Abstract
Polyglutamine (PolyQ)-related diseases are dominant late-onset genetic disorders that are manifested by progressive neurodegeneration, leading to behavioral and physical impairments. An increased body of evidence suggests that chromatin structure and epigenetic regulation are involved in disease pathology. PolyQ diseases often display an aberrant transcriptional regulation due to the disrupted function of histone-modifying complexes and altered interactions of the polyQ-extended proteins with chromatin-related factors. In this review we describe recent findings relating to the role of chromatin in polyQ diseases. We discuss the involvement of epigenetic-related factors and chromatin structure in genomic instability of CAG repeats; we describe changes in the expression and regulation of chromatin-related enzymes and in the levels and patterns of histone modifications in disease state; we illustrate the potential beneficial effects of different histone deacetylase (HDAC) inhibitors for the treatment of polyQ diseases, and we end by describing the potential use of human pluripotent stem cells and their differentiated derivatives for modeling polyQ diseases in vitro. Taken together, these accumulating studies strongly suggest that disrupted chromatin regulation may be directly involved with the pathophysiology of polyQ-related diseases.
Introduction
Polyglutamine-expansion disease family encompasses at least nine heritable disorders, including Huntington disease (HD) and the spinocerebellar ataxias SCA1, SCA2, SCA3, SCA6, SCA7 and SCA17 (reviewed in ref. Citation1). Each of these disorders results from the expansion of a CAG repeat, coding for a glutamine tract (polyQ) that is present in the wild-type protein. While in healthy individuals the polyQ tract varies between 35–50 repeats, depending on the disease, in patients or carriers, the polyQ tract reaches above 40 and often over 100 glutamines. These repeat expansion mutations are unstable, resulting in changes in repeat length between generations as well as between different cells and tissues of the same person. Curiously, polyQ-containing proteins are ubiquitously expressed throughout the body, while the pathology is primarily restricted to neuronal tissue. The mechanisms that are responsible for this specificity are largely unknown.
Recent studies provided strong evidence for the involvement of chromatin in the etiology of polyQ neurodegenerative pathogenesis. Chromatin, the eukaryotic scaffold for gene regulation, is primarily regulated by chemical modifications of histones, which are the basic structural proteins around which the genomic DNA is wrapped (see Box 1 for details). Histone modifications play essential roles in the regulation of chromatin structure by preventing or facilitating transcription. Notably, accumulating data suggest a potential therapeutic role for drugs that target chromatin, such as histone deacetylase (HDAC) inhibitors, in a number of polyQ models.
Box 1. Chromatin structure and function.
The genome in eukaryotic cells is packaged in the form of chromatin. Chromatin consists primarily of DNA and histone proteins. The fundamental unit of chromatin is the nucleosome, which is comprised of 146 bp of DNA wrapped around an histone octamer made up of 2 copies each of the core histones, H2A, H2B, H3 and H4. Protruding from the nucleosome are the histone N-terminal tails, which can be modified on multiple residues. The control of eukaryotic gene expression largely depends on histone tail modifications. These include (but not limited to) acetylation, methylation, phosphorylation, ubiquitination and sumoylation, all of which play important roles in the regulation of gene expression by influencing chromatin structure and by recruiting regulatory proteins and enzymes. Some modifications, such as histone acetylation, are strongly associated with gene activation, while others (e.g., H3 lysine 9 methylation) are associated with heterochromatin and gene suppression.
In this review we discuss different aspects of epigenetic-related mechanisms involved in the etiology of polyQ-related diseases: epigenetics of genomic instability; the histone milieu of the expanded repeat tract, changes in histone modifications and histone-modifying complexes, potential use of drugs that target chromatin, and finally, human pluripotent stem cell models as promising tools to study polyQ diseases in vitro.
Instability and Chromatin Structure in polyQ Diseases
Genomic instability is triggered by the tendency of trinucleotide repeats to form alternative structures, such as hairpins and slipped-strand duplexes. These abnormal structures, which lead to changes in repeat length, can occur during DNA replication, repair, recombination and transcription.Citation2
CAG repeat expansions occur during multiple stages of human development, both in the germline and in the soma. Long CAG repeats, resulting in long polyQ tracts disrupt neuronal function and exacerbate disease pathology. The dynamics of instability often differs in the male and female germline, and typically varies from tissue to tissue. In HD for example, large expansions occur mostly through paternal transmissions. The repeat length will usually determine the age of onset and disease severity, a correlation known as “anticipation.”
What are the mechanisms that affect repeat instability? Multiple studies in recent years suggest the involvement of bidirectional transcription (simultaneous sense and antisense transcription, a.k.a., convergent transcription), RNA interference (RNAi) machinery, and chromatin structure. Bidirectional transcription, through a long CAG tract, produces double stranded repeat RNA, which can be processed by the DICER pathway into single strand short CAG repeat RNAs (sCAGs). These sCAGs can form RNA hairpins, which can inhibit transcription by promoting formation of heterochromatin, which, in turn, can affect instability.Citation3,Citation4 Although the exact mechanism of RNAi-mediated silencing in human cells is not entirely clear, it was shown previously to involve promoter DNA methylationCitation5 and the action of Argonaute-1,Citation6 which likely also participate in the case of triple nucleotide repeat silencing. Bidirectional transcription that produces double stranded RNA of a long CAG repeat was also shown to induce stress response that leads to apoptosisCitation7 and was proven pathogenic in animal models of expanded polyQ diseases.Citation8 Together, these studies suggest that a complex interplay between bidirectional transcription, RNAi, and chromatin structure influences repeat instability.
Repeat instability is also affected by cis-regulatory elements and trans-acting factors. One example is the binding of CTCF to its target sequence (CCCTC). In SCA7 (see below) transgenic mice with CTCF binding-site mutations, CTCF binding is impaired, leading to increased somatic repeat instability. Instability was associated with DNA hypermethylation around the CTCF binding site at CpG loci, implying a role for epigenetic regulation in repeat instabilityCitation9 ().
Figure 1. Chromatin involvement in repeat instability. Some chromatin-related mechanisms that lead to repeat instability are depicted. Bidirectional transcription of long CAG repeats generates double stranded RNA, which is processed by the RNAi machinery into short RNAs that form hairpins. Those short CAG RNAs promote heterochromatin production, resulting in transcription inhibition (Refs. Citation3 and Citation4). Double stranded RNA of CAG repeats can also induce apoptosis, leading to polyQ-related pathology (Refs. Citation7 and Citation8). Perturbed CTCF binding and low Dnmt1 expression levels were also reported to promote instability (Refs. Citation9 and Citation10).
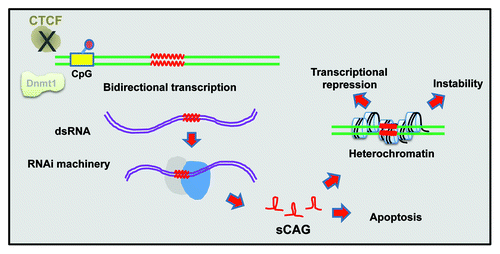
The involvement of DNA methylation in polyQ-related repeat instability was further demonstrated by knocking out one allele of the DNA methyltransferase Dnmt1 in a SCA1 (see below) mouse model. Partial Dnmt1 deficiency caused a significant increase in intergenerational expansions in the germline, without affecting somatic tissues. In addition, these mice displayed aberrant DNA methylation and elevated H3 lysine 9 (H3K9) methylation at sites within the CpG island around the expanded repeat tract, demonstrating a connection between local chromatin environment and repeat instability.Citation10
Repeat instability is also affected by the histone acetyltransferase (HAT) CBP, which was shown to interact with polyQ proteins in several polyQ diseases.Citation11 CBP depletion or inhibition contributes to polyQ pathology, and enhances repeat instability in SCA3 (see below) and HD fly models. Treatment with the HDAC inhibitor (HDACi) trichostatin A (TSA) suppressed repeat instability by resumption of acetylation levels.Citation12 Along the same lines, a screen for proteins involved in repeat expansion instability identified several HDACs, including HDAC3 that promoted expansion. HDAC3 inhibition, using either small molecules or RNAi, was able to partially relieve the expansion-associated gene silencing in vitro in both yeast and human cells, and to suppress somatic expansions that contribute to disease progression.Citation13
Taken together, these studies demonstrate the involvement of epigenetic regulation, both locally and globally, in repeat instability vis-à-vis disease pathology and progression.
Histone Modifications in polyQ Diseases
As indicated above, chromatin structure is greatly influenced by histone modifications. Histones can be methylated, acetylated, phosphorylated, ubiquitinated and sumoylated on multiple residues through the action of specialized enzymes. Several modifications, such as H3 acetylated on lysine 9 (H3K9ac) or H3K4me3, are correlated with active chromatin, while other modifications, e.g., H3K9me3 or H4K20me3 are associated with inactive chromatin.Citation14 The interplay between the different histone modifier complexes determine chromatin structure and function (Box 1).
Huntington’s disease
HD is a late-onset autosomal dominant neurodegenerative disorder, characterized by involuntary movements, progressive cognitive decline and emotional lability. The disorder is caused by an expanded CAG repeat (> 36) in the first exon of the HTT gene. Despite the expression of the mutant HTT protein in essentially all brain regions, there is a specific progressive loss of cortical and striatal neurons. Although neuronal death mechanisms are not entirely understood, transcriptional dysregulation and aberrant chromatin remodeling are central features in HD pathology,Citation15 and may provide some explanations. In support, the WT HTT protein interacts with a number of transcription factors and histone modifiers, including CBP, SP1, TBP, p300, Sin3a, and REST/NRSF.Citation16,Citation17
Most of what we know about HD in general, and particularly about the involvement of chromatin modifiers, comes from studies in animal models. Animal models, especially mouse and fly, recapitulate to a large extent, the behavioral, biochemical and neuropathological phenotypes that are characteristic of HD.Citation18,Citation19 Two of the most widely characterized HD transgenic mice are the R6/2 and N171–82Q (82Q) models, which express different lengths of the Htt N-terminal gene, containing 150 and 171 CAG repeats respectively.Citation20,Citation21 As noted above, CBP plays an important role in HD. The stronger interaction of CBP with the mutant HTT leads to CBP depletion and to hypoacetylation of histones H3 and H4 in brains of R6/2 mice.Citation22,Citation23This hypoacetylation is considered to occur late in HD pathology.Citation24 Moreover, monoallelic deletion of CBP triggers the induction of the H3K9 histone methyltransferase ESET and an increase in H3K9me3, leading to condensation of pericentromeric heterochromatin in neurons.Citation25 CBP depletion was also shown to be associated with cognitive dysfunction and long-term memory deficiency in HD.Citation26 In agreement, in a Drosophila model of HD, which contains the first 548 amino acids of the human HTT gene with a pathogenic 128 polyQ repeat tract,Citation27 upregulation of CBP expression rescued HD phenotypes.Citation28 However, partial depletion of CBP in HD transgenic mice did not contribute to HD pathogenesis, showing no clear impact on striatal degeneration, motor impairment, mutant HTT aggregation, or global levels of acetylated histones H3 or H4 in the brain.Citation29 Therefore, further studies are required to delineate the role of CBP in HD and decipher whether the different models used in these studies account for the seemingly contrasting results.
Aberrant histone modifications in HD are not restricted to acetylation and methylation. Monoubiquitination of histone H2A (uH2A), which is associated with gene repression,Citation30 was also shown to be implicated. Mutant HTT has been shown to mediate H2A monoubiquitination through a disrupted interaction with Bmi1, a component of the hPRC1L E3 ubiquitin ligase complex. This disrupted interaction leads to increased uH2A levels in a cell culture model system of HD and in R6/2 mice. Promoters of repressed genes in these mice show increased uH2A levels, while expressed genes display the opposite trend.Citation31 However, crossing the R6/2 mice with mice heterozygous for the polyubiquitin gene (Ubc), prevented uH2A accumulation, and no transcriptional and neurological improvements were recorded.Citation32 These data suggest a complex involvement of the ubiquitin system in the pathology of HD, which warrants further investigation.
Finally, an important factor that was implicated in the transcriptional dysregulation in HD is REST, which represses neuronal genes in non-neuronal tissues including undifferentiated neuronal progenitors, by binding to a DNA sequence element known as RE1.Citation33,Citation34 Intriguingly, in HD neurons, REST translocates from the cytoplasm to the nucleus, where it recruits several chromatin-modifying enzymes, inducing the repression of key target genes such as BDNF.Citation35-Citation38
Taken together, these data demonstrate the important role that histone modifying enzymes and histone modifications play in different stages of HD.
Ataxias
Spinocerebellar ataxia type 1 (SCA1) is a dominant neurodegenerative disease resulting from polyQ expansion (> 39) in the ataxin-1 (ATXN1) gene. ATXN1 interacts directly with Tip60, the catalytic subunit of a HAT complex, which is involved in transcriptional activation by acetylating H4 and H2A. Partial loss of Tip60, obtained by crossing SCA1–82Q transgenic mice with Tip60+/− mice, delayed ATXN1-mediated cerebellar degeneration during mid-stage disease progression.Citation39 These results suggest that Tip60 plays a transient role during SCA1 disease progression, although the exact mechanisms are not entirely understood.
Machado-Joseph disease (MJD; SCA3) is a dominantly inherited late-onset neurodegenerative disorder caused by polyQ expansion in the deubiquitinating enzyme ATXN3. Expanded polyQ fragments promote the generation of ATXN3-containing aggregates in brains of MJD patients and MJD mouse models. The formation of early aggregation intermediates is thought to have a critical role in disease initiation, but the precise pathogenic mechanism operating in MJD has remained largely elusive. Interestingly, ATXN3, both wild-type and mutant, was shown to interact with the histone acetyltransferases CBP, p300 and PCAF, and to inhibit transcriptional activation by these proteins,Citation40 linking it directly with chromatin regulation. It should also be noted that ATXN3 binds HDAC6,Citation41,Citation42 but since HDAC6 is a cytoplasmic HDAC which acts mostly on tubulin and does not bind histones in vivo, the relation to chromatin is indirect, and is perhaps confined to its inhibition by HDACi.
SCA7, caused by ATXN7 expansion, is characterized by cone-rod retinal degeneration, progressive neuronal dysfunction and cerebellar cell death. ATXN7 protein, both normal and polyQ-expanded, is a component of the mammalian SAGA and SLIK HAT complexes.Citation43 PolyQ-expanded ATXN7 disrupts the HAT activity of its associated complexes,Citation44 leading to retinal degeneration in SCA7 transgenic mice models.Citation45,Citation46 In addition, in a transgenic mouse model expressing polyQ-expanded ATXN7 in rod photoreceptors, the rod cells nuclei showed chromatin decondensation due to altered distribution of the linker histone H1c.Citation47 The loss of SAGA function by polyQ-ATXN7 can affect the time of onset and severity of SCA7 phenotypesCitation48 ().
Figure 2. Chromatin involvement in spinocerebellar ataxia type 7 (SCA7). CTCF binding at the promoter region of the ATXN7 gene promotes H3K27 methylation and antisense transcription from an alternative promoter. Impaired binding of CTCF abolishes H3K27 methylation and suppresses antisense transcription, along with induced polyQ-ATXN7 sense transcription, leading to disease phenotypes (Ref. Citation49). In addition, the ATXN7 protein (blue) is a component of both the SAGA and the SLIK HAT complexes. PolyQ-ATXN7 protein disrupts HAT activity of those complexes, resulting in chromatin disruption, retinal degeneration and SCA7 disease (Refs. Citation43Citation46).
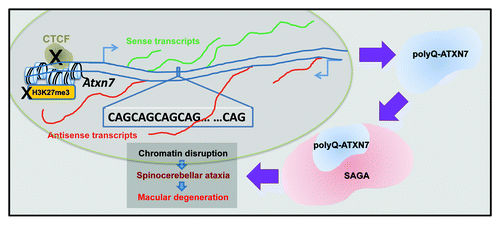
As noted above, the ATXN7 gene contains binding sites for CTCF. In addition to its effect on repeat instability as discussed earlier, CTCF binding promotes transcription of antisense noncoding RNA from an alternative promoter and mediates high levels of the repressive histone mark H3K27me3. Mice harboring mutations in the CTCF-recognition region of polyQ-Atxn7 demonstrated that the loss of antisense transcription induced Atxn7 sense transcription accompanied by reduced H3K27me3 and increased H3K9/H3K14 acetylation levels, leading to disease formation.Citation49 These data show the multifaceted contribution of the mutated ATXN7 to the pathology of SCA7, involving bidirectional transcription, altered chromatin regulation, distorted SAGA activity and dysregulated transcription of target genes in the nervous system. It also warrants investigation of potential similar mechanisms in other polyQ diseases, such as HD, in which a CTCF binding site in the HTT gene was identified,Citation50 and where an HTT antisense transcript was shown to regulate HTT transcription.Citation50
SCA8 is a late onset slowly progressing ataxia. The pathogenesis of SCA8 is complex and the clinical symptoms are broad. SCA8 involves a (CAG)n repeat in the ATXN8 gene (80–250 repeats), generating the rogue polyQ ATXN8 protein, and a (CTA)n (CTG)n trinucleotide repeat expansion in the ATXN8OS gene on the opposite strand, producing a non-coding RNA.Citation51,Citation52 The ATXN8 large triplet expansions (> 157) trigger H3K9 hypermethylation, H3K14 hypoacetylation and accumulation of repeat-expanded RNA in nuclear foci. These data demonstrate the intricate interplay between coding and non-coding transcription, and suggest the involvement of altered chromatin conformation in the regulation of ATXN8 expression. This example joins the growing list of cases, a few of which are depicted above, in which chromatin structure and epigenetic mechanisms are involved in the regulation of polyQ-expanded protein function. It was therefore natural to consider chromatin-directed compounds as potential drugs for polyQ-related diseases.
HDAC inhibitors as potential therapy for polyQ diseases
HDAC enzymes are divided into four different classes: class I (HDAC1, 2, 3 and 8), class IIa (HDAC4, 5, 7 and 9), class IIb (HDAC6 and 10), class III (SIRT1–7) and class IV (HDAC11). Classes I, II and IV are zinc-dependent, while the class III Sirtuins use NAD+ as a cofactor for their catalytic activity.Citation53 HDAC inhibitors (HDACi) block HDAC activity, resulting in increased histone acetylation, although much like the HDACs themselves, the selectivity of the HDACi are relatively poor and most available HDACi target a mixture of different HDACs. Of these, several have been examined for their potential beneficial effects in polyQ disease models (). Below we provide some notable examples.
Table 1. Beneficial effects of HDAC inhibition or suppression in polyQ disease models
Suberoylanilide hydroxamic acid (SAHA) is a potent HDAC inhibitor which targets class I HDACs, as well as the class II HDAC6, and which crosses the blood-brain-barrier (BBB).Citation54-Citation56 In the R6/2 HD mouse model, SAHA was shown to increase histone acetylation in the brain, and improve motor impairments.Citation57 Interestingly, SAHA chronic administration into the drinking water of R6/2 HD mice leads to HDAC2 and HDAC4 protein degradation, although their transcript levels remain unaltered. Moreover, in the same mouse model, Htt aggregation in the cortex and brain stem is reduced, along with normalization of Bdnf cortical transcript levels.Citation58 In another study, SAHA inhibited the HDAC3-dependent generation of neurotoxic metabolites by mutant Htt in both yeast and in microglia of R6/2 mice.Citation59 In light of these findings, SAHA seems to be a promising agent for the treatment of polyQ diseases, although because of its poor selectivity, it is not clear which of the different HDACs is responsible for the effects.
The HDACi Sodium Butyrate (NaBu), which inhibits HDAC1, 2, 4, 5 and 7, and akin to SAHA, also crosses the BBB, was shown to have beneficial effects in an HD and SCA3 mouse models. In SCA3 transgenic mice, NaBu was shown to reverse polyQ-ATXN3-mediated histone hypoacetylation and to improve neurological symptoms and survival of the mice.Citation60 In R6/2 HD mice, NaBu treatment increased life-expectancy and improved motor and neural symptoms. NaBu increased acetylation of histones and of the Sp1 transcription factor and restored the otherwise reduced expression of α and β globins as well as MKP1 in R6/2 mice.Citation22 This suggests that HDACi-mediated increased acetylation may relieve polyQ-mediated suppression of target genes. However, global changes in the levels of histone acetylation affect many other genes, likely causing additional, less beneficial effects.
Supporting this notion, several studies demonstrate the complexity of the relationship between histone modifications and gene expression changes. The HDAC inhibitor 4b shows therapeutic effects of motor, body-weight, brain and striatal degeneration in HD R6/2300Q transgenic mice. As expected, 4b reversed histone H3 hypoacetylation in association with restoration of expression levels of many transcripts, although interestingly, it also decreased the expression of specific genes associated with cell death, cell cycle, and the immune response.Citation61 Moreover, the non-toxic selective HDACi 4b-related compounds were found lately to specifically inhibit HDAC3 and HDAC1 effectively. This specific targeting led to an improvement of neurodegenerative effects in various cell culture, fly and mouse HD models.Citation62
In a Drosophila HD model, in which an N-terminal fragment of the human mutant (93Q) HTT is expressed in all neurons, Sir2 and Rpd3 (the mammalian HDAC3 ortholog) were found, among all the HDACs, to contribute to HD pathology. Reducing their levels individually in heterozygous flies, exerted neuroprotection effects, and their combined reduction augmented this observed protective effect further.Citation63 In contrast, in a mouse model of HD, mutant Htt binds Sirt1 (the mammalian Sir2 ortholog), inhibits its deacetylase activity, and leads to hyperacetylation of Sirt1 targets. Thus, increased Sirt1 level ameliorate motor impairment and neurodegeneration in mouse, and reduction of Sirt1 exacerbates mutant HTT toxicity.Citation64 This discrepancy is likely due to the different HD model systems used, and calls for investigating Sirt1 function in human models.
In addition to HDAC inhibitors, which have limited specificity and may have pleiotropic effects, the effects of targeting specific HDACs were also examined. Reducing the levels of HDAC3 or HDAC7 in R6/2 mice by crossing them with HDAC3+/− and HDAC7+/− heterozygous mice respectively, did not ameliorate HD neurodegenerative symptoms, although both HDAC3 and HDAC7 were shown to be affected by SAHA administration in the brains of R6/2 mice.Citation65,Citation66 Full knockout of HDAC6 in R6/2 mice also had no beneficial effects.Citation67 Thus, while SAHA and other HDACi may be promising agents in the treatment of polyQ diseases, their direct targets are usually still largely unknown, although as mentioned above, HDAC2 and HDAC4 are prime suspects in the case of HDAC inhibition by SAHA.Citation58
Taken together, these data demonstrate that although targeting HDACs in polyQ diseases is a promising avenue (), the lack of selective agents and the limited effect of targeting a specific HDAC diminish the overall initial enthusiasm.
To circumvent these issues, it may be necessary to examine a combinatorial approach of specific HDAC-targeting, or to develop more selective HDACi in order to fine-tune neuroprotection and improve neurodegenerative symptoms. In addition, the recently emerging human pluripotent model systems (see below) for studying neurodegenerative diseases are an important novel resource for studying disease mechanisms and ultimately for screening potential HDACi and other optional drugs.
Human pluripotent models for polyQ-related diseases
Studies of chromatin structure, repeat instability, RNA toxicity, polyQ function, neurodegeneration and disease phenotype improvement by chromatin-modifying enzymes have so far been performed, by and large, on other organisms than humans. The experimental models for neurodegenerative diseases based on other-than-human organisms such as flies and transgenic mice have many limitations in relation to disease mode characteristics, and often the results obtained in one model may not be recapitulated in another, let alone in humans. However, human tissue is a scarce reagent. Biopsies of human brain tissue are not easily accessible, and when available, neurons do not divide in vitro. Therefore, pluripotent, disease-bearing human cell lines offer great promise over existing models ().
Table 2. Phenotypes of polyQ-iPSC models
Pluripotent stem cells (PSCs) are defined by their ability to self-renew indefinitely in culture and to generate any cell type in vitro or in vivo. Human PSCs (hPSCs) can be derived from human in vitro fertilized (IVF) embryosCitation68 or reprogrammed from somatic cells using Oct4, Sox2 and Klf4 (with or without c-Myc) into so-called induced pluripotent stem cells (iPSC).Citation69 Both these pluripotent cellular systems can also be derived from patient cells. In the former case by using embryos which were found to contain a mutation in the now routine procedure of pre-implantation genetic diagnosis (PGD), and in the latter case using skin fibroblasts or lymphocytes (or any cell type that is available) from patients. Since polyQ diseases are late onset, and most of the polyQ diseases are dominant, and eventually lethal, there is a growing list of existing polyQ-containing PGD embryos, providing a powerful tool to model and study polyQ diseases in the lab. iPSCs offer a unique complementary platform, which is significantly easier to obtain, although changes between hESCs and iPSCs have been described at least for the fragile X (FX) syndrome.Citation70 In FX-hESCs derived from PGD embryos the FMR1 gene is active, and is silenced by promoter methylation and suppressive histone modifications upon differentiation. In contrast in FX-iPSCs derived from patient fibroblasts in which the FMR1 gene is heterochromatinized and silenced, FMR1 is not activated, and promoter methylation persists. These results suggest that while human pluripotent models offer unprecedented opportunities, each system must be carefully scrutinized to ensure that it properly recapitulates the relevant disease.
In addition, in order to study disease mechanisms in pluripotent cell models, they first must be differentiated into the relevant mature, post-mitotic, functional neuronal subtypes. Based on knowledge of embryogenesis and developmental studies, we can now use combinations of morphogens and neurotrophins to induce specific lineage committed cells, including neurons, from PSCs.Citation71 In HD, for example, loss of cortical and striatal neurons is the main pathological feature as disease progresses. Differentiation of pluripotent cells into striatal neurons has already been achieved, and their function was examined upon transplantation into rat or mouse brains.Citation72 The field of in vitro differentiation is continuously growing, and protocols for selective neuronal induction are constantly being established.Citation73-Citation76
iPSCs from a Rhesus monkey model of HD have recently been generated, characterized and differentiated into neurons. These monkey HD-iPSCs displayed characteristic features of HD, including the accumulation of mutant Htt aggregates and the formation of intranuclear inclusions upon neural differentiation in vitro,Citation77 suggesting that iPSC-derived neurons can be used successfully to model polyQ diseases.
More recently, several iPSC lines from HD patients and R6/2 transgenic mice were generated. Although CAG repeat expansion had no effect on reprogramming efficiency, proliferation rate, BDNF level, or neurogenic potential, the R6/2 derived iPSCs as well as their neuronal differentiated progeny showed increased lysosomal activity and altered cholesterol biosynthesis pathway, which are hallmarks of HD.Citation78,Citation79 In another study, human HD-iPSCs were differentiated into striatal neurons in a step-wise manner. The differentiating neurons showed enhanced caspase activity upon growth factor deprivation compared with normal differentiating cells.Citation80 In both these studies, the repeat length was shown to be stable and similar to that of the patients, and was not affected by the reprogramming or the differentiation process. In another recent study, HD-iPSCs displayed normal neuronal differentiation both in vitro and in vivo upon mouse brain transplantation, but interestingly, when differentiated into astrocytes, they showed progressive vacuolization, characteristic of HD astrocytes.Citation81 Finally, transplantation of neuronal precursors generated from HD-hiPSCs into the brain of an HD rat model, improved behavioral impairments associated with HD.Citation82
MJD has also been recently successfully modeled using iPSCs. Neurons generated from iPSCs of MJD patients revealed excitation-dependent accumulation of the ATXN3 aggregates. Glutamate-mediated neuronal induction caused calcium-induced proteolysis of ATXN3, leading to the formation of ATXN3 insoluble aggregates.Citation83 Taken together, these studies strongly support the idea that iPSCs can successfully model polyQ disorders () despite the late onset phenotypes observed in humans.
Despite the justified overwhelming enthusiasm for the use of pluripotent cell models to model and study diseases in vitro, caution should be practiced. Patient cell biopsies may differ significantly between one another; the passage of the cells may greatly influence the reprogramming process and the end result; additionally, since the iPSC derivation process itself is cumbersome, many variations among different lines may occur, including chromosomal aberrations,Citation70,Citation74,Citation84-Citation87 and it is therefore essential to compare several different lines; also, as is the case with the Fragile X example discussed above, hESCs and iPSCs may display major differences in chromatin structure and disease-related phenotypes; the passage of the pluripotent cells themselves may also create significant variation between experiments and lead to epigenetic instability, and finally, the lengthy neuronal differentiation process can also be a major cause for experimental variation and heterogeneity. Careful measures are therefore essential to ensure the creation of proper, reproducible pluripotent models.
Recently, several studies have demonstrated the ability to directly convert human fibroblasts into functional neurons without the need for pluripotent intermediates. In these studies, an expression of a combination of a small set (usually around 3) of defined transcription factors or microRNAs were sufficient to convert fibroblasts to neurons at an efficiency that greatly exceeds that of reprogramming fibroblasts to pluripotency.Citation88-Citation92 Since the pluripotent state is the Achilles heel of cell therapy, holding the terrifying potential to proliferate in vivo, the direct conversion approach (beyond the scope of this review) provides a promising alternative approach for regenerative medicine, and will no doubt attract significant attention is the years to come.
The use of human PSCs for regenerative medicine still faces some significant challenges in the clinical field, including safety, efficacy, and the establishment of the exact differentiation stage suitable for transplantation.Citation71 Regardless, human PSCs are already providing a unique and unmatched source for basic science. They are also gradually gaining momentum for the use of high-throughput drug screens, and for testing specific compounds, as demonstrated in several studies which focused on neuronal disorders.Citation93-Citation96 With the current pace, we anticipate that such reprogrammed or converted cells will become standard procedure for the treatment of neurodegenerative diseases in general and for polyQ disorders in particular.
Acknowledgments
We thank Dr Malka Nissim-Rafinia (Jerusalem) for comments; the Safra Foundation, the ISF Legacy-Heritage Bio-Medical Program (943/09 and 1252/12), the Israel Ministry of Science, the Israel Ministry of Health (6007) the Israel Cancer Research Foundation and the European Research Council (ERC-281781) for financial support. E.M. is the Joseph H. and Belle R. Braun senior Lecturer in Life Sciences. D.C.C. is an Eshkol Fellow of the Israel Ministry of Science.
References
- Orr HT, Zoghbi HY. Trinucleotide repeat disorders. Annu Rev Neurosci 2007; 30:575 - 621; http://dx.doi.org/10.1146/annurev.neuro.29.051605.113042; PMID: 17417937
- Liu Y, Wilson SH. DNA base excision repair: a mechanism of trinucleotide repeat expansion. Trends Biochem Sci 2012; 37:162 - 72; http://dx.doi.org/10.1016/j.tibs.2011.12.002; PMID: 22285516
- Dion V, Wilson JH. Instability and chromatin structure of expanded trinucleotide repeats. Trends Genet 2009; 25:288 - 97; http://dx.doi.org/10.1016/j.tig.2009.04.007; PMID: 19540013
- Grewal SI, Elgin SC. Transcription and RNA interference in the formation of heterochromatin. Nature 2007; 447:399 - 406; http://dx.doi.org/10.1038/nature05914; PMID: 17522672
- Morris KV, Chan SW, Jacobsen SE, Looney DJ. Small interfering RNA-induced transcriptional gene silencing in human cells. Science 2004; 305:1289 - 92; http://dx.doi.org/10.1126/science.1101372; PMID: 15297624
- Kim DH, Villeneuve LM, Morris KV, Rossi JJ. Argonaute-1 directs siRNA-mediated transcriptional gene silencing in human cells. Nat Struct Mol Biol 2006; 13:793 - 7; http://dx.doi.org/10.1038/nsmb1142; PMID: 16936726
- Lin Y, Leng M, Wan M, Wilson JH. Convergent transcription through a long CAG tract destabilizes repeats and induces apoptosis. Mol Cell Biol 2010; 30:4435 - 51; http://dx.doi.org/10.1128/MCB.00332-10; PMID: 20647539
- Lawlor KT, O’Keefe LV, Samaraweera SE, van Eyk CL, McLeod CJ, Maloney CA, et al. Double-stranded RNA is pathogenic in Drosophila models of expanded repeat neurodegenerative diseases. Hum Mol Genet 2011; 20:3757 - 68; http://dx.doi.org/10.1093/hmg/ddr292; PMID: 21724553
- Libby RT, Hagerman KA, Pineda VV, Lau R, Cho DH, Baccam SL, et al. CTCF cis-regulates trinucleotide repeat instability in an epigenetic manner: a novel basis for mutational hot spot determination. PLoS Genet 2008; 4:e1000257; http://dx.doi.org/10.1371/journal.pgen.1000257; PMID: 19008940
- Dion V, Lin Y, Hubert L Jr., Waterland RA, Wilson JH. Dnmt1 deficiency promotes CAG repeat expansion in the mouse germline. Hum Mol Genet 2008; 17:1306 - 17; http://dx.doi.org/10.1093/hmg/ddn019; PMID: 18252747
- Bodai L, Pallos J, Thompson LM, Marsh JL. Altered protein acetylation in polyglutamine diseases. Curr Med Chem 2003; 10:2577 - 87; http://dx.doi.org/10.2174/0929867033456530; PMID: 14529472
- Jung J, Bonini N. CREB-binding protein modulates repeat instability in a Drosophila model for polyQ disease. Science 2007; 315:1857 - 9; http://dx.doi.org/10.1126/science.1139517; PMID: 17332375
- Debacker K, Frizzell A, Gleeson O, Kirkham-McCarthy L, Mertz T, Lahue RS. Histone deacetylase complexes promote trinucleotide repeat expansions. PLoS Biol 2012; 10:e1001257; http://dx.doi.org/10.1371/journal.pbio.1001257; PMID: 22363205
- Nightingale KP, O’Neill LP, Turner BM. Histone modifications: signalling receptors and potential elements of a heritable epigenetic code. Curr Opin Genet Dev 2006; 16:125 - 36; http://dx.doi.org/10.1016/j.gde.2006.02.015; PMID: 16503131
- Benn CL, Sun T, Sadri-Vakili G, McFarland KN, DiRocco DP, Yohrling GJ, et al. Huntingtin modulates transcription, occupies gene promoters in vivo, and binds directly to DNA in a polyglutamine-dependent manner. J Neurosci 2008; 28:10720 - 33; http://dx.doi.org/10.1523/JNEUROSCI.2126-08.2008; PMID: 18923047
- Harjes P, Wanker EE. The hunt for huntingtin function: interaction partners tell many different stories. Trends Biochem Sci 2003; 28:425 - 33; http://dx.doi.org/10.1016/S0968-0004(03)00168-3; PMID: 12932731
- Li SH, Li XJ. Huntingtin-protein interactions and the pathogenesis of Huntington disease. Trends Genet 2004; 20:146 - 54; http://dx.doi.org/10.1016/j.tig.2004.01.008; PMID: 15036808
- Luthi-Carter R, Hanson SA, Strand AD, Bergstrom DA, Chun W, Peters NL, et al. Dysregulation of gene expression in the R6/2 model of polyglutamine disease: parallel changes in muscle and brain. Hum Mol Genet 2002; 11:1911 - 26; http://dx.doi.org/10.1093/hmg/11.17.1911; PMID: 12165554
- Stack EC, Kubilus JK, Smith K, Cormier K, Del Signore SJ, Guelin E, et al. Chronology of behavioral symptoms and neuropathological sequela in R6/2 Huntington’s disease transgenic mice. J Comp Neurol 2005; 490:354 - 70; http://dx.doi.org/10.1002/cne.20680; PMID: 16127709
- Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, et al. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell 1996; 87:493 - 506; http://dx.doi.org/10.1016/S0092-8674(00)81369-0; PMID: 8898202
- Schilling G, Becher MW, Sharp AH, Jinnah HA, Duan K, Kotzuk JA, et al. Intranuclear inclusions and neuritic aggregates in transgenic mice expressing a mutant N-terminal fragment of huntingtin. Hum Mol Genet 1999; 8:397 - 407; http://dx.doi.org/10.1093/hmg/8.3.397; PMID: 9949199
- Ferrante RJ, Kubilus JK, Lee J, Ryu H, Beesen A, Zucker B, et al. Histone deacetylase inhibition by sodium butyrate chemotherapy ameliorates the neurodegenerative phenotype in Huntington’s disease mice. J Neurosci 2003; 23:9418 - 27; PMID: 14561870
- Jiang H, Poirier MA, Liang Y, Pei Z, Weiskittel CE, Smith WW, et al. Depletion of CBP is directly linked with cellular toxicity caused by mutant huntingtin. Neurobiol Dis 2006; 23:543 - 51; http://dx.doi.org/10.1016/j.nbd.2006.04.011; PMID: 16766198
- Sadri-Vakili G, Bouzou B, Benn CL, Kim MO, Chawla P, Overland RP, et al. Histones associated with downregulated genes are hypo-acetylated in Huntington’s disease models. Hum Mol Genet 2007; 16:1293 - 306; http://dx.doi.org/10.1093/hmg/ddm078; PMID: 17409194
- Lee J, Hagerty S, Cormier KA, Kim J, Kung AL, Ferrante RJ, et al. Monoallele deletion of CBP leads to pericentromeric heterochromatin condensation through ESET expression and histone H3 (K9) methylation. Hum Mol Genet 2008; 17:1774 - 82; http://dx.doi.org/10.1093/hmg/ddn067; PMID: 18319327
- Giralt A, Puigdellívol M, Carretón O, Paoletti P, Valero J, Parra-Damas A, et al. Long-term memory deficits in Huntington’s disease are associated with reduced CBP histone acetylase activity. Hum Mol Genet 2012; 21:1203 - 16; http://dx.doi.org/10.1093/hmg/ddr552; PMID: 22116937
- Romero E, Cha GH, Verstreken P, Ly CV, Hughes RE, Bellen HJ, et al. Suppression of neurodegeneration and increased neurotransmission caused by expanded full-length huntingtin accumulating in the cytoplasm. Neuron 2008; 57:27 - 40; http://dx.doi.org/10.1016/j.neuron.2007.11.025; PMID: 18184562
- Taylor JP, Taye AA, Campbell C, Kazemi-Esfarjani P, Fischbeck KH, Min KT. Aberrant histone acetylation, altered transcription, and retinal degeneration in a Drosophila model of polyglutamine disease are rescued by CREB-binding protein. Genes Dev 2003; 17:1463 - 8; http://dx.doi.org/10.1101/gad.1087503; PMID: 12815067
- Klevytska AM, Tebbenkamp AT, Savonenko AV, Borchelt DR. Partial depletion of CREB-binding protein reduces life expectancy in a mouse model of Huntington disease. J Neuropathol Exp Neurol 2010; 69:396 - 404; http://dx.doi.org/10.1097/NEN.0b013e3181d6c436; PMID: 20448484
- Wang H, Wang L, Erdjument-Bromage H, Vidal M, Tempst P, Jones RS, et al. Role of histone H2A ubiquitination in Polycomb silencing. Nature 2004; 431:873 - 8; http://dx.doi.org/10.1038/nature02985; PMID: 15386022
- Kim MO, Chawla P, Overland RP, Xia E, Sadri-Vakili G, Cha JH. Altered histone monoubiquitylation mediated by mutant huntingtin induces transcriptional dysregulation. J Neurosci 2008; 28:3947 - 57; http://dx.doi.org/10.1523/JNEUROSCI.5667-07.2008; PMID: 18400894
- Bett JS, Benn CL, Ryu KY, Kopito RR, Bates GP. The polyubiquitin Ubc gene modulates histone H2A monoubiquitylation in the R6/2 mouse model of Huntington’s disease. J Cell Mol Med 2009; 13:8B 2645 - 57; http://dx.doi.org/10.1111/j.1582-4934.2008.00543.x; PMID: 19602042
- Chong JA, Tapia-Ramírez J, Kim S, Toledo-Aral JJ, Zheng Y, Boutros MC, et al. REST: a mammalian silencer protein that restricts sodium channel gene expression to neurons. Cell 1995; 80:949 - 57; http://dx.doi.org/10.1016/0092-8674(95)90298-8; PMID: 7697725
- Schoenherr CJ, Anderson DJ. The neuron-restrictive silencer factor (NRSF): a coordinate repressor of multiple neuron-specific genes. Science 1995; 267:1360 - 3; http://dx.doi.org/10.1126/science.7871435; PMID: 7871435
- Buckley NJ, Johnson R, Zuccato C, Bithell A, Cattaneo E. The role of REST in transcriptional and epigenetic dysregulation in Huntington’s disease. Neurobiol Dis 2010; 39:28 - 39; http://dx.doi.org/10.1016/j.nbd.2010.02.003; PMID: 20170730
- Ooi L, Wood IC. Chromatin crosstalk in development and disease: lessons from REST. Nat Rev Genet 2007; 8:544 - 54; http://dx.doi.org/10.1038/nrg2100; PMID: 17572692
- Zuccato C, Cattaneo E. Role of brain-derived neurotrophic factor in Huntington’s disease. Prog Neurobiol 2007; 81:294 - 330; http://dx.doi.org/10.1016/j.pneurobio.2007.01.003; PMID: 17379385
- Zuccato C, Cattaneo E. Brain-derived neurotrophic factor in neurodegenerative diseases. Nat Rev Neurol 2009; 5:311 - 22; http://dx.doi.org/10.1038/nrneurol.2009.54; PMID: 19498435
- Gehrking KM, Andresen JM, Duvick L, Lough J, Zoghbi HY, Orr HT. Partial loss of Tip60 slows mid-stage neurodegeneration in a spinocerebellar ataxia type 1 (SCA1) mouse model. Hum Mol Genet 2011; 20:2204 - 12; http://dx.doi.org/10.1093/hmg/ddr108; PMID: 21427130
- Li F, Macfarlan T, Pittman RN, Chakravarti D. Ataxin-3 is a histone-binding protein with two independent transcriptional corepressor activities. J Biol Chem 2002; 277:45004 - 12; http://dx.doi.org/10.1074/jbc.M205259200; PMID: 12297501
- Fiesel FC, Schurr C, Weber SS, Kahle PJ. TDP-43 knockdown impairs neurite outgrowth dependent on its target histone deacetylase 6. Mol Neurodegener 2011; 6:64; http://dx.doi.org/10.1186/1750-1326-6-64; PMID: 21878116
- Fiesel FC, Voigt A, Weber SS, Van den Haute C, Waldenmaier A, Görner K, et al. Knockdown of transactive response DNA-binding protein (TDP-43) downregulates histone deacetylase 6. EMBO J 2010; 29:209 - 21; http://dx.doi.org/10.1038/emboj.2009.324; PMID: 19910924
- Helmlinger D, Hardy S, Eberlin A, Devys D, Tora L. Both normal and polyglutamine- expanded ataxin-7 are components of TFTC-type GCN5 histone acetyltransferase- containing complexes. Biochem Soc Symp 2006; 73:155 - 63; PMID: 16626296
- McMahon SJ, Pray-Grant MG, Schieltz D, Yates JR 3rd, Grant PA. Polyglutamine-expanded spinocerebellar ataxia-7 protein disrupts normal SAGA and SLIK histone acetyltransferase activity. Proc Natl Acad Sci USA 2005; 102:8478 - 82; http://dx.doi.org/10.1073/pnas.0503493102; PMID: 15932941
- Helmlinger D, Hardy S, Abou-Sleymane G, Eberlin A, Bowman AB, Gansmüller A, et al. Glutamine-expanded ataxin-7 alters TFTC/STAGA recruitment and chromatin structure leading to photoreceptor dysfunction. PLoS Biol 2006; 4:e67; http://dx.doi.org/10.1371/journal.pbio.0040067; PMID: 16494529
- Palhan VB, Chen S, Peng GH, Tjernberg A, Gamper AM, Fan Y, et al. Polyglutamine-expanded ataxin-7 inhibits STAGA histone acetyltransferase activity to produce retinal degeneration. Proc Natl Acad Sci USA 2005; 102:8472 - 7; http://dx.doi.org/10.1073/pnas.0503505102; PMID: 15932940
- Kizilyaprak C, Spehner D, Devys D, Schultz P. The linker histone H1C contributes to the SCA7 nuclear phenotype. Nucleus 2011; 2:444 - 54; http://dx.doi.org/10.4161/nucl.2.5.17843; PMID: 21970987
- Chen YC, Gatchel JR, Lewis RW, Mao CA, Grant PA, Zoghbi HY, et al. Gcn5 loss-of-function accelerates cerebellar and retinal degeneration in a SCA7 mouse model. Hum Mol Genet 2012; 21:394 - 405; http://dx.doi.org/10.1093/hmg/ddr474; PMID: 22002997
- Sopher BL, Ladd PD, Pineda VV, Libby RT, Sunkin SM, Hurley JB, et al. CTCF regulates ataxin-7 expression through promotion of a convergently transcribed, antisense noncoding RNA. Neuron 2011; 70:1071 - 84; http://dx.doi.org/10.1016/j.neuron.2011.05.027; PMID: 21689595
- Chung DW, Rudnicki DD, Yu L, Margolis RL. A natural antisense transcript at the Huntington’s disease repeat locus regulates HTT expression. Hum Mol Genet 2011; 20:3467 - 77; http://dx.doi.org/10.1093/hmg/ddr263; PMID: 21672921
- Ikeda Y, Daughters RS, Ranum LP. Bidirectional expression of the SCA8 expansion mutation: one mutation, two genes. Cerebellum 2008; 7:150 - 8; http://dx.doi.org/10.1007/s12311-008-0010-7; PMID: 18418692
- Moseley ML, Zu T, Ikeda Y, Gao W, Mosemiller AK, Daughters RS, et al. Bidirectional expression of CUG and CAG expansion transcripts and intranuclear polyglutamine inclusions in spinocerebellar ataxia type 8. Nat Genet 2006; 38:758 - 69; http://dx.doi.org/10.1038/ng1827; PMID: 16804541
- de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J 2003; 370:737 - 49; http://dx.doi.org/10.1042/BJ20021321; PMID: 12429021
- Marks PA, Xu WS. Histone deacetylase inhibitors: Potential in cancer therapy. J Cell Biochem 2009; 107:600 - 8; http://dx.doi.org/10.1002/jcb.22185; PMID: 19459166
- Parmigiani RB, Xu WS, Venta-Perez G, Erdjument-Bromage H, Yaneva M, Tempst P, et al. HDAC6 is a specific deacetylase of peroxiredoxins and is involved in redox regulation. Proc Natl Acad Sci USA 2008; 105:9633 - 8; http://dx.doi.org/10.1073/pnas.0803749105; PMID: 18606987
- Richon VM, Zhou X, Rifkind RA, Marks PA. Histone deacetylase inhibitors: development of suberoylanilide hydroxamic acid (SAHA) for the treatment of cancers. Blood Cells Mol Dis 2001; 27:260 - 4; http://dx.doi.org/10.1006/bcmd.2000.0376; PMID: 11358386
- Hockly E, Richon VM, Woodman B, Smith DL, Zhou X, Rosa E, et al. Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, ameliorates motor deficits in a mouse model of Huntington’s disease. Proc Natl Acad Sci USA 2003; 100:2041 - 6; http://dx.doi.org/10.1073/pnas.0437870100; PMID: 12576549
- Mielcarek M, Benn CL, Franklin SA, Smith DL, Woodman B, Marks PA, et al. SAHA decreases HDAC 2 and 4 levels in vivo and improves molecular phenotypes in the R6/2 mouse model of Huntington’s disease. PLoS ONE 2011; 6:e27746; http://dx.doi.org/10.1371/journal.pone.0027746; PMID: 22140466
- Giorgini F, Möller T, Kwan W, Zwilling D, Wacker JL, Hong S, et al. Histone deacetylase inhibition modulates kynurenine pathway activation in yeast, microglia, and mice expressing a mutant huntingtin fragment. J Biol Chem 2008; 283:7390 - 400; http://dx.doi.org/10.1074/jbc.M708192200; PMID: 18079112
- Chou AH, Chen SY, Yeh TH, Weng YH, Wang HL. HDAC inhibitor sodium butyrate reverses transcriptional downregulation and ameliorates ataxic symptoms in a transgenic mouse model of SCA3. Neurobiol Dis 2011; 41:481 - 8; http://dx.doi.org/10.1016/j.nbd.2010.10.019; PMID: 21047555
- Thomas EA, Coppola G, Desplats PA, Tang B, Soragni E, Burnett R, et al. The HDAC inhibitor 4b ameliorates the disease phenotype and transcriptional abnormalities in Huntington’s disease transgenic mice. Proc Natl Acad Sci USA 2008; 105:15564 - 9; http://dx.doi.org/10.1073/pnas.0804249105; PMID: 18829438
- Jia H, Pallos J, Jacques V, Lau A, Tang B, Cooper A, et al. Histone deacetylase (HDAC) inhibitors targeting HDAC3 and HDAC1 ameliorate polyglutamine-elicited phenotypes in model systems of Huntington’s disease. Neurobiol Dis 2012; 46:351 - 61; http://dx.doi.org/10.1016/j.nbd.2012.01.016; PMID: 22590724
- Pallos J, Bodai L, Lukacsovich T, Purcell JM, Steffan JS, Thompson LM, et al. Inhibition of specific HDACs and sirtuins suppresses pathogenesis in a Drosophila model of Huntington’s disease. Hum Mol Genet 2008; 17:3767 - 75; http://dx.doi.org/10.1093/hmg/ddn273; PMID: 18762557
- Jiang M, Wang J, Fu J, Du L, Jeong H, West T, et al. Neuroprotective role of Sirt1 in mammalian models of Huntington’s disease through activation of multiple Sirt1 targets. Nat Med 2012; 18:153 - 8; http://dx.doi.org/10.1038/nm.2558; PMID: 22179319
- Benn CL, Butler R, Mariner L, Nixon J, Moffitt H, Mielcarek M, et al. Genetic knock-down of HDAC7 does not ameliorate disease pathogenesis in the R6/2 mouse model of Huntington’s disease. PLoS ONE 2009; 4:e5747; http://dx.doi.org/10.1371/journal.pone.0005747; PMID: 19484127
- Moumné L, Campbell K, Howland D, Ouyang Y, Bates GP. Genetic knock-down of HDAC3 does not modify disease-related phenotypes in a mouse model of Huntington’s disease. PLoS ONE 2012; 7:e31080; http://dx.doi.org/10.1371/journal.pone.0031080; PMID: 22347433
- Bobrowska A, Paganetti P, Matthias P, Bates GP. Hdac6 knock-out increases tubulin acetylation but does not modify disease progression in the R6/2 mouse model of Huntington’s disease. PLoS ONE 2011; 6:e20696; http://dx.doi.org/10.1371/journal.pone.0020696; PMID: 21677773
- Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, et al. Embryonic stem cell lines derived from human blastocysts. Science 1998; 282:1145 - 7; http://dx.doi.org/10.1126/science.282.5391.1145; PMID: 9804556
- Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006; 126:663 - 76; http://dx.doi.org/10.1016/j.cell.2006.07.024; PMID: 16904174
- Urbach A, Bar-Nur O, Daley GQ, Benvenisty N. Differential modeling of fragile X syndrome by human embryonic stem cells and induced pluripotent stem cells. Cell Stem Cell 2010; 6:407 - 11; http://dx.doi.org/10.1016/j.stem.2010.04.005; PMID: 20452313
- Benchoua A, Onteniente B. Intracerebral transplantation for neurological disorders. Lessons from developmental, experimental, and clinical studies. Front Cell Neurosci 2011; 6:2; PMID: 22319470
- Aubry L, Bugi A, Lefort N, Rousseau F, Peschanski M, Perrier AL. Striatal progenitors derived from human ES cells mature into DARPP32 neurons in vitro and in quinolinic acid-lesioned rats. Proc Natl Acad Sci USA 2008; 105:16707 - 12; http://dx.doi.org/10.1073/pnas.0808488105; PMID: 18922775
- Chambers SM, Fasano CA, Papapetrou EP, Tomishima M, Sadelain M, Studer L. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat Biotechnol 2009; 27:275 - 80; http://dx.doi.org/10.1038/nbt.1529; PMID: 19252484
- Hu BY, Weick JP, Yu J, Ma LX, Zhang XQ, Thomson JA, et al. Neural differentiation of human induced pluripotent stem cells follows developmental principles but with variable potency. Proc Natl Acad Sci USA 2010; 107:4335 - 40; http://dx.doi.org/10.1073/pnas.0910012107; PMID: 20160098
- Kozhich OA, Hamilton RS, Mallon BS. Standardized Generation and Differentiation of Neural Precursor Cells from Human Pluripotent Stem Cells. Stem Cell Rev 2012; http://dx.doi.org/10.1007/s12015-012-9357-8; PMID: 22388559
- Lie KH, Chung HC, Sidhu KS. Derivation, propagation, and characterization of neuroprogenitors from pluripotent stem cells (hESCs and hiPSCs). Methods Mol Biol 2012; 873:237 - 46; http://dx.doi.org/10.1007/978-1-61779-794-1_15; PMID: 22528359
- Chan AW, Cheng PH, Neumann A, Yang JJ. Reprogramming Huntington monkey skin cells into pluripotent stem cells. Cell Reprogram 2010; 12:509 - 17; http://dx.doi.org/10.1089/cell.2010.0019; PMID: 20936902
- Camnasio S, Carri AD, Lombardo A, Grad I, Mariotti C, Castucci A, et al. The first reported generation of several induced pluripotent stem cell lines from homozygous and heterozygous Huntington’s disease patients demonstrates mutation related enhanced lysosomal activity. Neurobiol Dis 2012; 46:41 - 51; http://dx.doi.org/10.1016/j.nbd.2011.12.042; PMID: 22405424
- Castiglioni V, Onorati M, Rochon C, Cattaneo E. Induced pluripotent stem cell lines from Huntington’s disease mice undergo neuronal differentiation while showing alterations in the lysosomal pathway. Neurobiol Dis 2012; 46:30 - 40; http://dx.doi.org/10.1016/j.nbd.2011.12.032; PMID: 22227000
- Zhang N, An MC, Montoro D, Ellerby LM. Characterization of Human Huntington’s Disease Cell Model from Induced Pluripotent Stem Cells. PLoS Curr 2010; 2:RRN1193; http://dx.doi.org/10.1371/currents.RRN1193; PMID: 21037797
- Juopperi TA, Kim WR, Chiang CH, Yu H, Margolis RL, Ross CA, et al. Astrocytes generated from patient induced pluripotent stem cells recapitulate features of Huntington’s disease patient cells. Mol Brain 2012; 5:17; http://dx.doi.org/10.1186/1756-6606-5-17; PMID: 22613578
- Jeon I, Lee N, Li JY, Park IH, Park KS, Moon J, et al. Neuronal Properties, In Vivo Effects and Pathology of a Huntington’s Disease Patient-Derived Induced Pluripotent Stem Cells. Stem Cells 2012; http://dx.doi.org/10.1002/stem.1135; PMID: 22628015
- Koch P, Breuer P, Peitz M, Jungverdorben J, Kesavan J, Poppe D, et al. Excitation-induced ataxin-3 aggregation in neurons from patients with Machado-Joseph disease. Nature 2011; 480:543 - 6; PMID: 22113611
- Narsinh KH, Plews J, Wu JC. Comparison of human induced pluripotent and embryonic stem cells: fraternal or identical twins?. Mol Ther 2011; 19:635 - 8; http://dx.doi.org/10.1038/mt.2011.41; PMID: 21455209
- Osafune K, Caron L, Borowiak M, Martinez RJ, Fitz-Gerald CS, Sato Y, et al. Marked differences in differentiation propensity among human embryonic stem cell lines. Nat Biotechnol 2008; 26:313 - 5; http://dx.doi.org/10.1038/nbt1383; PMID: 18278034
- Pick M, Stelzer Y, Bar-Nur O, Mayshar Y, Eden A, Benvenisty N. Clone- and gene-specific aberrations of parental imprinting in human induced pluripotent stem cells. Stem Cells 2009; 27:2686 - 90; http://dx.doi.org/10.1002/stem.205; PMID: 19711451
- Rugg-Gunn PJ, Ferguson-Smith AC, Pedersen RA. Status of genomic imprinting in human embryonic stem cells as revealed by a large cohort of independently derived and maintained lines. Hum Mol Genet 2007; 16 Spec No. 2:R243 - 51; http://dx.doi.org/10.1093/hmg/ddm245; PMID: 17911167
- Caiazzo M, Dell’Anno MT, Dvoretskova E, Lazarevic D, Taverna S, Leo D, et al. Direct generation of functional dopaminergic neurons from mouse and human fibroblasts. Nature 2011; 476:224 - 7; http://dx.doi.org/10.1038/nature10284; PMID: 21725324
- Pang ZP, Yang N, Vierbuchen T, Ostermeier A, Fuentes DR, Yang TQ, et al. Induction of human neuronal cells by defined transcription factors. Nature 2011; 476:220 - 3; PMID: 21617644
- Pfisterer U, Wood J, Nihlberg K, Hallgren O, Bjermer L, Westergren-Thorsson G, et al. Efficient induction of functional neurons from adult human fibroblasts. Cell Cycle 2011; 10:3311 - 6; http://dx.doi.org/10.4161/cc.10.19.17584; PMID: 21934358
- Qiang L, Fujita R, Yamashita T, Angulo S, Rhinn H, Rhee D, et al. Directed conversion of Alzheimer’s disease patient skin fibroblasts into functional neurons. Cell 2011; 146:359 - 71; http://dx.doi.org/10.1016/j.cell.2011.07.007; PMID: 21816272
- Yoo AS, Sun AX, Li L, Shcheglovitov A, Portmann T, Li Y, et al. MicroRNA-mediated conversion of human fibroblasts to neurons. Nature 2011; 476:228 - 31; http://dx.doi.org/10.1038/nature10323; PMID: 21753754
- Brennand KJ, Simone A, Jou J, Gelboin-Burkhart C, Tran N, Sangar S, et al. Modelling schizophrenia using human induced pluripotent stem cells. Nature 2011; 473:221 - 5; http://dx.doi.org/10.1038/nature09915; PMID: 21490598
- Di Giorgio FP, Boulting GL, Bobrowicz S, Eggan KC. Human embryonic stem cell-derived motor neurons are sensitive to the toxic effect of glial cells carrying an ALS-causing mutation. Cell Stem Cell 2008; 3:637 - 48; http://dx.doi.org/10.1016/j.stem.2008.09.017; PMID: 19041780
- Lee G, Papapetrou EP, Kim H, Chambers SM, Tomishima MJ, Fasano CA, et al. Modelling pathogenesis and treatment of familial dysautonomia using patient-specific iPSCs. Nature 2009; 461:402 - 6; http://dx.doi.org/10.1038/nature08320; PMID: 19693009
- Marchetto MC, Muotri AR, Mu Y, Smith AM, Cezar GG, Gage FH. Non-cell-autonomous effect of human SOD1 G37R astrocytes on motor neurons derived from human embryonic stem cells. Cell Stem Cell 2008; 3:649 - 57; http://dx.doi.org/10.1016/j.stem.2008.10.001; PMID: 19041781