Abstract
Progeroid phenotypes are mainly encountered in 2 types of syndromes: in laminopathies, which are characterized by nuclear shape abnormalities due to lamin A alteration, and in DNA damage response defect syndromes. Because lamin A dysregulation leads to DNA damages, it has been proposed that senescence occurs in both types of syndromes through the accumulation of damages. We recently showed that elevated oxidative stress is responsible for lamin B1 accumulation, nuclear shape alteration and senescence in the DDR syndrome, ataxia telangiectasia (A-T). Interestingly, overexpression of lamin B1 in wild type cells is sufficient to induce senescence without the induction of DNA damages. Here, we will discuss the importance of controlling the lamins level in order for maintenance nuclear architecture and we will comment the relationships of lamins with other senescence mechanisms. Finally, we will describe emerging data reporting redox control by lamins, leading us to propose a general mechanism by which reactive oxygen species can induce senescence through lamin dysregulation and NSA.
Introduction
It has been commonly proposed that senescence prevents the proliferation of cells bearing damaged DNA, thus constituting a barrier against tumor development. However, senescence is a double-edged sword, as recent data have proposed that senescent cells could favor tumor proliferation by secreting inflammatory factors.Citation1 Thus, the different pathways controlling senescence should be tightly controlled and coordinated.
Since the free radical theory of aging proposed by Harman in the 1950s, oxidative stress (OS) remains one of the most frequently cited causes for aging.Citation2 However, the precise molecular control of senescence induced by OS is far from being fully elucidated.Citation3,Citation4 In addition to OS, telomere erosion, defects in the DNA damage response (DDR) and alterations in the nuclear architecture are also associated with premature aging.Citation5 The potential interplay between these different processes leading to senescence remains poorly understood, and no unifying model can be constructed.
The most severe premature aging syndromes, such as Hutchinson-Gilford progeria syndrome (HGPS) or atypical Werner syndrome, are associated with alterations in nuclear shape resulting from the deregulation of lamin A/C.Citation6-Citation8 Lamins A/C, B1 and B2 are the major constituents of the lamina, which lines the inner nuclear membrane and determines its shape and integrity.Citation9-Citation11 Based on their localization at the nuclear periphery, lamins modulate gene expression either by interacting with chromatin or by sequestering transcription factors. Additionally, other roles for lamina in the control of mitosis, DNA replication or the DNA damage response have more recently emerged.Citation11,Citation12
Progeroid syndromes have often been classified into two categories: laminopathies, such as HGPS, which are associated with defects in lamin A and nuclear shape alterations (NSA), and the DDR defect syndromes, which we will refer to here as “DDR-pathies.” Because defects in lamin A result in the alteration of DDR, it was proposed that the two types of syndromes both undergo senescence through the accumulation of unprocessed DNA damage.Citation5 According to this model, DNA damage accumulation would constitute the common pivotal process for senescence induction. Similarly, since OS also generates oxidative DNA damage, it was also proposed that OS induces senescence through the accumulation of DNA damage.
Recently, in the DDR-pathy ataxia telangiectasia (A-T), we identified lamin B1 accumulation leading to NSA.Citation13 A-T is a rare genetic autosomal recessive disorder characterized by cerebellar ataxia, oculomotor apraxia, oculotaneous telangiectasia, immune deficiency, elevated α-fetoprotein level, hypersensitivity to ionizing radiation (IR), genetic instability and, increased risk of cancer. Importantly, premature senescence and elevated oxidative stress are observed in A-T cells. A-T is caused by the loss of function mutations in the ataxia telangiectasia mutated (ATM) gene, which encodes a serine/threonine protein kinase that regulates the early steps of the DNA damages signaling pathway and thereby controls the DDR.Citation14-Citation16 Consequently, mutations in ATM lead to DDR defects and easily account for some of the clinical features of A-T including radiation sensitivity, genetic instability, immunodeficiency and cancer predisposition. However, the clinical picture of A-T is more complex, and the relationships between DDR defects, neurological disorders and premature aging remain elusive. Because A-T cells exhibit NSA through the misregulation of lamin B1, A-T can also be classified as a laminopathy. Importantly, normalizing the lamin B1 levels in A-T cells to the level of wild-type cells decreased both NSA and senescence, although the ATM mutation (and thus, spontaneous DNA damage accumulation) was not corrected. Reciprocally, overexpressing lamin B1 in wild-type cells leads to NSA and senescence without increasing the level of reactive oxygen species (ROS) and without activating DDR at detectable levels.Citation13 Taken together, these data suggest that NSA is the actual crossing road for senescence induction, which can bypass the requirement for DNA damages accumulation. Thus, this raises the question of the relative role of DNA damages accumulation vs. NSA in the induction of senescence in both laminopathies and in A-T. More generally, lamin B1 accumulation results from endogenous or exogenous OS; thus, it may also represent a pivotal mediator in the induction of senescence by OS through NSA.Citation13
Here, we will discuss the importance of the respective levels of lamin A and lamin B1 in the maintenance of nuclear architecture and examine how lamin misregulation and nuclear shape alteration could lead to senescence. Finally, we will describe emerging data reporting the control of oxidative stress by lamins, leading us to propose a new general mechanism by which ROS can induce senescence.
Lamin Levels Must be Tightly Controlled for the Maintenance of Nuclear Architecture and for Protection Against Senescence
The alteration of nuclear shape seems to be a common occurrence during senescence. Indeed, in progeroid syndromes, NSA is associated with laminopathies (such as HGPS) and in A-T. More generally, nuclear abnormalities and changes in lamins also occur during the aging process of wild-type cells, illustrating the importance of such alterations in the general senescence process.Citation17,Citation18 Importantly, the regular sphericity of the nucleus appears to depend on the relative level of Lamin A/C and Lamin B. Indeed, overexpression as well as a silencing of either lamin A or lamin B1 leads to NSA and senescence.Citation13,Citation19-Citation21
Lamin A dysregulation leads to NSA and senescence
The silencing of lamin A results in NSA and senescence. On the other hand, the overexpression of the full length wild-type cDNA results also in growth defects, dysmorphic nuclei and senescence,Citation21-Citation23 however the picture is more complex due to maturation of the lamin A protein.
Lamin A is synthesized as a precursor molecule (prelamin A), which is then post-translationally modified to generate mature lamin A lacking the carboxyl-terminal tail. The steps of the maturation are as follows: prelamin A is first modified by a farnesyltransferase. This farnesylated protein is modified, by Rce1 (or ZMPSTE24), a prenyl-CAAX-specific endoprotease, by removal of the last three C-terminal amino acids. This intermediate is then carboxymethylated. Finally, farnesylated prelamin A is processed by ZMPSTE24, which cleaves 15 C-terminal amino acids and releases mature, unfarnesylated lamin A.Citation11 The most frequent mutation in HGPS patients is a de novo heterozygous C to T transition at nucleotide 1824 of the LMNA gene, which activates a cryptic splicing site; splicing at this site causes the production of a mutant lamin A protein that contains an internal deletion of a region, 50 amino acids in length, containing the internal proteolytic cleavage site used for lamin A maturation (i.e., the removal of the last carboxyl-terminal amino acids).Citation24,Citation25 Thus, HGPS cells contain an immature intermediate of the lamin A protein, termed progerin, which is farnesylated but not cleaved.
HGPS cells, or cells expressing progerin, are characterized by an atypical nuclear membrane morphology (i.e., blebs) and poor growth. Recent data report the presence of small amount of progerin in cells from older individuals, suggesting a role for this lamin A variant in normal aging. It has been reported that the overexpression of wild-type lamin A results in phenotypes resembling those of cells expressing progerin (growth defects, dysmorphic nuclei, premature senescence).Citation21 Because inhibitors of farnesyl transferase (FTIs) reversed these phenotypes, it is likely that abnormal prelamin A-processing intermediates are responsible for these defects. Candelario et al. recently reported that the accumulation of any partially processed prelamin A protein alters cellular homeostasis to some degree, but the most dramatic effects are caused by variants with a permanently farnesylated carboxyl-terminal tail.Citation26
Overexpression as well as decrease of lamin B1 level leads to NSA and senescence
Different studies showed that if lamin B1 level is altered, NSA is observed. It has been reported that deficiency in lamin B1 leads to NSACitation19,Citation20 and senescence.Citation27 We also confirmed an induction of senescence after extinction of lamin B1 by siRNA, in two types of human fibroblasts (embryonic or postnatal) (unpublished data). Notably, downregulation of lamin B1 was reported during replicative senescence and in a patient with atypical progeria.Citation27,Citation29 According to these reports, A-T cells that exhibit premature senescence also present a decreased level of lamin B1 mRNA.Citation13 However, the stabilization of lamin B1 protein by elevated oxidative stress results in fine to an increase of protein level.Citation13
Lamin B1 undergoes modifications; however, in contrast with lamin A, the modified form of lamin B1 is not cleaved and keeps its farnesylated C-terminal tail. This poses the question as to whether overexpression of wild type lamin B1 resumes some phenotypes of progerin-expressing cells. Interestingly, we, and other have shown that the overexpression of lamin B1 leads to NSA.Citation13,Citation28,Citation30 Particularly, A-T cells exhibit an accumulation of lamin B1 protein, alterations in nuclear architecture and premature senescence. Importantly, we show that the overexpression of lamin B1 in normal cells is sufficient to induce NSA and senescence. Importantly, the normalization of the lamin B1 level in A-T cells rescues both NSA and senescence.Citation13
The unifying hypothesis could be that nuclear shape change, presumably reflecting a change in lamina organization, appears to drive senescence.Citation31 However, as nuclear abnormalities occur in other laminopathies without premature aging, the changes in the organization of the lamina, which are at the origin of senescence, must to be precisely clarified. We could argue that different mutations of lamin A, deletion (or silencing) or accumulation of lamins (of A- or B-type), would not have the same consequences on cellular metabolism, on chromatin structure, on telomeres, on proliferation control or on level of shape abnormality. A comparative study would be very useful to understand what kind of shape abnormalities or what level of shape abnormalities is necessary to induce senescence.
Finally, the expression of one type of lamin could perturb the general localization of the other lamins, regardless of any change in its protein level. For example, it was reported that the expression of lamin A (E145K progeria mutation) induced the formation of blebs in the nuclear membrane in which lamin B1 was excluded.Citation30 Another studies show that deletion of lamin B1 increases the size of the Lamin A/C meshwork and induces the formation of lamin A/C-rich and lamin B2-deficient NE blebsCitation32 (and for review, see ref. Citation11).
Taken together, these data underscore the importance of the stoichiometric balance of different lamins (and possibly their respective locations) for proper nuclear shape and for the control of senescence.
Lamin, NSA and Senescence: Relationships with Other Senescence Mechanisms
Relationships with telomere dysfunction and DDR defects
Telomeres shortening are known to induce a premature cell cycle arrest via the activation of P53, thereby promoting senescence. Interestingly, telomere shortening has been reported in HGPS, in several human fibroblast cell lines overexpressing different mutant LMNA variants and in LMNA knockout mouse fibroblasts.Citation23,Citation33-Citation36 In contrast with HGP fibroblasts, the telomere length in hematopoietic cells, which typically do not express lamin A, was within the normal range in different HGPS patients’ samples. These results suggest that expression of mutant lamin A is necessary for telomere loss in HGPS.Citation36
Although that telomere shortening has been thought to participate in senescence in laminopathies, this assumption is still debated, since apparent contradictory results were reported. Some reports showed an extend of life span in HGP cells by hTERT expression,Citation34,Citation37,Citation38 when other report showed that fibroblasts clones from HGPS patients can senesce despite the restoration of telomerase activity and the stabilization of telomere length.Citation39
In addition, not only telomeres erosion was reported in progerin expressing cells or HGPS cells, but also damages at telomeres.Citation36,Citation38 A failure to repair DNA damage at telomeres, causing loss of variable amounts of DNA could explain both the high variability in telomere length observed between chromosomes and the increase frequency of signal ends in HGPS.Citation36 Unrepaired DNA damage could also prevent the cells from completing cell division leading to apoptosis or senescence. These damages at telomeres could be signaled and can trigger cell cycle arrest through DNA damage response (DDR) activation. It has been proposed that DNA damages observed in HGP or progerin expressing cells reflect the telomeres dysfunction.Citation38 However, the impact of hTERT on the DNA damages is still debated, one study shows that hTERT expression decreases the progerin-induced DNA damages and this reduction is correlated with the onset of increased proliferative capacity of HGP fibroblasts from one HGP patientCitation38 and, in contrast another report indicates that telomerase fails to protect against progerin-induced DNA damage.Citation40
Following lamin B1 overexpression, senescence occurs in one or two generations after.Citation13 Thus, telomere shortening is unlikely to account for senescence in such a short time frame. Moreover, although ATM regulates telomeric maintenance, h-TERT-expressing A-T fibroblasts experience premature senescence, demonstrating that the senescence of A-T cells is not explained by telomeric shortening alone.Citation41 Taken together, these data show that lamin B1 might trigger senescence through alternative pathways aside from telomere attrition.
Cells from HGPS patients and HGPS animal models, cells expressing different mutants of lamin A or MEF defective for the lamin A maturation protease ZMPSTE24 display persistent DNA damages. These damages could result from alterations in the localization, expression or stability of different proteins involved in the DNA damage response. In cells defective for lamin A that were treated with genotoxic agents, aberrant ATR localization, fewer γ-H2AX or 53BP1 damage-induced foci were observed. Moreover, an increase in 53BP1 degradation and a defect in homologous recombination due to a decrease in the expression of RAD51 and BRCA1 proteins were reported (for reviewCitation12). In contrast with silencing of lamin A, after the silencing of lamin B1, no γ-H2AX foci were observed, although an increase in ATM, Chk1 and Chk2 were detected.Citation27 Moreover, neither the presence or accumulation of DNA damages nor the activation of DDR can explain the senescence induction upon lamin B1 overexpression because no change in the levels of p-Chk1, p-Chk2 and γ-H2AX and no change in the number of γ-H2AX foci were observed 48 h after transfection of primary fibroblasts with lamin B1, which corresponds to the time of senescence induction.Citation13 Taken together, these data suggest that senescence mechanisms induced by the absence of lamin A or the dysregulation of lamin B1 may be different, and that senescence could occur in absence of DNA damage accumulation in case of lamin B1 accumulation. In line with this, there are an increasing number of reports showing in different situations of senescence an absence of DNA damage signaling activation.Citation42 In accordance with this, data have shown that although farnesyl-transferase inhibitors (FTI) can improve the nuclear shape of HGPS cells, the DNA damage is still persistent after treatment.Citation43 This suggests that either the phenotypes of NSA and DNA damage can be separable or induction of DNA damage is irreversible even if the initial defect is fixed.
Lamins, NSA and oxidative stress
In Ataxia telangiectasia cells, which exhibit premature senescence, an increase of lamin B1 protein level is measured by western-blot and immunofluorescence analysis.Citation13 This increase in protein level, corresponds to a p38-MAPK-dependent stabilization of lamin B1 protein, due to elevated oxidative stress. The senescence was reduced in A-T cells, when lamin B1 level was normalized by anti-oxydant or p38-MAPK inhibitor treatment. These data show that the increase of lamin B1 due to oxidative stress is at least partly responsible of senescence in A-T.Citation13 To generalize our data, on the impact of oxidative stress on lamin B1, we have examined its level in two senescence situations in which ROS induction was previously described: exposure to a high dose of ionizing radiation (IR) and oncogene-induced senescence (OIS). Notably, it has been report that ROS induction is important to trigger senescence under RAS activation.Citation44 To analyze the impact of OIS, we used a stable cellular model in which Ras V12 was induced by the addition of 4-TAM to the medium.Citation45 This model requires neither lentiviral infection nor selection for several days. In both models, we showed an increase of lamin B1 level associated with both NSA and senescence. However, these data are challenged by new reports showing a decrease in the lamin B1 level after IR or during OIS.Citation27,Citation29 These apparent discrepancies could be due to the cellular models, culture conditions (O2, confluency, etc.) or experimental conditions used; for example, viral infection was used to express RAS, which contrasted with our study. In our hands, the increased of level of lamin B1 followed the activation of p38-MAPK in response to oxidative stress. Differences in sensitivity to oxidative stress could exist between embryonic, neonatal or adult fibroblasts and may explain the apparent discrepancies between the different studies. In this context, it can be underscored that in a pathophysiological model of A-T primary fibroblast cells, NSA and senescence can be rescued by antioxidant treatment or by p38 MAPK inhibition, which normalizes the level of lamin B1. This illustrates the importance of oxidative stress in controlling the level of endogenous lamin B1 protein and the importance of the level of lamin B1 for senescence induction. Indeed, the direct reduction of lamin B1 by RNA interference, which normalized the level of lamin B1, also decreases senescence.Citation13 Interestingly, senescence is not totally abolished. Since ATM defect is still present, the persistent DNA damages could explain the remaining senescence.
Altogether these results lead to the hypothesis that two phenomenon could occur in senescence conditions: a p38 MAPK-independent decrease of gene expression of lamin B1 (probably due to the action of pRb or p53 activationCitation27,Citation29), and, in case of oxidative stress, a p38 MAPK-dependent stabilization of lamin B1 protein. The increase of protein level could even counteract or outweigh the decrease of gene expression, in case of important or persistent oxidative stress (as in case of A-T). To induce senescence by RAS activation, we performed the experiments at 3% oxygen. Thus, although oxygen conditions of cell culture may not explain all the difference between different experimental conditions, it would be important, to reconcile all data (apparently contradictory), to measure ROS and potential p38 MAPK activation in the different senescence situations explored in the different reports.
In mouse embryonic fibroblasts (MEFs) lamin B1 has been shown to sequester the transcription/repression factor Oct1 at the nuclear periphery. This sequestration hampers the transcriptional repression by Oct1 of genes involved in ROS detoxification such as PRDX2, GPX3 or SOD2. Consistently, increased ROS levels are observed in MEFs that express a non-functional lamin B1 (LMNB1Δ/Δ) associated with a higher sensitivity to hydrogen peroxide (H202).Citation46 These data propose that lamin, especially lamin B1, could participate in redox control and could suggest that oxidative stress may be partly responsible of senescence induction under lamin B1 extinction conditions. However, in human fibroblasts, senescence triggered either by overexpression or suppression of lamin B1, cannot be explained by an elevated oxidative stress. In fact, both higher or lower lamin B1 level lead to a decrease in ROS.Citation13,Citation27 In the case of overexpression of lamin B1, this reduction is likely to be due to the retention of Oct1 to nuclear periphery and consequently the inhibition of its transcriptional repression activity on detoxifying genes. In condition of lamin B1 defect or silencing, ROS reduction could result from activation of p53, which is known to control detoxifying genes.Citation47 Indeed, antioxidant genes such as SOD2, GPX1 and others were increased upon lamin B1 silencing. Because the senescence induced by lamin B1 deletion cannot be explained by an increased in oxidative stress, it has been proposed that senescence is likely to be due to the activation of p53 and subsequent upregulation of the p21 gene.Citation27 Moreover, the apparent discrepancy between the data obtained in MEFs and those obtained in human fibroblasts may be due to the differential susceptibility to oxidative stress between mouse and human cells.Citation48
We propose a model in which the accumulation of lamin B1 decreases the level of ROS and increases viability as part of a transient response to oxidative stress. However, when oxidative stress is chronic and persistent, such as in AT or when the mechanism of detoxification is overwhelmed, the accumulation of lamin B1, like a double-edged sword, leads to NSA and senescence.Citation13
Beside their role in redox control in sequestering transcription factors, other functions of lamins could account for an elevated oxidative stress associated with NSA.Citation49 Indeed, the lamina or the nuclear envelope may constitute a nuclear shield with an elevated perinuclear concentration of detoxification enzymes.Citation50 The function of these detoxification enzymes might be altered during NSA, thus increasing the risk of damage to essential nuclear components. Indeed, it has been reported that a disruption in the integrity of the nuclear membrane allows the passage of components and even organelles from the cytoplasm to the nucleus.Citation51 Consequently, the presence of mitochondria in the nucleoplasm should increase nuclear ROS to a level that can damage DNA.
Finally, mitochondrial dysfunction (decreased expression of COX2) has been reported in cells expressing pre-lamin A, leading to an elevated ROS level. This elevated ROS phenotype was reversed by FTI treatment, confirming the toxicity of the farnesylated form of lamin A.Citation52 The alteration of mitochondrial metabolism was also suggested by a proteomic analysis of zmpste24−/− cells.Citation53
A unifying model can be proposed in which oxidative stress, regardless of the cause, affects lamins proteins functions and/or levels, resulting in both alteration of nuclear envelop structure and in loss of redox control, which amplify the oxidative stress. In agreement with this model, it has recently been reported that the tail domain of lamin A is a target of oxidative damage, and its irreversible oxidation led to a loss of lamin A function and entry into senescence.Citation54 Oxidative stress is known to affect telomeresCitation55 and cause DNA damage; the increase in oxidative stress could therefore also be accountable for other cellular phenotypes encountered in laminopathies (). Indeed, the accumulation of DNA damage in laminopathy progeria seems to be due to ROS generation, as an antioxidant treatment reduced the basal level of double-stranded DNA breaks.Citation56 Consistently, other studies showed accumulation of prelamin A after oxidative stress and the accumulation of prelamin A disrupt mitosis and induce DNA damage.Citation57 Additionally, our data suggest that oxidative stress, regardless of the cause, increases the level of lamin B1 to protect against ROS.Citation13 However, prolonged lamin B1 overexpression leads to NSA, which can trigger senescence even without DNA damages ().
Figure 1. Interplay between oxidative stress, telomere shortening, and DNA damage for the induction of senescence. Oxidative stress, regardless of the cause, is known to alter telomeres and to cause DNA damage. Telomere shortening and DNA damage are involved in the induction of senescence. Our group and others have reported that oxidative stress can alter lamins functions (by oxidation of lamin A, accumulation of pre-lamin A or lamin B1) leading to nuclear shape. All these alterations are associated with senescence. Interestingly, dysregulation of lamins is also associated with telomere shortening and DNA damage.
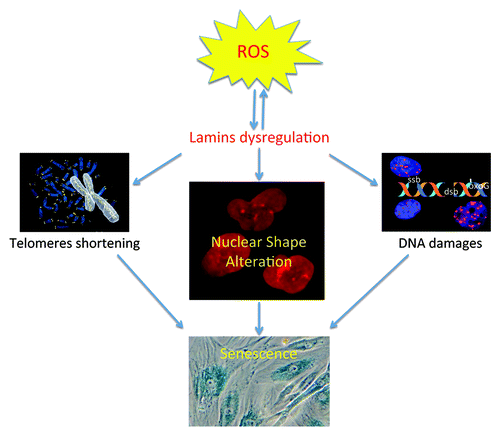
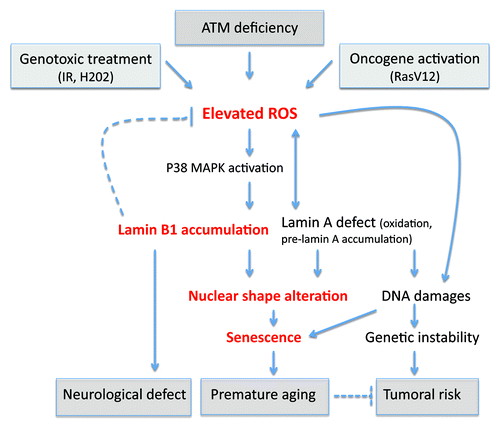
Figure 2. Consequences of oxidative stress through lamins dyregulation. An elevated ROS level has been observed after genotoxic stress, oncogene activation, or deficiency of ATM. We found that in ataxia telangiectasia, the elevated ROS leads to an accumulation of lamin B1 by p38 MAPK activation. This accumulation leads to nuclear shape alteration and senescence. Interestingly, it has been proposed that the duplication of lamin B1 is responsible for demyelination of centraln nervous system in adult-onset autosomal dominant leukodystrohy (ADLD) and might potentially explain other neurological defects. Redox regulation is altered in Hutchinson-Gilford progeria cells or in lamin B1-deficient cells. Oxidative stress affects the function of lamin A by oxidation. Altered levels of lamin accumulation and defects in lamins cause alterations in nuclear shape and lead to senescence. Lamin A dysregulation affects DNA damage signaling and repair, leading to genetic instability. This genetic instability can partially participate in premature aging and may also cause the tumoral predisposition observed in DDR pathies. However, in laminopathies, no cancer predisposition is observed, suggesting in this case that senescence hampers the proliferation of cells harbouring DNA damage.
Taken together, the data described here highlight the importance of redox control in the etiology of laminopathy pathologies. Additionally, these data could encourage the development of therapeutic strategies combined with antioxidant administration for the treatment of HGPS patients. Unraveling the intricacy of the molecular mechanisms through which the maintenance of nuclear architecture controls fundamental cellular functions that affect senescence induction constitutes an exciting challenge for future research on aging and cancer.
Acknowledgments
We thank the members of the laboratory for helpful discussions. This work was supported by grants from Association pour la Recherche sur le Cancer (ARC); Comité des Hauts de Seine de la Ligue Nationale Contre le Cancer; Division of Life Science (DSV), CEA and Centre National de Recherche Scientifique (CNRS). We apologize to authors who cannot be cited due to space limitation.
References
- Rodier F, Campisi J. Four faces of cellular senescence. J Cell Biol 2011; 192:547 - 56; http://dx.doi.org/10.1083/jcb.201009094; PMID: 21321098
- Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol 1956; 11:298 - 300; PMID: 13332224
- Muller FL, Lustgarten MS, Jang Y, Richardson A, Van Remmen H. Trends in oxidative aging theories. Free Radic Biol Med 2007; 43:477 - 503; http://dx.doi.org/10.1016/j.freeradbiomed.2007.03.034; PMID: 17640558
- Gil Del Valle L. Oxidative stress in aging: Theoretical outcomes and clinical evidences in humans. Biomed Pharmacother 2010; In Press http://dx.doi.org/10.1016/j.biopha.2010.09.010; PMID: 20950991
- Lans H, Hoeijmakers JH. Cell biology: ageing nucleus gets out of shape. Nature 2006; 440:32 - 4; http://dx.doi.org/10.1038/440032a; PMID: 16511477
- Misteli T, Scaffidi P. Genome instability in progeria: when repair gets old. Nat Med 2005; 11:718 - 9; http://dx.doi.org/10.1038/nm0705-718; PMID: 16015360
- Kudlow BA, Kennedy BK, Monnat RJJ Jr.. Werner and Hutchinson-Gilford progeria syndromes: mechanistic basis of human progeroid diseases. Nat Rev Mol Cell Biol 2007; 8:394 - 404; http://dx.doi.org/10.1038/nrm2161; PMID: 17450177
- Worman HJ, Bonne G. “Laminopathies”: a wide spectrum of human diseases. Exp Cell Res 2007; 313:2121 - 33; http://dx.doi.org/10.1016/j.yexcr.2007.03.028; PMID: 17467691
- Broers JL, Ramaekers FC, Bonne G, Yaou RB, Hutchison CJ. Nuclear lamins: laminopathies and their role in premature ageing. Physiol Rev 2006; 86:967 - 1008; http://dx.doi.org/10.1152/physrev.00047.2005; PMID: 16816143
- Prokocimer M, Davidovich M, Nissim-Rafinia M, Wiesel-Motiuk N, Bar DZ, Barkan R, et al. Nuclear lamins: key regulators of nuclear structure and activities. J Cell Mol Med 2009; 13:1059 - 85; http://dx.doi.org/10.1111/j.1582-4934.2008.00676.x; PMID: 19210577
- Dechat T, Adam SA, Taimen P, Shimi T, Goldman RD. Nuclear lamins. Cold Spring Harb Perspect Biol 2010; 2:a000547; http://dx.doi.org/10.1101/cshperspect.a000547; PMID: 20826548
- Warren DT, Shanahan CM. Defective DNA-damage repair induced by nuclear lamina dysfunction is a key mediator of smooth muscle cell aging. Biochem Soc Trans 2011; 39:1780 - 5; http://dx.doi.org/10.1042/BST20110703; PMID: 22103525
- Barascu A, Le Chalony C, Pennarun G, Genet D, Imam N, Lopez B, et al. Oxidative stress induces an ATM-independent senescence pathway through p38 MAPK-mediated lamin B1 accumulation. EMBO J 2012; 31:1080 - 94; http://dx.doi.org/10.1038/emboj.2011.492; PMID: 22246186
- Shiloh Y. The ATM-mediated DNA-damage response: taking shape. Trends Biochem Sci 2006; 31:402 - 10; http://dx.doi.org/10.1016/j.tibs.2006.05.004; PMID: 16774833
- Lavin MF, Kozlov S. ATM activation and DNA damage response. Cell Cycle 2007; 6:931 - 42; http://dx.doi.org/10.4161/cc.6.8.4180; PMID: 17457059
- Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER 3rd, Hurov KE, Luo J, et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007; 316:1160 - 6; http://dx.doi.org/10.1126/science.1140321; PMID: 17525332
- Scaffidi P, Misteli T. Lamin A-dependent nuclear defects in human aging. Science 2006; 312:1059 - 63; http://dx.doi.org/10.1126/science.1127168; PMID: 16645051
- McClintock D, Ratner D, Lokuge M, Owens DM, Gordon LB, Collins FS, et al. The mutant form of lamin A that causes Hutchinson-Gilford progeria is a biomarker of cellular aging in human skin. PLoS One 2007; 2:e1269; http://dx.doi.org/10.1371/journal.pone.0001269; PMID: 18060063
- Vergnes L, Péterfy M, Bergo MO, Young SG, Reue K. Lamin B1 is required for mouse development and nuclear integrity. Proc Natl Acad Sci U S A 2004; 101:10428 - 33; http://dx.doi.org/10.1073/pnas.0401424101; PMID: 15232008
- Lammerding J, Fong LG, Ji JY, Reue K, Stewart CL, Young SG, et al. Lamins A and C but not lamin B1 regulate nuclear mechanics. J Biol Chem 2006; 281:25768 - 80; http://dx.doi.org/10.1074/jbc.M513511200; PMID: 16825190
- Candelario J, Sudhakar S, Navarro S, Reddy S, Comai L. Perturbation of wild-type lamin A metabolism results in a progeroid phenotype. Aging Cell 2008; 7:355 - 67; http://dx.doi.org/10.1111/j.1474-9726.2008.00393.x; PMID: 18363904
- Lammerding J, Hsiao J, Schulze PC, Kozlov S, Stewart CL, Lee RT. Abnormal nuclear shape and impaired mechanotransduction in emerin-deficient cells. J Cell Biol 2005; 170:781 - 91; http://dx.doi.org/10.1083/jcb.200502148; PMID: 16115958
- Huang S, Risques RA, Martin GM, Rabinovitch PS, Oshima J. Accelerated telomere shortening and replicative senescence in human fibroblasts overexpressing mutant and wild-type lamin A. Exp Cell Res 2008; 314:82 - 91; http://dx.doi.org/10.1016/j.yexcr.2007.08.004; PMID: 17870066
- De Sandre-Giovannoli A, Bernard R, Cau P, Navarro C, Amiel J, Boccaccio I, et al. Lamin a truncation in Hutchinson-Gilford progeria. Science 2003; 300:2055; http://dx.doi.org/10.1126/science.1084125; PMID: 12702809
- Eriksson M, Brown WT, Gordon LB, Glynn MW, Singer J, Scott L, et al. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature 2003; 423:293 - 8; http://dx.doi.org/10.1038/nature01629; PMID: 12714972
- Candelario J, Borrego S, Reddy S, Comai L. Accumulation of distinct prelamin A variants in human diploid fibroblasts differentially affects cell homeostasis. Exp Cell Res 2011; 317:319 - 29; http://dx.doi.org/10.1016/j.yexcr.2010.10.014; PMID: 20974128
- Shimi T, Butin-Israeli V, Adam SA, Hamanaka RB, Goldman AE, Lucas CA, et al. The role of nuclear lamin B1 in cell proliferation and senescence. Genes Dev 2011; 25:2579 - 93; http://dx.doi.org/10.1101/gad.179515.111; PMID: 22155925
- Padiath QS, Saigoh K, Schiffmann R, Asahara H, Yamada T, Koeppen A, et al. Lamin B1 duplications cause autosomal dominant leukodystrophy. Nat Genet 2006; 38:1114 - 23; http://dx.doi.org/10.1038/ng1872; PMID: 16951681
- Freund A, Laberge RM, Demaria M, Campisi J. Lamin B1 loss is a senescence-associated biomarker. Mol Biol Cell 2012; 23:2066 - 75; http://dx.doi.org/10.1091/mbc.E11-10-0884; PMID: 22496421
- Taimen P, Pfleghaar K, Shimi T, Möller D, Ben-Harush K, Erdos MR, et al. A progeria mutation reveals functions for lamin A in nuclear assembly, architecture, and chromosome organization. Proc Natl Acad Sci U S A 2009; 106:20788 - 93; http://dx.doi.org/10.1073/pnas.0911895106; PMID: 19926845
- Hutchison CJ. B-type lamins and their elusive roles in metazoan cell proliferation and senescence. EMBO J 2012; 31:1058 - 9; http://dx.doi.org/10.1038/emboj.2012.39; PMID: 22343942
- Shimi T, Pfleghaar K, Kojima S, Pack CG, Solovei I, Goldman AE, et al. The A- and B-type nuclear lamin networks: microdomains involved in chromatin organization and transcription. Genes Dev 2008; 22:3409 - 21; http://dx.doi.org/10.1101/gad.1735208; PMID: 19141474
- Allsopp RC, Vaziri H, Patterson C, Goldstein S, Younglai EV, Futcher AB, et al. Telomere length predicts replicative capacity of human fibroblasts. Proc Natl Acad Sci U S A 1992; 89:10114 - 8; http://dx.doi.org/10.1073/pnas.89.21.10114; PMID: 1438199
- Kudlow BA, Stanfel MN, Burtner CR, Johnston ED, Kennedy BK. Suppression of proliferative defects associated with processing-defective lamin A mutants by hTERT or inactivation of p53. Mol Biol Cell 2008; 19:5238 - 48; http://dx.doi.org/10.1091/mbc.E08-05-0492; PMID: 18843043
- Gonzalez-Suarez I, Gonzalo S. Nurturing the genome: A-type lamins preserve genomic stability. Nucleus 2010; 1:129 - 35; http://dx.doi.org/10.4161/nucl.1.2.10797; PMID: 21326943
- Decker ML, Chavez E, Vulto I, Lansdorp PM. Telomere length in Hutchinson-Gilford progeria syndrome. Mech Ageing Dev 2009; 130:377 - 83; http://dx.doi.org/10.1016/j.mad.2009.03.001; PMID: 19428457
- Ouellette MM, McDaniel LD, Wright WE, Shay JW, Schultz RA. The establishment of telomerase-immortalized cell lines representing human chromosome instability syndromes. Hum Mol Genet 2000; 9:403 - 11; http://dx.doi.org/10.1093/hmg/9.3.403; PMID: 10655550
- Benson EK, Lee SW, Aaronson SA. Role of progerin-induced telomere dysfunction in HGPS premature cellular senescence. J Cell Sci 2010; 123:2605 - 12; http://dx.doi.org/10.1242/jcs.067306; PMID: 20605919
- Wallis CV, Sheerin AN, Green MH, Jones CJ, Kipling D, Faragher RG. Fibroblast clones from patients with Hutchinson-Gilford progeria can senesce despite the presence of telomerase. Exp Gerontol 2004; 39:461 - 7; http://dx.doi.org/10.1016/j.exger.2003.12.015; PMID: 15050279
- Scaffidi P, Misteli T. Lamin A-dependent misregulation of adult stem cells associated with accelerated ageing. Nat Cell Biol 2008; 10:452 - 9; http://dx.doi.org/10.1038/ncb1708; PMID: 18311132
- Naka K, Tachibana A, Ikeda K, Motoyama N. Stress-induced premature senescence in hTERT-expressing ataxia telangiectasia fibroblasts. J Biol Chem 2004; 279:2030 - 7; http://dx.doi.org/10.1074/jbc.M309457200; PMID: 14570874
- Ogrunc M, d’Adda di Fagagna F. Never-ageing cellular senescence. Eur J Cancer 2011; 47:1616 - 22; http://dx.doi.org/10.1016/j.ejca.2011.04.003; PMID: 21561762
- Liu Y, Rusinol A, Sinensky M, Wang Y, Zou Y. DNA damage responses in progeroid syndromes arise from defective maturation of prelamin A. J Cell Sci 2006; 119:4644 - 9; http://dx.doi.org/10.1242/jcs.03263; PMID: 17062639
- Weyemi U, Lagente-Chevallier O, Boufraqech M, Prenois F, Courtin F, Caillou B, et al. ROS-generating NADPH oxidase NOX4 is a critical mediator in oncogenic H-Ras-induced DNA damage and subsequent senescence. Oncogene 2012; 31:1117 - 29; http://dx.doi.org/10.1038/onc.2011.327; PMID: 21841825
- Narita M, Young AR, Narita M. Autophagy facilitates oncogene-induced senescence. Autophagy 2009; 5:1046 - 7; http://dx.doi.org/10.4161/auto.5.7.9444; PMID: 19652542
- Malhas AN, Lee CF, Vaux DJ. Lamin B1 controls oxidative stress responses via Oct-1. J Cell Biol 2009; 184:45 - 55; http://dx.doi.org/10.1083/jcb.200804155; PMID: 19139261
- Sablina AA, Budanov AV, Ilyinskaya GV, Agapova LS, Kravchenko JE, Chumakov PM. The antioxidant function of the p53 tumor suppressor. Nat Med 2005; 11:1306 - 13; http://dx.doi.org/10.1038/nm1320; PMID: 16286925
- Parrinello S, Samper E, Krtolica A, Goldstein J, Melov S, Campisi J. Oxygen sensitivity severely limits the replicative lifespan of murine fibroblasts. Nat Cell Biol 2003; 5:741 - 7; http://dx.doi.org/10.1038/ncb1024; PMID: 12855956
- Sieprath T, Darwiche R, De Vos WH. Lamins as mediators of oxidative stress. Biochem Biophys Res Commun 2012; 421:635 - 9; http://dx.doi.org/10.1016/j.bbrc.2012.04.058; PMID: 22538370
- Fabrini R, Bocedi A, Pallottini V, Canuti L, De Canio M, Urbani A, et al. Nuclear shield: a multi-enzyme task-force for nucleus protection. PLoS One 2010; 5:e14125; http://dx.doi.org/10.1371/journal.pone.0014125; PMID: 21170318
- De Vos WH, Houben F, Kamps M, Malhas A, Verheyen F, Cox J, et al. Repetitive disruptions of the nuclear envelope invoke temporary loss of cellular compartmentalization in laminopathies. Hum Mol Genet 2011; 20:4175 - 86; http://dx.doi.org/10.1093/hmg/ddr344; PMID: 21831885
- Caron M, Auclairt M, Vissian A, Vigouroux C, Capeau J. Contribution of mitochondrial dysfunction and oxidative stress to cellular premature senescence induced by antiretroviral thymidine analogues. Antivir Ther 2008; 13:27 - 38; PMID: 18389896
- Peinado JR, Quirós PM, Pulido MR, Mariño G, Martínez-Chantar ML, Vázquez-Martínez R, et al. Proteomic profiling of adipose tissue from Zmpste24-/- mice, a model of lipodystrophy and premature aging, reveals major changes in mitochondrial function and vimentin processing. Mol Cell Proteomics 2011;; 10:M111 - , 008094; http://dx.doi.org/10.1074/mcp.M111.008094; PMID: 21828285
- Pekovic V, Gibbs-Seymour I, Markiewicz E, Alzoghaibi F, Benham AM, Edwards R, et al. Conserved cysteine residues in the mammalian lamin A tail are essential for cellular responses to ROS generation. Aging Cell 2011; 10:1067 - 79; http://dx.doi.org/10.1111/j.1474-9726.2011.00750.x; PMID: 21951640
- Buckingham EM, Klingelhutz AJ. The role of telomeres in the ageing of human skin. Exp Dermatol 2011; 20:297 - 302; http://dx.doi.org/10.1111/j.1600-0625.2010.01242.x; PMID: 21371125
- Richards SA, Muter J, Ritchie P, Lattanzi G, Hutchison CJ. The accumulation of un-repairable DNA damage in laminopathy progeria fibroblasts is caused by ROS generation and is prevented by treatment with N-acetyl cysteine. Hum Mol Genet 2011; 20:3997 - 4004; http://dx.doi.org/10.1093/hmg/ddr327; PMID: 21807766
- Ragnauth CD, Warren DT, Liu Y, McNair R, Tajsic T, Figg N, et al. Prelamin A acts to accelerate smooth muscle cell senescence and is a novel biomarker of human vascular aging. Circulation 2010; 121:2200 - 10; http://dx.doi.org/10.1161/CIRCULATIONAHA.109.902056; PMID: 20458013