein spots were removed from the gel and placed in a 96 well microtitre plate. Gel plugs were equilibrated in 50 µl of 50 mM ammonium bicarbonate, reduced and alkylated with 10 mM DTT and 100 mM iodoacetamide and destained and dessicated with acetonitrile. 50 mM ammonium bicarbonate containing 5% (w/v) trypsin (Promega) was used to rehydrate the gel plugs and digest the proteins for 12 hours at 37°C. Following digestion peptides were eluted with 50% (v/v) acetonitrile, 0.1% (v/v) trifluoroacetic acid into a final volume of 50 µl, vacuum dried and re-suspended in 10 µl 0.1% (v/v) formic acid for mass spectrometer analysis.
Mass spectrometry.
MALDI-ToF-ToF mass spectrometry was performed on a 4800 Plus MALDI TOF/TOF Analyser (Applied Biosystems, Warrington, UK). 1 µl of matrix solution (saturated α-cyano-4-hydroxy-cinnamic acid in 50% (v/v) acetonitrile, 0.1% (v/v) trifluoroacetic acid and 10 mM ammonium acetate) was spotted onto the MALDI target. 1 µl peptide solution was then added to each position and left to dry for 1 hour. TOF-MS analysis was performed using automated data acquisition and processing with the Applied Biosystems 4000 series Explorer software (v3.5). Spectra were then noise-corrected, peak de-isotoped and internally calibrated. The eight most abundant precursor ions seen in each were selected for fragmentation and MS-MS analysis using a 1 kV CID fragmentation method.
Combined peak lists of MS and MS-MS data were generated by GPS Explorer software (v3.6 Applied Biosciences) and matched to theoretical trypsic digests of proteins in the NCBInr database (www.ncbi.nlm.nih.gov) using MASCOT software (v2.2, Matrix Science). A precursor mass tolerance of 50 ppm, a MS-MS tolerance of 0.2 Daa single missed cleavage, oxidised methionines and carboxylmethyl cysteines as potential modifications were parameters used in the search. Results were ranked by the MOWSE probability score,Citation50 with a score of >82 considered successful.
Disclosure of Potential Conflict of Interest
No potential conflicts of interest were disclosed.
Abbreviations
| 2D DiGE | = | 2D difference in-gel electrophoresis |
| 2-D GE | = | 2D gel electrophoresis |
| BSA | = | bovine serum albumin |
| CRC | = | colorectal cancer |
| KASH | = | klarsicht/ANC-1/syne homology |
| HRP | = | horseradish peroxidase |
| IEF | = | isoelectric focussing |
| IF | = | intermediate filament |
| LINC | = | linker of nucleoskeleton and cytoskeleton |
| MALDI ToF-ToF | = | matrix-assisted laser-desorption/ionization time-of-flight/time-of-flight |
| MS | = | mass spectrometry |
| NEM | = | N-ethylmaleimide |
| SDS-PAGE | = | sodium dodecyl sulphate polyacrylamide gel electrophoresis |
| SILAC | = | stable isotope labelling with amino acids in cell culture |
| SUN | = | Sad1 and UNC-84 homology |
| TG2 | = | tissue transglutaminase 2 |
Figures and Tables
Figure 1 Expression of lamin A in CRC cells causes increased cell motility. (A) Whole cell extracts of SW480/lamA and SW480/cntl cells were resolved on a 10% SDS-PA GE gel, transferred to nitrocellulose and probed with Jol2 (anti-lamin A/C) antibody. Actin antibody was used as a loading control. (B) Densitometric analysis of endogenous lamin A expression in SW480/lamA cells relative to SW480/cntl cells. Images were taken with Fujifilm Intelligent Dark Box II and relative densities were measured with Image J software. (C) Densitometric analysis of GFP-lamin A plus lamin A expression in SW480/lamA cells relative to lamin A expression in SW480/cntl cells were performed as above. Error bars in (B and C) show the standard error calculated in replicate experiments, *p < 0.05. (D) Representative phase-contrast images of the start and end time points of a cell wounding assay, in which scratch wounds were made in 100% confluent cultures of SW480/lamA or SW480/cntl cells and wound closure was subsequently measured over 24 h using a Zeiss Cell Imager. Scale bars = 20 µm. (E) The mean distance moved by cells over 24 hours in scratch-wounding assays was calculated from phase-contrast images. In each experiment, three wound locations were chosen for each of three biological replicates and images were taken every 15 minutes for 24 hours. Error bars represent the standard error calculated from the biological replicates. SW480/lamA cells moved 20% further than SW480/cntl cells in 24 hours (p < 0.05). (F) Immunoblot of whole cell extracts from SW480/lamA cells transfected with si-laminA (si-l) or scrambled negative control siRNA (si-c), were resolved on a 10% SDS-PA GE gel and probed with Jol2 (anti-lamin A/C) antibody. Anti-actin antibody was used as a loading control. Cell pellets were harvested at 24, 48, 72, 96 and 120 hours post transfection with siRNA. (G) Representative phase-contrast images of the start and end time points of a cell wounding assay, in which SW480/lamA cells were grown to 100% confluency and wounded at 96 hours post-transfection with si-lamin A or si-cntl. Wound closure was measured over 24 h using a Zeiss Cell Imager. Scale bars = 20 µm. (H) The mean distance moved by cells in 24 hours was calculated from the phase-contrast images. In each experiment, four wound locations were chosen for each of three biological replicates and images were taken every 15 minutes for 24 hour. Error bars represent the standard error calculated from the biological replicates performed for each siRNA type. Si-cntl cells moved 40% further than si-laminA cells over 24 h (p < 0.0005).
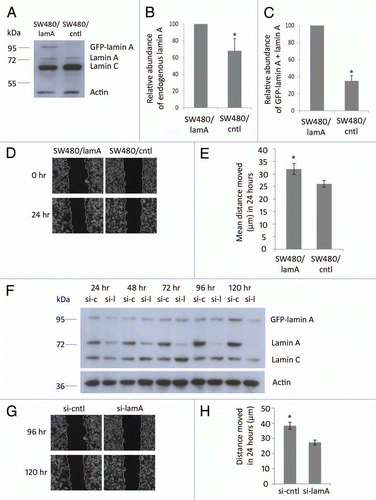
Figure 2 The membrane-bound protein vinculin is lost from detergent/high salt resistant N/CSK during biochemical fractionation of SW480 cells, whereas cytoskeletal proteins remain mostly insoluble. Pellets and supernatants were prepared from SW480/lamA and SW480/cntl cells at 70% confluency as described in materials and methods. Each fraction was resolved on 12% SDS-PAGE gels and transferred to nitrocellulose. Blots were probed with anti-actin (A and B), anti-α-tubulin (C and D), anti-keratin 18 (E and F) or anti-vinculin (G and H) antibodies.
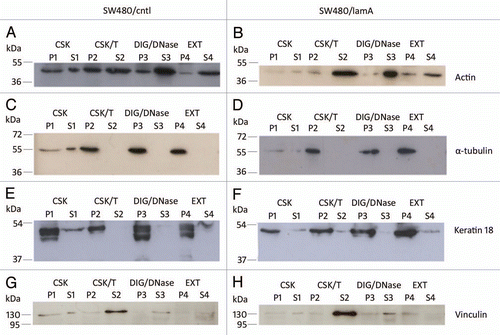
Figure 3 2D mini-format gels confirm reproducibility of the final cytoskeleton fraction prepared from SW480/lamA and SW480/ctl cells. Detergent/high salt resistant N/CSKs were isolated from SW480/lamA and SW480/cntl cells in six independent experiments. Samples were acetone precipitated and resolubilised in lysis buffer. 100 µg of protein was loaded using in-gel rehydration into 7 cm pH 4–7 IPG strips. Proteins were resolved in the first dimension using isoelectric focusing and in the second dimension using 10% mini-format SDS-PA GE gels. The resolved proteins were visualised using Coomassie blue staining. Images show mini-format gels run with replicate samples from SW480/cntl cells (A, C, E, G, I and K) and from SW480/lamA cells (B, D, F, H, J and L).

Figure 4 Differences in protein abundances in detergent/high salt resistant N/CSK isolated from SW480/lamA and SW480/cntl cells (A) 2-D DIGE gel comparing detergent/high salt resistant N/CSK from SW480/lamA and SW480/cntl cells. Arrows represent protein spots selected for analysis by MALDI-ToF-ToF. Differentially expressed spots were excised from a high protein load pick gel. Spots are annotated with their spot number for cross-referencing with Tables 1 and 2. Spots circled in red represent proteins over-represented in the fraction from SW480/lamA cells. Spots circled in green represent proteins under-represented in the fraction from SW480/lamA cells. (B) Cropped 2-D DIGE gel as above, annotated to show the identities of the proteins determined by MALDI-ToF-ToF.
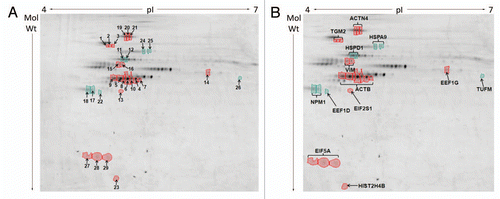
Table 1A Proteins over- and under-represented in detergent/ high salt resistant N/CSKs of SW480/lamA cells; (A) Proteins over-represented in the cytoskeleton preparations of SW480/lamA cells
Table 1B Proteins over- and under-represented in detergent/ high salt resistant N/CSKs of SW480/lamA cells; (B) Proteins under-represented in the cytoskeleton preparations of SW480/lamA cells
Additional material
Download Zip (11.5 MB)Acknowledgments
The authors are grateful to Dr. Naomi Willis for her advice in culturing SW480 cell lines. This work was supported by grants from the J.G.W. Patterson Foundation and from the South Teesside NHS Trust.
References
- Hutchison CJ, Worman HJ. A-type lamins: guardians of the soma?. Nat Cell Biol 2004; 6:1062
- Broers JL, Raymond Y, Rot MK, Kuijpers H, Wagenaar SS, Ramaekers FC. Nuclear A-type lamins are differentially expressed in human lung cancer subtypes. Am J Pathol 1993; 143:211 - 220
- Moss SF, Krivosheyev V, de Souza A, Chin K, Gaetz HP, Chaudhary N, et al. Decreased and aberrant nuclear lamin expression in gastrointestinal tract neoplasms. Gut 1999; 45:723 - 729
- Venables RS, McLean S, Luny D, Moteleb E, Morley S, Quinlan RA, et al. Expression of individual lamins in basal cell carcinomas of the skin. Br J Cancer 2001; 84:512 - 519
- Tilli CM, Ramaekers FC, Broers JL, Hutchison CJ, Neumann HA. Lamin expression in normal human skin, actinic keratosis, squamous cell carcinoma and basal cell carcinoma. Br J Dermatol 2003; 148:102 - 109
- Hudson ME, Pozdnyakova I, Haines K, Mor G, Snyder M. Identification of differentially expressed proteins in ovarian cancer using high-density protein microarrays. Proc Natl Acad Sci USA 2007; 104:17494 - 17499
- Willis ND, Cox TR, Rahman-Casans SF, Smits K, Przyborski SA, van den Brandt P, et al. Lamin A/C is a risk biomarker in colorectal cancer. PLoS One 2008; 3:2988
- Agrelo R, Setien F, Espada J, Artiga MJ, Rodriguez M, Perez-Rosado A, et al. Inactivation of the lamin A/C gene by CpG island promoter hypermethylation in hematologic malignancies, and its association with poor survival in nodal diffuse large B-cell lymphoma. J Clin Oncol 2005; 23:3940 - 3947
- Wu Z, Wu L, Weng D, Xu D, Geng J, Zhao F. Reduced expression of lamin A/C correlates with poor histological differentiation and prognosis in primary gastric carcinoma. J Exp Clin Cancer Res 2009; 28:8
- Foran E, McWilliam P, Kelleher D, Croke DT, Long A. The leukocyte protein L-plastin induces proliferation, invasion and loss of E-cadherin expression in colon cancer cells. Int J Cancer 2006; 118:2098 - 2104
- Perl AK, Wilgenbus P, Dahl U, Semb H, Christofori G. A causal role for E-cadherin in the transition from adenoma to carcinoma. Nature 1998; 392:190 - 193
- Starr DA, Han M. ANChors away: an actin based mechanism of nuclear positioning. J Cell Sci 2003; 116:211 - 216
- Crisp M, Liu Q, Roux K, Rattner JB, Shanahan C, Burke B, et al. Coupling of the nucleus and cytoplasm: role of the LINC complex. J Cell Biol 2006; 172:41 - 53
- Haque F, Lloyd DJ, Smallwood DT, Dent CL, Shanahan CM, Fry AM, et al. SUN1 interacts with nuclear lamin A and cytoplasmic nesprins to provide a physical connection between the nuclear lamina and the cytoskeleton. Mol Cell Biol 2006; 26:3738 - 3751
- Zhen YY, Libotte T, Munck M, Noegel AA, Korenbaum E. NUANCE, a giant protein connecting the nucleus and actin cytoskeleton. J Cell Sci 2002; 115:3207 - 3222
- Wilhelmsen K, Litjens SH, Kuikman I, Tshimbalanga N, Janssen H, van den Bout I, et al. Nesprin-3, a novel outer nuclear membrane protein, associates with the cytoskeletal linker protein plectin. J Cell Biol 2005; 171:799 - 810
- Mislow JM, Holaska JM, Kim MS, Lee KK, Segura-Totten M, Wilson KL, et al. Nesprin-1alpha self-associates and binds directly to emerin and lamin A in vitro. FEBS Lett 2002; 525:135 - 140
- Libotte T, Zaim H, Abraham S, Padmakumar VC, Schneider M, Lu W, et al. Lamin A/C-dependent localization of Nesprin-2, a giant scaffolder at the nuclear envelope. Mol Biol Cell 2005; 16:3411 - 3424
- Dyer JA, Kill IR, Pugh G, Quinlan RA, Lane EB, Hutchison CJ. Cell cycle changes in A-type lamin associations detected in human dermal fibroblasts using monoclonal antibodies. Chromosome Res 1997; 5:383 - 394
- Boisvert FM, Lamond AI. p53-Dependent subcellular proteome localization following DNA damage. Proteomics 2010; 10:4087 - 4097
- Nowak D, Krawczenko A, Dus D, Malicka-Blaszkiewicz M. Actin in human colon adenocarcinoma cells with different metastatic potential. Acta Biochim Pol 2002; 49:823 - 828
- Wang W, Goswami S, Lapidus K, Wells AL, Wyckoff JB, Sahai E, et al. Identification and testing of a gene expression signature of invasive carcinoma cells within primary mammary tumors. Cancer Res 2004; 64:8585 - 8594
- Honda K, Yamada T, Hayashida Y, Idogawa M, Sato S, Hasegawa F, et al. Actinin-4 increases cell motility and promotes lymph node metastasis of colorectal cancer. Gastroenterology 2005; 128:51 - 62
- Kikuchi S, Honda K, Tsuda H, Hiraoka N, Imoto I, Kosuge T, et al. Expression and gene amplification of actinin-4 in invasive ductal carcinoma of the pancreas. Clin Cancer Res 2008; 14:5348 - 5356
- Yamamoto S, Tsuda H, Honda K, Onozato K, Takano M, Tamai S, et al. Actinin-4 gene amplification in ovarian cancer: a candidate oncogene associated with poor patient prognosis and tumor chemoresistance. Mod Pathol 2009; 22:499 - 507
- Nagaraja GM, Othman M, Fox BP, Alsaber R, Pellegrino CM, Zeng Y, et al. Gene expression signatures and biomarkers of noninvasive and invasive breast cancer cells: comprehensive profiles by representational difference analysis, microarrays and proteomics. Oncogene 2006; 25:2328 - 2338
- McInroy L, Maatta A. Downregulation of vimentin expression inhibits carcinoma cell migration and adhesion. Biochem Biophys Res Commun 2007; 360:109 - 114
- Nemes Z Jr, Adany R, Balazs M, Boross P, Fesus L. Identification of cytoplasmic actin as an abundant glutaminyl substrate for tissue transglutaminase in HL-60 and U937 cells undergoing apoptosis. J Biol Chem 1997; 272:20577 - 20583
- Orru S, Caputo I, D'Amato A, Ruoppolo M, Esposito C. Proteomics identification of acyl-acceptor and acyldonor substrates for transglutaminase in a human intestinal epithelial cell line. Implications for celiac disease. J Biol Chem 2003; 278:31766 - 31773
- Robinson NJ, Baker PN, Jones CJ, Aplin JD. A role for tissue transglutaminase in stabilization of membrane-cytoskeletal particles shed from the human placenta. Biol Reprod 2007; 77:648 - 657
- Gupta M, Greenberg CS, Eckman DM, Sane DC. Arterial vimentin is a transglutaminase substrate: a link between vasomotor activity and remodeling?. J Vasc Res 2007; 44:339 - 344
- Cappello F, David S, Rappa F, Bucchieri F, Marasa L, Bartolotta TE, et al. The expression of HSP60 and HSP10 in large bowel carcinomas with lymph node metastase. BMC Cancer 2005; 5:139
- Dundas SR, Lawrie LC, Rooney PH, Murray GI. Mortalin is overexpressed by colorectal adenocarcinomas and correlates with poor survival. J Pathol 2005; 205:74 - 81
- Lim MJ, Wang XW. Nucleophosmin and human cancer. Cancer Detect Prev 2006; 30:481 - 490
- Gache V, Louwagie M, Garin J, Caudron N, Lafanechere L, Valiron O. Identification of proteins binding the native tubulin dimer. Biochem Biophys Res Commun 2005; 327:35 - 42
- Staubach S, Razawi H, Hanisch FG. Proteomics of MUC1-containing lipid rafts from plasma membranes and exosomes of human breast carcinoma cells MCF-7. Proteomics 2009; 9:2820 - 2835
- Sandsmark DK, Zhang H, Hegedus B, Pelletier CL, Weber JD, Gutmann DH. Nucleophosmin mediates mammalian target of rapamycin-dependent actin cytoskeleton dynamics and proliferation in neurofibromin-deficient astrocytes. Cancer Res 2007; 67:4790 - 4799
- Yi H, Li XH, Yi B, Zheng J, Zhu G, Li C, et al. Identification of Rack1, EF-Tu and Rhodanese as aging-related proteins in human colonic epithelium by proteomic analysis. J Proteome Res 2010; 9:1416 - 1423
- Taylor CA, Sun Z, Cliche DO, Ming H, Eshaque B, Jin S, et al. Eukaryotic translation initiation factor 5A induces apoptosis in colon cancer cells and associates with the nucleus in response to tumour necrosis factor alpha signalling. Exp Cell Res 2007; 313:437 - 449
- Rosenwald IB, Pechet L, Han A, Lu L, Pihan G, Woda B, et al. Expression of translation initiation factors elF-4E and elF-2alpha and a potential physiologic role of continuous protein synthesis in human platelets. Thromb Haemost 2001; 85:142 - 151
- Ogawa K, Utsunomiya T, Mimori K, Tanaka Y, Tanaka F, Inoue H, et al. Clinical significance of elongation factor-1 delta mRNA expression in oesophageal carcinoma. Br J Cancer 2004; 91:282 - 286
- Mimori K, Mori M, Inoue H, Ueo H, Mafune K, Akiyoshi T, et al. Elongation factor 1 gamma mRNA expression in oesophageal carcinoma. Gut 1996; 38:66 - 70
- Jufvas A, Stralfors P, Vener AV. Histone variants and their post-translational modifications in primary human fat cells. PLoS One 2011; 6:15960
- Ngan CY, Yamamoto H, Seshimo I, Tsujino T, Man-i M, Ikeda JI, et al. Quantitative evaluation of vimentin expression in tumour stroma of colorectal cancer. Br J Cancer 2007; 96:986 - 992
- Miettinen PJ, Ebner R, Lopez AR, Derynck R. TGF-beta induced transdifferentiation of mammary epithelial cells to mesenchymal cells: involvement of type I receptors. J Cell Biol 1994; 127:2021 - 2036
- Zavadil J, Bitzer M, Liang D, Yang YC, Massimi A, Kneitz S, et al. Genetic programs of epithelial cell plasticity directed by transforming growth factor-beta. Proc Natl Acad Sci USA 2001; 98:6686 - 6691
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970; 227:680 - 685
- Ramagli LS, Rodriguez LV. Quantitation of microgram amounts of protein in two-dimensional polyacrylamide gel electrophoresis sample buffer. ELECTROPHORESIS 1985; 6:559 - 563
- Karp NA, Lilley KS. Investigating sample pooling strategies for DIGE experiments to address biological variability. Proteomics 2009; 9:388 - 397
- Pappin DJ, Hojrup P, Bleasby AJ. Rapid identification of proteins by peptide-mass fingerprinting. Curr Biol 1993; 3:327 - 332