Abstract
During recent years a number of severe clinical syndromes, collectively termed laminopathies, turned out to be caused by various, distinct mutations in the human LMNA gene. Arising from this, remarkable progress has been made to unravel the molecular pathophysiology underlying these disorders. A great benefit in this context was the generation of an A-type lamin deficient mouse line (Lmna−/−) by Sullivan and others,Citation1 which has become one of the most frequently used models in the field and provided profound insights to many different aspects of A-type lamin function. Here, we report the unexpected finding that these mice express a truncated Lmna gene product on both transcriptional and protein level. Combining different approaches including mass spectrometry, we precisely define this product as a C-terminally truncated lamin A mutant that lacks domains important for protein interactions and post-translational processing. Based on our findings we discuss implications for the interpretation of previous studies using Lmna−/− mice and the concept of human laminopathies.
Introduction
The nuclear lamina is a dense protein meshwork that lines the inner nuclear membrane and serves as a nuclear scaffold. It is composed of a distinct group of intermediate filaments collectively termed lamins.Citation2,Citation3 Commonly, lamins are subdivided in A- and B-type primarily according to their biochemical properties. Mammalian genomes include two genes (Lmnb1 and Lmnb2) encoding the three B-type lamins B1, B2 and B3.Citation4-Citation6 In contrast, all mammalian A-type lamins (A, A∆10, C and C2) are alternative splice variants arising from a single Lmna gene.Citation7-Citation10
In mice, the expression of A-type lamins is developmentally regulated in a tissue-specific manner.Citation11,Citation12 With regard to this, A-type lamins were proposed to serve tissue-specific functions and to be implicated in terminal tissue differentiation. In recent years, this notion has found support by the finding that distinct mutations in the LMNA gene induce severe human diseases with tissue-specific dysfunctions, collectively classified as laminopathies.Citation13-Citation19 Arising from these observations, novel physiological roles are emerging that extend the significance of the lamina far beyond nuclear scaffolding. Thus, lamins are currently considered as key regulators of gene expression by their involvement in signaling, transcription and chromatin organization.Citation20-Citation22
Ongoing characterization of the relationship between physiological functions of nuclear lamins and their contribution to the molecular pathophysiology of laminopathies has greatly benefited from genetically modified mouse models. One of the most versatile models providing insights to various issues of A-type lamin function was provided by Sullivan and colleaguesCitation1 who created a mouse line with targeted disruption of the Lmna locus. Initially, these mice (Lmna−/−) were found to develop severe cardiac and skeletal myopathy by 3–4 weeks of post-natal development resulting in premature death between weeks 6 and 8. On the cellular level, embryonic fibroblasts (MEFs) and tissues from Lmna−/− mice were characterized by aberrant nuclear morphology, partial loss of peripheral heterochromatin and, most notably, mislocalization of emerin, thus bearing strong resemblance to abnormalities associated with human Emery-Dreifuss muscular dystrophy.Citation1 Additional reports demonstrated the emergence of dilated cardiomyopathy in both Lmna−/− and Lmna+/− animals with considerable gene dosage-dependent differences in disease onset and progressionCitation23,Citation24 as well as the disruption of spermatogenesis in Lmna−/− males.Citation25 A multitude of further studies used Lmna−/− MEFs to investigate the contribution of A-type lamins to nucleo-cytoskeletal integrity, nuclear mechanics and mechanotransductionCitation26-Citation32 as well as their impact on cell-cycle control, differentiation and genome stability.Citation33-Citation37
Here, we demonstrate the expression of a truncated and as yet unnoticed Lmna gene product in Lmna−/− mice and MEFs on mRNA and protein level. Despite the widespread use of this mouse line and its careful initial characterization, this gene product - referred to as lamin A∆8–11 - has apparently been overlooked continuously. Taking into account recent data provided by reports using Lmna−/− mice, we discuss implications of our findings for current models of lamin function and the concept of human laminopathies.
Results
Reactivity of three different A-type lamin-specific antibodies with a nuclear envelope antigen expressed in Lmna−/− MEFs and tissues
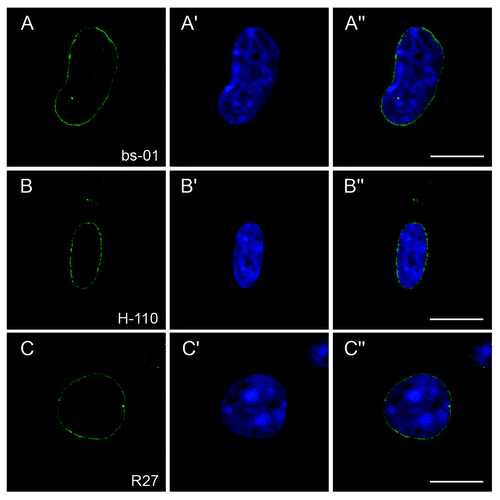
Having Lmna−/− MEFsCitation1 on hand, we intended to use this cell line as negative control to validate the specificity of new polyclonal antibodies (pAb bs-01) against murine lamin A/C raised in our laboratory. Unexpectedly, though affinity purified against the pure immunized epitope, these antibodies showed distinct reactivity with an antigen localized at the nuclear envelope (NE) of Lmna−/− MEFs in immunocytochemistry (−A”). Assuming non-specific cross-reactivity at first, we were surprised to make the same observation with the established and frequently used A-type lamin antibodies pAb H-110 (Santa Cruz Biotechnology, Santa Cruz, USA) and mAb R27Citation38 since both of them specifically stained the NE of Lmna−/− MEFs as well (−B” and C−C”).
Figure 1. Reactivity of three different A-type lamin-specific antibodies with a NE antigen present in Lmna−/− MEFs. Lmna−/− MEFs were stained with either pAb bs-01 (A), pAb H-110 (B) or mAb R27 (C). DNA was stained with Hoechst 33258 (A’-C’). Specimens were analyzed by confocal microscopy and individual channels were merged (A”-C”). All three antibodies stained the NE of Lmna−/− MEFs. Bars, 10 µm.
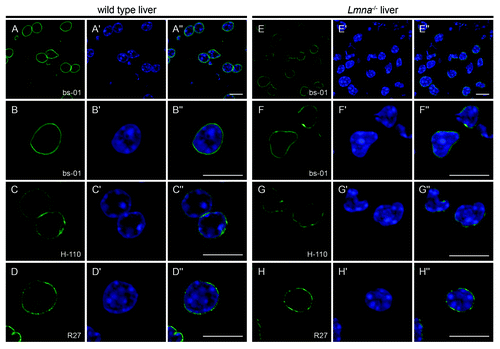
To verify these unexpected findings we performed immunohistochemistry on liver cryosections from Lmna−/− mice (). In accordance with the results obtained with MEFs, all three antibodies (pAb bs-01, pAb H-110 and mAb R27) stained the NE of Lmna−/− hepatocytes, although signal intensities were slightly reduced compared with wild type controls (compare panels A and E). Consistent with the initial characterization by Sullivan et al.,Citation1 nuclei of Lmna−/− cells frequently displayed considerable morphological aberrations (F and G).
Figure 2. The NE of cells from Lmna−/− liver tissue is stained with A-type lamin-specific antibodies. Liver cryosections of wild type (A-D) and Lmna−/− (E-H) mice were stained with either pAb bs-01 (A, B, E, F), pAb H-110 (C and G) or mAb R27 (D and H). DNA was stained with Hoechst 33258 (A’-H’). Specimens were analyzed by confocal microscopy and individual channels were merged (A”-H”). All three antibodies stained the NE of Lmna−/− hepatocytes, although signal intensities were slightly reduced compared with wild type controls (compare overviews in A and E). Additionally, Lmna−/− nuclei were frequently misshapen (see panels F-F” and G-G”). Bars, 10 µm.
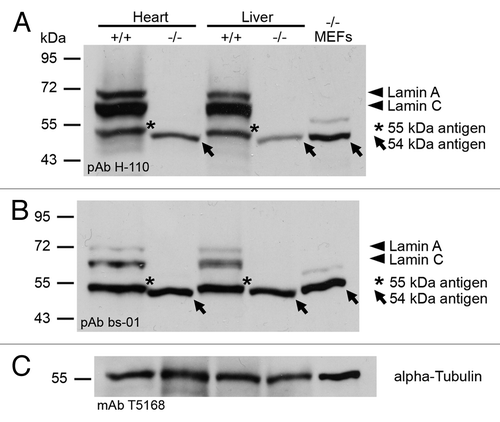
In a second step, we analyzed for the actual presence of an A-type lamin-related antigen in Lmna−/− MEFs and tissues interacting with pAb bs-01 and pAb H-110 by immunoblotting (). In line with the findings reported previously,Citation1 both antibodies proved the absence of full-length lamins A (70 kDa) and C (60 kDa) in Lmna−/− heart, liver and MEFs. However, in these samples both antibodies clearly reacted with a distinct antigen migrating at about 54 kDa. Although both pAb bs-01 and pAb H-110 detected bands of similar molecular weight (~55 kDa) in wild type controls as well (heart and liver), these antigens migrated slightly higher indicating that they represent proteins different from those found in Lmna−/− samples (see also below). To define the relative expression of the 54 kDa antigen in Lmna−/− tissues in comparison to the amount of A-type lamins expressed in wild type mice, we quantified the corresponding protein bands detected with pAb H-110 from heart and liver. This revealed that the relative expression of the 54 kDa antigen is reduced to 24% in Lmna−/− heart and 25% in Lmna−/− liver compared with the total amount of A-type lamins (A and C) in the corresponding wild type controls. When specifically compared with wild type lamin A, however, the relative expression of the 54 kDa antigen was not significantly altered (87% in heart and 110% in liver). Together these data indicated that, although full-length lamins A and C are absent in Lmna−/− mice, these animals apparently express an A-type lamin-related 54 kDa antigen at a level that is similar to the expression of lamin A in wild type tissues.
Figure 3. Detection of a 54 kDa antigen in Lmna−/− samples with different A-type lamin-specific antibodies by immunoblotting. Lmna−/− and wild type samples were subjected to SDS-PAGE followed by western blotting. Membranes were stained with either anti-lamin A/C pAb H-110 (A) or anti-lamin A/C pAb bs-01 (B). Sample loading was controlled with anti-α-tubulin mAb T5168 (C). Lamins A (70 kDa) and C (60 kDa) are indicated by arrowheads. The 54 kDa antigen of Lmna−/− samples is marked by arrows. Asterisks indicate a 55 kDa antigen present in wild type samples.
Detection of a cDNA encoding a yet unnoticed Lmna gene product in Lmna−/− MEFs and tissues
Implicating apparent inconsistencies with the initial studyCitation1 that reported absence of both full-length and truncated lamin A/C proteins in Lmna−/− mice, the results described above appeared rather puzzling thus far. One possible explanation for the divergent observations might be the use of different antibodies. To resolve this issue, we considered it necessary to perform additional experiments independent of the use of further antibodies.
To this, we claimed that if a truncated lamin A/C protein would indeed be present in Lmna−/− mice, a corresponding mRNA should be detectable as well. With regard to the targeted disruption of the Lmna gene performed by Sullivan et al. who replaced exons 8 to part of 11 by a neomycin resistance cassette, we computationally generated a cDNA encoding a putative Lmna gene product consisting of exons 1 to 7 and 12 but lacking exons 8 to 11 (therefore referred to as lamin A∆8–11). A corresponding lamin A∆8–11 mRNA, which could be produced by alternative splicing in vivo, would exclude the gene knockout construct and would therefore probably give rise to a stable polypeptide. In silico translation (http://expasy.org/tools/pi_tool.html) revealed a predicted mass of a putative lamin A∆8–11 protein of 53.5 kDa. This calculated mass would in fact correspond well to the size of the antigen detected in Lmna−/− samples by immunoblotting.
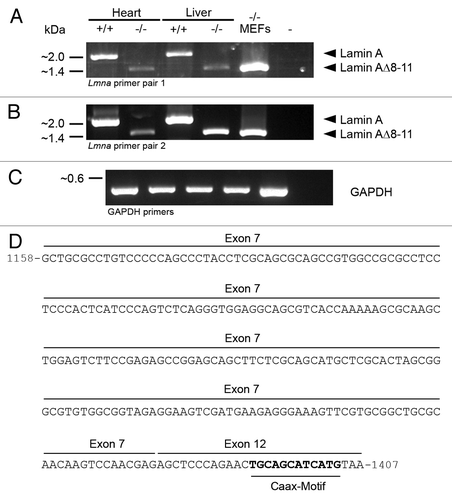
To test for the existence of an mRNA encoding lamin A∆8–11 in vivo, we performed RT-PCR with Lmna−/− MEFs and tissues using two different lamin A-specific primer sets with binding in exons 1 (5′) and 12 (3′) each (). Using these primers, amplicons corresponding to full-length lamin A (~2 kbp) were readily detectable in wild type controls (heart and liver). As expected from our immunoblots and in line with the data provided by Sullivan et al.Citation1 these amplicons were absent in MEFs and tissues isolated from Lmna−/− mice. Instead, in all Lmna−/− samples PCR products well matching the size of the putative lamin A∆8–11 message (~1.4 kbp) could be amplified. Consistent with the notion that the 54 kDa antigen of Lmna−/− samples and the 55 kDa antigen of wild type controls detected by immunoblotting are different proteins, a corresponding ~1.4 kbp band was not detected in RT-PCR reactions of wild type controls (see also below). To verify the predicted splicing from exon 7 to 12 in Lmna−/− mice, the obtained ~1.4 kbp PCR products were cloned and sequenced (). This revealed the expected nucleotide sequence of lamin A∆8–11 comprising exons 1–7 and 12 but lacking exons 8–11 of Lmna. Together, these data unequivocally demonstrate that the truncated lamin A∆8–11 is in fact expressed in Lmna−/− MEFs and tissues at least on the transcriptional level.
Figure 4. RT-PCR reveals the presence of an mRNA encoding the truncated Lmna gene product lamin A∆8–11 in Lmna−/− heart, liver and MEFs. Two different primer sets were used to analyze the expression of Lmna gene products in Lmna−/− samples and wild type controls. Primers of pair 1 (A) were complementary to the protein coding sequence of lamin A including the start codon encoded by exon 1 and the stop codon encoded by exon 12. Primers of pair 2 (B) specifically bound in the 5′UTR and 3′UTR of lamin A encoded by exons 1 and 12, respectively. Positive controls were performed with GAPDH-specific primers (C). The amplicons corresponding to full-length lamin A (~2 kbp) and lamin A∆8–11 (~1.4 kbp) are indicated by arrowheads. (D) To verify the specificity of RT-PCR amplification, PCR products were cloned and sequenced. The 3′-end of the sequence of the 1.4 kbp amplicons obtained from Lmna−/− samples (i.e., lamin A∆8–11) is depicted. Here, nucleotides encoded by exon 7 of Lmna are spliced to exon 12. Nucleotides 1–1157 (not depicted) are encoded by exons 1–6 of Lmna. Nucleotides coding for a CaaX-motif at the C-terminus of the corresponding polypeptide are given in bold.
Lamin A∆8–11 is a truncated Lmna gene product that is present as a stable protein in Lmna−/− MEFs
Since expression of an mRNA is not sufficient evidence for the presence of the corresponding protein we had to go back to the protein level for further analysis. To validate whether lamin A∆8–11 is present as a stable polypeptide in Lmna−/− cells and to determine whether this is identical with the 54 kDa antigen detected in immunoblots of Lmna−/− samples, this antigen was purified by biochemical approaches and analyzed by mass spectrometry.
To this end, Lmna−/− MEFs were fractionated by successive extraction with 1% Triton X-100, DNaseI, 1 M sodium chloride and 8 M urea according to a protocol described previously.Citation39 As expected, the vast majority of cellular proteins were extractable with 1% Triton X-100, DNaseI and 1 M sodium chloride (). In contrast, the 54 kDa antigen of Lmna−/− MEFs was almost completely retained in the insoluble fraction under these conditions and could not be solubilized until treated with 8 M urea (). Since resistance to extraction with non-ionic detergents, DNaseI and high salt is a common feature of karyoskeletal structures,Citation40-Citation43 this result was consistent with the possibility that the 54 kDa antigen found in Lmna−/− cells might in fact be a nuclear lamin.
Figure 5. Purification of the 54 kDa antigen of Lmna−/− MEFs by salt extraction and ion exchange chromatography. (A and B) Lmna−/− MEFs were successively extracted with 1% Triton X-100 (ST), DNaseI and 1 M NaCl (SNaCl). Salt resistant pellets were solubilized in 8 M urea buffer (Surea). Equivalents of respective supernatants (ST, SNaCl and Surea), the final pellet (Purea) as well as whole cells (control; C) were analyzed by SDS-PAGE followed by staining with Coomassie brilliant blue (A) or immunoblotting with pAb bs-01 (B). As evidenced by Coomassie staining, the vast majority of cellular proteins were solubilized by extraction with Triton X-100, DNaseI and 1 M NaCl. In contrast, the 54 kDa antigen of Lmna−/− MEFs was not solubilized until extracted with 8 M urea. The abundant protein band of about 34 kDa (lane SNaCl; marked by arrow) corresponds to the supplemented DNaseI. (C) Salt resistant fractions obtained from cell fractionation were further processed by ion exchange chromatography on a CM-Sepharose matrix. Elution was performed by incremental increase of the NaCl concentration. For each concentration 20 fractions with a volume of 1.5 ml each were used. Names of fractions are given as “x.y.” Here, x indicates the NaCl concentration (mM) and y indicates the number of the fraction (1–20). Eluted fractions were analyzed with regard to the presence of the 54 kDa antigen by western blotting with pAb bs-01. The majority of the 54 kDa antigen eluted at 60 mM of NaCl in a reproducible manner. (D) Protein fractions containing the 54 kDa antigen were separated by SDS-PAGE followed by silver staining. The bands corresponding to the 54 kDa antigen (as judged by the elution profile; see arrows) were recovered from the gels and subjected to mass spectrometry.
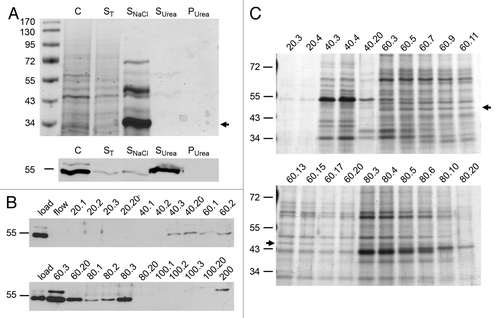
Salt resistant fractions were used for further purification by ion exchange chromatography according to Krohne.Citation44 Protein binding to a carboxymethyl (CM) sepharose matrix was performed at pH 6.0. As verified by immunoblotting, binding of the 54 kDa antigen to the matrix occurred quantitatively under these conditions (), indicating that its isoelectric point was above 6.0 which was in line with the theoretical value (6.75) of a putative lamin A∆8–11 obtained by computational analysis (http://expasy.org/tools/pi_tool.html). Elution of bound proteins was performed by incremental increase of sodium chloride concentration and the elution profile of the 54 kDa antigen was monitored by immunoblotting. The majority of the 54 kDa antigen eluted at 60 mM of sodium chloride in a reproducible manner (). Protein fractions were analyzed by SDS-PAGE followed by silver staining (). The protein band corresponding to the 54 kDa antigen (as judged by the elution profile) was recovered from the gel and subjected to mass spectrometry.
Since we assumed that the 54 kDa antigen isolated from Lmna−/− cells constitutes lamin A∆8–11, our analyses should reveal the presence of peptides encoded by exons 1–7 of Lmna (i.e., residues 1–460 of lamin A/C) whereas peptides encoded by exons 8–11 (i.e., residues 461–657 of pre-lamin A) should consistently be absent. In fact, this proved correct in five independent runs with samples of three independent salt extractions (34% to 70% sequence coverage; Fig. S1). In contrast, analysis of full-length lamin A that was extracted from wild type cells and used as a positive control revealed multiple peptides (sequence coverage 69%) including peptides specific for the C-terminal part of lamin A encoded by exons 8–11. Moreover, mass spectrometric analysis of the 55 kDa antigen that was detected in immunoblots of wild type controls with A-type lamin-specific antibodies revealed a peptide encoded by exon 8 which is clearly absent from lamin AΔ8–11 (Fig. S2). This is consistent with the notion that the 55 kDa antigen of wild type cells is not identical with lamin AΔ8–11 and rather reflects a lamin A/C degradation product.Citation45,Citation46 Thus, by our analyses we could clearly demonstrate the presence of the truncated lamin A mutant Δ8–11 in the Lmna−/− mouse line which was assumed to completely lack A-type lamin expression so far.
Careful analysis of the lamin A∆8–11 sequence revealed further significant implications. The C-terminal CaaX-motif (CSIM) of pre-lamin A is target of extensive post-translational processing including cysteine S-farnesylation, endoproteolytic cleavage of the tripeptide SIM and carboxy-methylation of the remaining farnesyl-cysteine. Finally, by proteolytic cleavage at tyrosine-647 a 15-mer peptide including the farnesylated cysteine is removed from the very C-terminus by metalloprotease Zmpste24 thus producing mature lamin A.Citation47-Citation55 Interestingly, all sequence requirements for C-terminal CaaX-processing of pre-lamin A excluding the final proteolytic maturation step performed by Zmpste24 are encoded by exon 12 of Lmna which is present in the lamin AΔ8–11 mRNA. The cleavage site surrounding tyrosine-647 encoded by exon 11, however, is clearly absent from lamin AΔ8–11. This suggested that lamin A∆8–11 might undergo uncommon post-translational processing resulting in the persistence of a permanently farnesylated A-type lamin in Lmna−/− mice. Unfortunately, we could not detect a farnesylated C-terminal peptide of lamin AΔ8–11 in our mass spectrometry, probably due to high hydrophobicity caused by farnesylation. A non-farnesylated peptide encoded by exon 12, however, was neither detectable. Moreover, several studies demonstrate that a C-terminal CaaX-motif is clearly sufficient for post-translational farnesylation of target proteins.Citation56-Citation58 This, together with the fact that Zmpste24 is the only known enzyme able to cleave farnesylated pre-lamin A, indicates that lamin A∆8–11 probably constitutes a permanently farnesylated lamin A mutant.
Discussion
In summary, the data reported in our study reveal that the truncated Lmna mutant lamin AΔ8–11 persists in the Lmna−/− mouse line which has previously been considered as completely deficient for A-type lamins.Citation1 This is an unexpected but important finding that sheds new light on the interpretation of phenotypes that have been observed in Lmna−/− mice (and derived cell lines) and their underlying molecular mechanism. Due to the expression of a truncated Lmna gene product, Lmna−/− mice should no longer be considered as “null.” Thus, taking up an issue that has recently been discussed for other Lmna alleles,Citation59 two scenarios, which are not mutually exclusive, are conceivable to explain the phenotypes of Lmna−/− mice: First, resulting from the deletion of exons 8–11 and the consequent loss of significant domains, lamin A∆8–11 could act as a functionally hypoactive protein carrying out certain functions but failing to perform others (loss-of-function model). Second, if uncommon post-translational processing leading to potential permanent farnesylation of lamin A∆8–11 should in fact occur, it could also act as a toxic molecule with a dominant-negative effect (gain-of-function model).
The latter notion (gain-of-function model) would be in line with multiple reports demonstrating that disturbance of proper pre-lamin A processing elicits severe disorders in various mouse models and in humans.Citation16,Citation47,Citation54,Citation60-Citation65 In the case of lamin A∆8–11, however, several findings indicate that its toxicity is relatively low. First, mice heterozygous for an Lmna allele encoding the truncated lamin A variant progerin (LmnaHG/+) - which in fact is a toxic lamin - suffer from severe progeroid syndromes with relatively broad phenotypes (reduced growth rates, bone abnormalities, loss of body fat) and die prematurely.Citation62 In contrast, mice heterozygous for lamin A∆8–11 (Lmna+/−) have a more or less normal life span and feature a much milder and more subtle phenotype leading to late-onset cardiomyopathy.Citation1,Citation24 Second, the devastating disease phenotype of Zmpste24-deficient mice (Zmpste24−/−, Lmna+/+) that are unable to perform the final cleavage of farnesylated pre-lamin A to produce mature lamin A is completely eliminated in a heterozygous Lmna background (Zmpste24−/−, Lmna+/−; a situation which is equivalent to heterozygosity for lamin A∆8–11).Citation66 Thus, we assume lamin A∆8–11 to be less toxic than both progerin and farnesylated pre-lamin A in vivo. Despite this, we of course cannot rule out at present that lamin A∆8–11 features some sort of toxicity on the cellular level. A slight toxic effect emanating from lamin A∆8–11 would rather provide a viable explanation why transfection of Lmna−/− MEFs with lamin A/C constructs failed to fully restore normal nuclear morphology and did not rescue certain mechanical stress phenotypes in various in vitro approaches.Citation27,Citation32 Supporting this aspect, the phenotypic comparison of heterozygous Lmna+/− miceCitation24 with heterozygous animals of a novel Lmna mouse modelCitation67 could be interpreted as a further in vivo indication for a moderate dominant-negative effect associated with lamin A∆8–11 (see below).
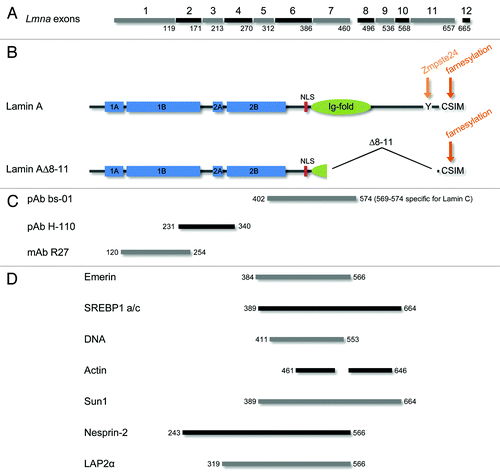
Nonetheless, many of the phenotypes observed in Lmna−/− mice and MEFs are in line with the assumption that, due to the lack of significant domains encoded by exons 8–11, lamin A∆8–11 primarily acts as a hypoactive protein (loss-of-function model). Concerning their molecular structure, lamins are composed of a central α-helical rod domain flanked by a non-helical N-terminal head and a C-terminal tail that contains a nuclear localization signal (NLS) and adopts a conserved immunoglobulin-like fold (Ig-fold).Citation68-Citation70 Compared with the structure of wild type lamin A, the integrity of both the N-terminal head and the α-helical rod are completely unaffected by the truncation of lamin A∆8–11 (). Moreover, the region of the C-terminal lamin tail directly adjacent to the α-helical rod (including the NLS) is still present in the mutated lamin. Thus, lamin A∆8–11 comprises the protein domains that are currently considered as most crucial for normal lamin assembly into higher-order nuclear structures.Citation71-Citation73 In contrast, most of the residues in the lamin tail forming the conserved Ig-fold are absent from lamin A∆8–11. Since the C-terminal Ig-fold is an important domain for interactions of lamins with multiple binding partners,Citation22,Citation74 its deletion predicts considerable perturbations of lamin binding to other proteins. In fact, a number of studies that used Lmna−/− mice and MEFs reported phenotypes that can consistently be ascribed to disturbed protein interactions of laminA∆8–11 and therefore strongly support the loss-of-function model (see also ): Lmna−/− mice were initially characterized by severe muscular dystrophy and mislocalization of emerin.Citation1 Since most of the emerin binding site which has been mapped to residues 384–566 of lamins A/CCitation75 is absent from lamin A∆8–11, loss of emerin binding and consequent emerin mislocalization in Lmna−/− cells seems plausible. Moreover, residues 389–664 of lamin A were found to interact with SREBP1 a/c which is known to promote adipocyte differentiation and lipid synthesis. Disturbance of lamin A-SREBP1 interactions is associated with certain types of lipodystrophy.Citation76,Citation77 Thus, absence of a major portion of the SREBP-binding domain from lamin A∆8–11 could at least in part account for the loss of white body fat reported for Lmna−/− mice.Citation1 In a similar manner, loss of DNA binding in lamin A∆8–11 (residues 411–553 of lamins A/C)Citation78 could cause the loss of peripheral heterochromatin observed in Lmna−/− cells.Citation1 Additionally, the C-terminal tail domains of lamins A/C were found to contribute to their interactions with several structural proteins including actin,Citation79,Citation80 SUN-domain proteinsCitation30,Citation81 and Nesprin-2 29. Consistent with the fact that the C-terminal tail is virtually completely absent from lamin A∆8–11, integrity and functionality of complexes formed with these binding partners were altered in Lmna−/− cells and mice.Citation28-Citation30,Citation82-Citation84 Finally, the LAP2α binding domain (residues 319–566 of lamin A/C)Citation85 is partially deleted in lamin A∆8–11. Since nucleoplasmic LAP2α-lamin A complexes were found to regulate retinoblastoma protein function,Citation86 reduced binding of LAP2α to lamin A∆8–11 could thus contribute to the abnormalities of retinoblastoma protein function and cell-cycle control observed in Lmna−/− MEFs.Citation33,Citation35,Citation36
Figure 6. Lamin A∆8–11 is a truncated A-type lamin that lacks domains important for protein interactions and post-translation processing. (A) Lamin A encoding exons of the murine Lmna gene are marked by numbers 1–12. The last residue of the lamin A protein sequence encoded by each exon is indicated. Full-length pre-lamin A comprises residues 1–665 encoded by exons 1–12. In contrast, residues 461–657 of full-length lamin A encoded by exons 8–11 are absent from lamin A∆8–11. (B) Important domains and sequence motifs of full-length lamin A and lamin A∆8–11 are depicted. Neither the integrity of the central α-helical rod domain (comprising coils 1A, 1B, 2A and 2B; blue) nor the presence of the nuclear localization signal (NLS; red) is altered in lamin A∆8–11. However, the majority of residues forming the Ig-fold (green) are absent. Notably, the C-terminal sequence motifs implicated in post-translational processing (orange) of pre-lamin A are partly deleted in laminA∆8–11 as well. While the C-terminal CSIM (target of protein farnesylation) appears to be present in this truncated A-type lamin as judged from its mRNA sequence, the site of cleavage by metalloprotease Zmpste24 around tyrosine-647 is clearly absent. This suggests that lamin A∆8–11 could constitute a permanently farnesylated A-type lamin. (C) Epitopes of the anti-lamin A antibodies used in this study reside within the depicted regions. (D) Mapped binding epitopes of the indicated A-type lamin-binding partners (emerin, SREBP1 a/c, DNA, actin, Sun1, Nesprin-2 and LAP2α) are completely or in part absent from lamin A∆8–11. Respective residues of full-length lamin A are given by numbers. For references see text.
Recently, Kubben et al.Citation67 developed a novel Lmna null mouse based on genetrap technology (LmnaGT−/−) that was characterized by post-natal maturation defects of adipose, cardiac and muscle tissues. Compared with Lmna−/− mice the phenotype of LmnaGT−/− animals was similar with regard to the tissues affected but was more severe in terms of temporal onset and progression of the symptoms. Consequently, premature death occurred much earlier in the LmnaGT−/− model (2–3 weeks pp vs. 6–8 weeks pp). Obvious differences concerning the applied gene knockout strategies were discussed as a most probable reason for the different phenotypes of both mouse models.Citation67 Our current data strongly supports this notion, since the persistence of a truncated, hypoactive A-type lamin in the Lmna−/− strain plausibly explains its relatively milder phenotype. Interestingly, comparison of both Lmna models regarding the characteristics of heterozygous animals draws a different picture. Here, Wolf et al.Citation24 reported progressive electrophysiological abnormalities commencing around 4 weeks after birth and the emergence of late-onset cardiomyopathy in aged (50 weeks) heterozygous Lmna+/− mice. In contrast, no such defects were detectable in heterozygous LmnaGT+/− animals up to one year in age.Citation67 Certainly, genetic background-dependent lamin A/C haploinsufficiency provides one possible explanation for these differences. Alternatively, however, based on our data, the emergence of specific age-dependent cardiac abnormalities in heterozygous Lmna+/− mice could also be due to a moderate toxic effect associated with lamin A∆8–11. In this case, the molecular properties of the truncated lamin mutant A∆8–11 would combine pronounced loss-of-function features with a moderate dominant-negative effect.
In conclusion, our current study demonstrates the expression of the lamin deletion mutant A∆8–11 in the Lmna−/− mouse line which was initially thought to completely lack A-type lamin expression. To avoid potential misconceptions we therefore propose this mouse line to be rather considered as Lmna∆8–11 for future discussion. With regard to the considerations discussed above, Lmna∆8–11 mice and derived MEFs seem to remain a viable loss-of-function model to study A-type lamin function in ongoing experiments. Nonetheless, our findings set obvious limitations to future studies and the expression of lamin A∆8–11 in Lmna∆8–11 mice should be carefully taken into account for their interpretation.
Materials and Methods
Lmna−/− mice and MEFs
Generation of Lmna−/− mice was described previously.Citation1 Lmna−/− MEFs were cultured in DMEM Glutamax (Gibco) with 10% (v/v) FCS, 1% (v/v) Penicillin/Streptomycin and 1% (v/v) L-Glutamin at 37°C and 5% CO2 according to standard procedures.
Antibodies
Polyclonal antibodies bs-01 were produced in rabbit. To this, a His-tagged fusion construct comprising the C-terminal part of murine lamin C (aa 402–574) was expressed in E. coli Rosetta (Novagen), purified on Ni-NTA agarose resin (Qiagen) and used for immunization of rabbits (Seqlab, Göttingen, Germany). Sera obtained from final bleedings were affinity-purified against the antigen coupled to a NHS-activated HiTrap column according to the manufacturer’s instructions (GE Healthcare). Affinity-purified bs-01 was diluted 1:100 for immunohistochemistry (IC) or 1:10.000 for immunoblotting (IB). Further primary antibodies were rabbit pAb H-110 (IC 1:50; IB 1:10.000; Santa Cruz Biotechnology), mAb R27 (IC 1:100;Citation38) and mAb T5168 (IB 1:20.000; Sigma-Aldrich). Secondary antibodies used in this study were purchased from Dianova or LI-COR and used according to the manufacturer’s instructions.
Immunofluorescence microscopy
MEFs and liver cryosections (5 µm) were fixed with 1% formaldehyde in PBS (140 mM NaCl, 2.6 mM KCl, 6.4 mM Na2HPO4, 1.4 mM KH2PO4, pH 7.4) and permeabilized with 0.1% Triton X-100 in PBS. Specimens were blocked with PBT (PBS, 1.5% BSA, 0.1% Tween-20) followed by successive incubation with primary and secondary antibodies (Dianova) for 2 h and 30 min, each. DNA was counterstained with Hoechst 33258. Specimens were embedded in Glycerol-PBS (1:1) and analyzed by confocal microscopy on a TCS-SP2 (Leica Microsystems). Fluorochromes were scanned sequentially using appropriate laser settings (Hoechst 33258: excitation 405 nm, detection 420–500 nm; Cy2: excitation 488 nm, detection 500–580 nm). Single section scans were taken with a 63×/1.40 HCX PL APO lbd.BL oil-immersion objective, pinhole at 1.00 P AU and 2-fold accumulation. Depending on the experiment, either three or ten sequenced single section scans were used to calculate maximum 2D projection with Leica TCS-SP2 microscope software. Image data were processed with Adobe Photoshop CS2 (Adobe Systems). Laser intensities and gain settings during image acquisition as well as image processing protocols were equivalent for Lmna−/− and control samples.
SDS-PAGE and immunoblotting
SDS-PAGE was performed on 10% polyacrylamide gels according to LaemmliCitation87 or Thomas and KornbergCitation88 depending on the experiment. Proteins were either stained with Coomassie brilliant blue G 250 (Serva) or silver nitrate (Applichem). For immunoblotting, proteins were transferred to nitrocellulose membranes as described by Matsudaira.Citation89 Membranes were blocked over night at 4°C in TBST buffer (10 mM Tris/HCl, 150 mM NaCl, 0.1% Tween 20, pH 7.4) containing 10% milk powder and subsequently incubated with primary antibodies for 1–2 h at room temperature. For conventional immunodetection, membranes were then incubated with respective peroxidase-conjugated secondary antibodies (Dianova; 1 h at room temperature) and bound antibodies were detected using Western Lightning Plus-ECL (Perkin Elmer). For quantitative immunoblotting, secondary antibodies were goat anti-rabbit IRDye 800 CW and goat anti-mouse IRDye 680 LT (LI-COR; incubation for 1 h at room temperature in the dark). Here, blots were scanned with the LI-COR Odyssey infrared imager and protein bands were quantified with the manufacturer’s software. For each sample, the expression of A-type lamins (A, C and A∆8–11) was normalized to α-tubulin. This allowed direct comparison of the relative lamin expression levels in wild type and corresponding Lmna−/− tissues.
RT-PCR
Total RNA was prepared from MEFs and tissues using TriFAST (Peqlab Biotechnology). cDNA was produced from 1 µg of total RNA using oligo-dT primers (Fermentas) and M-MLV reverse transcriptase (Promege). Lamin A-specific primers were 5′-ATGGAGACCCCGTCACA-3′ and 5′-TTACATGATGCTGCAGTTCT-3′ (primer set 1) or 5′-AGCACTGTCACCCTGCCGG-3′ and 5′-CTGCCTGGCAGGTCCCAGA-3′ (primer set 2), respectively. Positive controls were performed with GAPDH-specific primers (5′- GGGCCCACTTGAAGGGTGGAGC-3′ and 5′- GTCAGATCCACGACGGACACATTGG-3′). To control specificity of amplification, PCR products were cloned into pSC-B-amp/kan (StrataClone Blunt PCR Cloning Kit; Agilent Technologies) and sequenced by GATC Biotech.
Cell fractionation and ion exchange chromatography
Cell fractionation was essentially performed as described previously.Citation39 Salt resistant pellets were solubilized in urea buffer (8 M urea, 50 mM sodium acetate, 2 mM DTT, pH 6.0). Ion exchange chromatography was performed on CM Sepharose Fast Flow (GE Healthcare) according to Krohne.Citation44 Bound proteins were eluted with urea buffer supplemented with NaCl. NaCl concentrations were successively raised by 20 mM increments. Eluted fractions were analyzed by SDS-PAGE and immunoblotting.
Mass Spectrometry
ESI mass spectrometry was performed as follows. Protein bands were excised from the gel and proteins in the gel pieces were reduced with 10 mM DTT for 1 h at 56°C and alkylated with 55 mM iodoacetamide for 30 min at RT. The gel pieces were washed three times alternately with 40 mM NH4HCO3 and ethanol. After drying with acetonitrile an in-gel trypsin digestion was performed by adding 100 ng trypsin in 40 mM NH4HCO3 and incubation overnight at 37°C. Tryptic peptides were extracted from the gel plugs with acetonitrile/0.1% TFA and analyzed by nanoLC-ESI-MS/MS. Tryptic peptides were separated using a nanoAcquity UPLC system (Waters GmbH). Liquid chromatography separation was performed using a C18 trap column (180 µm × 20 mm) with a particle size of 5 µm and a BEH130 C18 main- column (100 µm × 100 mm) with a particle size of 1.7µm (Waters GmbH). Flow rate was 0.4 µl / min and the gradient was from 0 to 90% acetonitrile in 0.1 formic acid over 90 min. The UPLC system was coupled online to an LTQ Orbitrap XL mass spectrometer (Thermo Scientific). Data were acquired by scan cycles of one FTMS scan with a resolution of 60000 at m/z 400 and a range from 300 to 2000 m/z in parallel with six MS/MS scans in the ion trap of the most abundant precursor ions. Database searches were performed using the MASCOT search engine (Matrix Science; version 2.2) against the NCBInr database (release 2011–10–14). Peptide mass tolerance was 5 ppm and fragment mass tolerance 0.4 Da. Carbamidomethylation of cysteine was set as fixed modification. Variable modifications included oxidation of methionine and deamidation of asparagine and glutamine. One missed cleavage site in case of incomplete trypsin hydrolysis was allowed.
Alternatively, tandem mass spectrometry was performed using online Q-ToF in the following manner. Protein bands were excised from SDS-PAGE stained with Coomassie Blue and treated as described in Testerink et al.Citation90 The resulting slices were reduced and then S-alkylated using dithiothreitol and iodoacetamide, respectively. After vacuum-drying, the proteins were digested in gel using Trypsin Gold (Promega) overnight before peptides were extracted according to Shevchenko et al.Citation91 The peptide containing eluates were evaporated and reconstituted in 6 µl 60% Acetonitrile and 1% formic acid. The resulting solutions were injected onto an Ultimate 2000 nano-HPLC system (LC Packings, Amsterdam, Netherlands) equipped with a PepMap100 C18 reverse phase column (25 cm × 75 µm i.d.; Dionex).The peptides were eluated using a 45 min linear gradient with increasing acetonitrile concentration and a flow rate of 0.3 µl/min and directly ionized by electrospray in a Q-ToF (Micromass). The most intense ions in survey scans acquired from m/z 350–1400 were selected in a data dependent mode for low energy collision induced dissociation (MS/MS). The generated spectra were processed using the MaxEnt3 algorithm in the Masslynx Proteinlynx software. The generated peak lists were submitted to an internally licensed version of MASCOT (Matrix Science) and searched against a complete orf translation from mouse (MSIPI_mouse mouse_3.67).Citation92 Two miscleavages, variable oxidation of methionine and a tolerance of 0.6 Da were allowed for peptides and MS/MS. Probabilistic MASCOT scoring was used to evaluate the identified peptides and proteins. For each sample at least 2 biological and technical replicates were analyzed.
| Abbreviations: | ||
| ESI | = | electrospray ionization |
| mAb | = | monoclonal antibody |
| MEF | = | murine embryonic fibroblast |
| NE | = | nuclear envelope |
| NLS | = | nuclear localization signal |
| pAb | = | polyclonal antibodies |
Additional material
Download Zip (696.7 KB)Acknowledgments
We thank Colin L. Stewart, Werner W. Franke and Georg Krohne for fruitful discussions, and Uwe Warnken and Kerstin Kammerer for their help with the mass spectrometry analyses. mAb R27 was kindly provided by Georg Krohne. The work was supported by DFG grant Al1090/1–1 to M.A., the Graduate School GK 1048 “Molecular basis of organ development in vertebrates” and the Elitenetzwerk Bayern.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Material
Supplemental material may be found here: www.landesbioscience.com/journals/nucleus/article/21676
References
- Sullivan T, Escalante-Alcalde D, Bhatt H, Anver M, Bhat N, Nagashima K, et al. Loss of A-type lamin expression compromises nuclear envelope integrity leading to muscular dystrophy. J Cell Biol 1999; 147:913 - 20; http://dx.doi.org/10.1083/jcb.147.5.913; PMID: 10579712
- Fisher DZ, Chaudhary N, Blobel G. cDNA sequencing of nuclear lamins A and C reveals primary and secondary structural homology to intermediate filament proteins. Proc Natl Acad Sci USA 1986; 83:6450 - 4; http://dx.doi.org/10.1073/pnas.83.17.6450; PMID: 3462705
- McKeon FD, Kirschner MW, Caput D. Homologies in both primary and secondary structure between nuclear envelope and intermediate filament proteins. Nature 1986; 319:463 - 8; http://dx.doi.org/10.1038/319463a0; PMID: 3453101
- Höger TH, Krohne G, Franke WW. Amino acid sequence and molecular characterization of murine lamin B as deduced from cDNA clones. Eur J Cell Biol 1988; 47:283 - 90; PMID: 3243285
- Zewe M, Höger TH, Fink T, Lichter P, Krohne G, Franke WW. Gene structure and chromosomal localization of the murine lamin B2 gene. Eur J Cell Biol 1991; 56:342 - 50; PMID: 1802718
- Furukawa K, Hotta Y. cDNA cloning of a germ cell specific lamin B3 from mouse spermatocytes and analysis of its function by ectopic expression in somatic cells. EMBO J 1993; 12:97 - 106; PMID: 8094052
- Furukawa K, Inagaki H, Hotta Y. Identification and cloning of an mRNA coding for a germ cell-specific A-type lamin in mice. Exp Cell Res 1994; 212:426 - 30; http://dx.doi.org/10.1006/excr.1994.1164; PMID: 8187835
- Lin F, Worman HJ. Structural organization of the human gene encoding nuclear lamin A and nuclear lamin C. J Biol Chem 1993; 268:16321 - 6; PMID: 8344919
- Machiels BM, Zorenc AH, Endert JM, Kuijpers HJ, van Eys GJ, Ramaekers FC, et al. An alternative splicing product of the lamin A/C gene lacks exon 10. J Biol Chem 1996; 271:9249 - 53; http://dx.doi.org/10.1074/jbc.271.16.9249; PMID: 8621584
- Nakajima N, Abe K. Genomic structure of the mouse A-type lamin gene locus encoding somatic and germ cell-specific lamins. FEBS Lett 1995; 365:108 - 14; http://dx.doi.org/10.1016/0014-5793(95)00453-G; PMID: 7781761
- Röber RA, Weber K, Osborn M. Differential timing of nuclear lamin A/C expression in the various organs of the mouse embryo and the young animal: a developmental study. Development 1989; 105:365 - 78; PMID: 2680424
- Stewart C, Burke B. Teratocarcinoma stem cells and early mouse embryos contain only a single major lamin polypeptide closely resembling lamin B. Cell 1987; 51:383 - 92; http://dx.doi.org/10.1016/0092-8674(87)90634-9; PMID: 3311384
- Bonne G, Di Barletta MR, Varnous S, Bécane HM, Hammouda EH, Merlini L, et al. Mutations in the gene encoding lamin A/C cause autosomal dominant Emery-Dreifuss muscular dystrophy. Nat Genet 1999; 21:285 - 8; http://dx.doi.org/10.1038/6799; PMID: 10080180
- Shackleton S, Lloyd DJ, Jackson SN, Evans R, Niermeijer MF, Singh BM, et al. LMNA, encoding lamin A/C, is mutated in partial lipodystrophy. Nat Genet 2000; 24:153 - 6; http://dx.doi.org/10.1038/72807; PMID: 10655060
- De Sandre-Giovannoli A, Chaouch M, Kozlov S, Vallat JM, Tazir M, Kassouri N, et al. Homozygous defects in LMNA, encoding lamin A/C nuclear-envelope proteins, cause autosomal recessive axonal neuropathy in human (Charcot-Marie-Tooth disorder type 2) and mouse. Am J Hum Genet 2002; 70:726 - 36; http://dx.doi.org/10.1086/339274; PMID: 11799477
- Eriksson M, Brown WT, Gordon LB, Glynn MW, Singer J, Scott L, et al. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature 2003; 423:293 - 8; http://dx.doi.org/10.1038/nature01629; PMID: 12714972
- Worman HJ, Fong LG, Muchir A, Young SG. Laminopathies and the long strange trip from basic cell biology to therapy. J Clin Invest 2009; 119:1825 - 36; http://dx.doi.org/10.1172/JCI37679; PMID: 19587457
- Muchir A, Bonne G, van der Kooi AJ, van Meegen M, Baas F, Bolhuis PA, et al. Identification of mutations in the gene encoding lamins A/C in autosomal dominant limb girdle muscular dystrophy with atrioventricular conduction disturbances (LGMD1B). Hum Mol Genet 2000; 9:1453 - 9; http://dx.doi.org/10.1093/hmg/9.9.1453; PMID: 10814726
- De Sandre-Giovannoli A, Bernard R, Cau P, Navarro C, Amiel J, Boccaccio I, et al. Lamin A truncation in Hutchinson-Gilford progeria. Science 2003; 300:2055; http://dx.doi.org/10.1126/science.1084125; PMID: 12702809
- Andrés V, González JM. Role of A-type lamins in signaling, transcription, and chromatin organization. J Cell Biol 2009; 187:945 - 57; http://dx.doi.org/10.1083/jcb.200904124; PMID: 20038676
- Dauer WT, Worman HJ. The nuclear envelope as a signaling node in development and disease. Dev Cell 2009; 17:626 - 38; http://dx.doi.org/10.1016/j.devcel.2009.10.016; PMID: 19922868
- Wilson KL, Foisner R. Lamin-binding Proteins. Cold Spring Harb Perspect Biol 2010; 2:a000554; http://dx.doi.org/10.1101/cshperspect.a000554; PMID: 20452940
- Nikolova V, Leimena C, McMahon AC, Tan JC, Chandar S, Jogia D, et al. Defects in nuclear structure and function promote dilated cardiomyopathy in lamin A/C-deficient mice. J Clin Invest 2004; 113:357 - 69; PMID: 14755333
- Wolf CM, Wang L, Alcalai R, Pizard A, Burgon PG, Ahmad F, et al. Lamin A/C haploinsufficiency causes dilated cardiomyopathy and apoptosis-triggered cardiac conduction system disease. J Mol Cell Cardiol 2008; 44:293 - 303; http://dx.doi.org/10.1016/j.yjmcc.2007.11.008; PMID: 18182166
- Alsheimer M, Liebe B, Sewell L, Stewart CL, Scherthan H, Benavente R. Disruption of spermatogenesis in mice lacking A-type lamins. J Cell Sci 2004; 117:1173 - 8; http://dx.doi.org/10.1242/jcs.00975; PMID: 14996939
- Lammerding J, Schulze PC, Takahashi T, Kozlov S, Sullivan T, Kamm RD, et al. Lamin A/C deficiency causes defective nuclear mechanics and mechanotransduction. J Clin Invest 2004; 113:370 - 8; PMID: 14755334
- Broers JL, Peeters EA, Kuijpers HJ, Endert J, Bouten CV, Oomens CW, et al. Decreased mechanical stiffness in LMNA-/- cells is caused by defective nucleo-cytoskeletal integrity: implications for the development of laminopathies. Hum Mol Genet 2004; 13:2567 - 80; http://dx.doi.org/10.1093/hmg/ddh295; PMID: 15367494
- Crisp M, Liu Q, Roux K, Rattner JB, Shanahan C, Burke B, et al. Coupling of the nucleus and cytoplasm: role of the LINC complex. J Cell Biol 2006; 172:41 - 53; http://dx.doi.org/10.1083/jcb.200509124; PMID: 16380439
- Libotte T, Zaim H, Abraham S, Padmakumar VC, Schneider M, Lu W, et al. Lamin A/C-dependent localization of Nesprin-2, a giant scaffolder at the nuclear envelope. Mol Biol Cell 2005; 16:3411 - 24; http://dx.doi.org/10.1091/mbc.E04-11-1009; PMID: 15843432
- Haque F, Lloyd DJ, Smallwood DT, Dent CL, Shanahan CM, Fry AM, et al. SUN1 interacts with nuclear lamin A and cytoplasmic nesprins to provide a physical connection between the nuclear lamina and the cytoskeleton. Mol Cell Biol 2006; 26:3738 - 51; http://dx.doi.org/10.1128/MCB.26.10.3738-3751.2006; PMID: 16648470
- Lee JS, Hale CM, Panorchan P, Khatau SB, George JP, Tseng Y, et al. Nuclear lamin A/C deficiency induces defects in cell mechanics, polarization, and migration. Biophys J 2007; 93:2542 - 52; http://dx.doi.org/10.1529/biophysj.106.102426; PMID: 17631533
- Lammerding J, Fong LG, Ji JY, Reue K, Stewart CL, Young SG, et al. Lamins A and C but not lamin B1 regulate nuclear mechanics. J Biol Chem 2006; 281:25768 - 80; http://dx.doi.org/10.1074/jbc.M513511200; PMID: 16825190
- Johnson BR, Nitta RT, Frock RL, Mounkes L, Barbie DA, Stewart CL, et al. A-type lamins regulate retinoblastoma protein function by promoting subnuclear localization and preventing proteasomal degradation. Proc Natl Acad Sci USA 2004; 101:9677 - 82; http://dx.doi.org/10.1073/pnas.0403250101; PMID: 15210943
- Frock RL, Kudlow BA, Evans AM, Jameson SA, Hauschka SD, Kennedy BK. Lamin A/C and emerin are critical for skeletal muscle satellite cell differentiation. Genes Dev 2006; 20:486 - 500; http://dx.doi.org/10.1101/gad.1364906; PMID: 16481476
- Nitta RT, Jameson SA, Kudlow BA, Conlan LA, Kennedy BK. Stabilization of the retinoblastoma protein by A-type nuclear lamins is required for INK4A-mediated cell cycle arrest. Mol Cell Biol 2006; 26:5360 - 72; http://dx.doi.org/10.1128/MCB.02464-05; PMID: 16809772
- Rodríguez J, Calvo F, González JM, Casar B, Andrés V, Crespo P. ERK1/2 MAP kinases promote cell cycle entry by rapid, kinase-independent disruption of retinoblastoma-lamin A complexes. J Cell Biol 2010; 191:967 - 79; http://dx.doi.org/10.1083/jcb.201004067; PMID: 21115804
- Gonzalez-Suarez I, Redwood AB, Perkins SM, Vermolen B, Lichtensztejin D, Grotsky DA, et al. Novel roles for A-type lamins in telomere biology and the DNA damage response pathway. EMBO J 2009; 28:2414 - 27; http://dx.doi.org/10.1038/emboj.2009.196; PMID: 19629036
- Höger TH, Grund C, Franke WW, Krohne G. Immunolocalization of lamins in the thick nuclear lamina of human synovial cells. Eur J Cell Biol 1991; 54:150 - 6; PMID: 2032545
- Alsheimer M, von Glasenapp E, Hock R, Benavente R. Architecture of the nuclear periphery of rat pachytene spermatocytes: distribution of nuclear envelope proteins in relation to synaptonemal complex attachment sites. Mol Biol Cell 1999; 10:1235 - 45; PMID: 10198069
- Dwyer N, Blobel G. A modified procedure for the isolation of a pore complex-lamina fraction from rat liver nuclei. J Cell Biol 1976; 70:581 - 91; http://dx.doi.org/10.1083/jcb.70.3.581; PMID: 986398
- Senior A, Gerace L. Integral membrane proteins specific to the inner nuclear membrane and associated with the nuclear lamina. J Cell Biol 1988; 107:2029 - 36; http://dx.doi.org/10.1083/jcb.107.6.2029; PMID: 3058715
- Dechat T, Gotzmann J, Stockinger A, Harris CA, Talle MA, Siekierka JJ, et al. Detergent-salt resistance of LAP2alpha in interphase nuclei and phosphorylation-dependent association with chromosomes early in nuclear assembly implies functions in nuclear structure dynamics. EMBO J 1998; 17:4887 - 902; http://dx.doi.org/10.1093/emboj/17.16.4887; PMID: 9707448
- Foisner R, Gerace L. Integral membrane proteins of the nuclear envelope interact with lamins and chromosomes, and binding is modulated by mitotic phosphorylation. Cell 1993; 73:1267 - 79; http://dx.doi.org/10.1016/0092-8674(93)90355-T; PMID: 8324822
- Krohne G. Lamins. Methods Cell Biol 2004; 78:573 - 96; http://dx.doi.org/10.1016/S0091-679X(04)78020-6; PMID: 15646632
- Schirmer EC, Gerace L. The stability of the nuclear lamina polymer changes with the composition of lamin subtypes according to their individual binding strengths. J Biol Chem 2004; 279:42811 - 7; http://dx.doi.org/10.1074/jbc.M407705200; PMID: 15284226
- Clawson GA, Wang YF, Schwartz AM, Hatem CL. The Mr 46,000 nuclear scaffold ATP-binding protein: identification of the putative nucleoside triphosphatase by proteolysis and monoclonal antibodies directed against lamins A/C. Cell Growth Differ 1990; 1:559 - 68; PMID: 1965140
- Bergo MO, Gavino B, Ross J, Schmidt WK, Hong C, Kendall LV, et al. Zmpste24 deficiency in mice causes spontaneous bone fractures, muscle weakness, and a prelamin A processing defect. Proc Natl Acad Sci USA 2002; 99:13049 - 54; http://dx.doi.org/10.1073/pnas.192460799; PMID: 12235369
- Young SG, Fong LG, Michaelis S, Prelamin A. Prelamin A, Zmpste24, misshapen cell nuclei, and progeria--new evidence suggesting that protein farnesylation could be important for disease pathogenesis. J Lipid Res 2005; 46:2531 - 58; http://dx.doi.org/10.1194/jlr.R500011-JLR200; PMID: 16207929
- Corrigan DP, Kuszczak D, Rusinol AE, Thewke DP, Hrycyna CA, Michaelis S, et al. Prelamin A endoproteolytic processing in vitro by recombinant Zmpste24. Biochem J 2005; 387:129 - 38; http://dx.doi.org/10.1042/BJ20041359; PMID: 15479156
- Clarke S, Vogel JP, Deschenes RJ, Stock J. Posttranslational modification of the Ha-ras oncogene protein: evidence for a third class of protein carboxyl methyltransferases. Proc Natl Acad Sci USA 1988; 85:4643 - 7; http://dx.doi.org/10.1073/pnas.85.13.4643; PMID: 3290900
- Dai Q, Choy E, Chiu V, Romano J, Slivka SR, Steitz SA, et al. Mammalian prenylcysteine carboxyl methyltransferase is in the endoplasmic reticulum. J Biol Chem 1998; 273:15030 - 4; http://dx.doi.org/10.1074/jbc.273.24.15030; PMID: 9614111
- Weber K, Plessmann U, Traub P. Maturation of nuclear lamin A involves a specific carboxy-terminal trimming, which removes the polyisoprenylation site from the precursor; implications for the structure of the nuclear lamina. FEBS Lett 1989; 257:411 - 4; http://dx.doi.org/10.1016/0014-5793(89)81584-4; PMID: 2583287
- Beck LA, Hosick TJ, Sinensky M. Isoprenylation is required for the processing of the lamin A precursor. J Cell Biol 1990; 110:1489 - 99; http://dx.doi.org/10.1083/jcb.110.5.1489; PMID: 2335559
- Pendás AM, Zhou Z, Cadiñanos J, Freije JM, Wang J, Hultenby K, et al. Defective prelamin A processing and muscular and adipocyte alterations in Zmpste24 metalloproteinase-deficient mice. Nat Genet 2002; 31:94 - 9; PMID: 11923874
- Kilic F, Dalton MB, Burrell SK, Mayer JP, Patterson SD, Sinensky M. In vitro assay and characterization of the farnesylation-dependent prelamin A endoprotease. J Biol Chem 1997; 272:5298 - 304; http://dx.doi.org/10.1074/jbc.272.8.5298; PMID: 9030603
- Reiss Y, Goldstein JL, Seabra MC, Casey PJ, Brown MS. Inhibition of purified p21ras farnesyl:protein transferase by Cys-AAX tetrapeptides. Cell 1990; 62:81 - 8; http://dx.doi.org/10.1016/0092-8674(90)90242-7; PMID: 2194674
- Wiedłocha A, Falnes PO, Rapak A, Klingenberg O, Muñoz R, Olsnes S. Translocation of cytosol of exogenous, CAAX-tagged acidic fibroblast growth factor. J Biol Chem 1995; 270:30680 - 5; http://dx.doi.org/10.1074/jbc.270.51.30680; PMID: 8530506
- Goldstein JL, Brown MS, Stradley SJ, Reiss Y, Gierasch LM. Nonfarnesylated tetrapeptide inhibitors of protein farnesyltransferase. J Biol Chem 1991; 266:15575 - 8; PMID: 1874715
- Davies BSJ, Coffinier C, Yang SH, Barnes RH 2nd, Jung H-J, Young SG, et al. Investigating the purpose of prelamin A processing. Nucleus 2011; 2:4 - 9; http://dx.doi.org/10.4161/nucl.2.1.13723; PMID: 21647293
- Dechat T, Shimi T, Adam SA, Rusinol AE, Andres DA, Spielmann HP, et al. Alterations in mitosis and cell cycle progression caused by a mutant lamin A known to accelerate human aging. Proc Natl Acad Sci USA 2007; 104:4955 - 60; http://dx.doi.org/10.1073/pnas.0700854104; PMID: 17360326
- Davies BS, Barnes RH 2nd, Tu Y, Ren S, Andres DA, Spielmann HP, et al. An accumulation of non-farnesylated prelamin A causes cardiomyopathy but not progeria. Hum Mol Genet 2010; 19:2682 - 94; http://dx.doi.org/10.1093/hmg/ddq158; PMID: 20421363
- Yang SH, Meta M, Qiao X, Frost D, Bauch J, Coffinier C, et al. A farnesyltransferase inhibitor improves disease phenotypes in mice with a Hutchinson-Gilford progeria syndrome mutation. J Clin Invest 2006; 116:2115 - 21; http://dx.doi.org/10.1172/JCI28968; PMID: 16862216
- Moulson CL, Go G, Gardner JM, van der Wal AC, Smitt JH, van Hagen JM, et al. Homozygous and compound heterozygous mutations in ZMPSTE24 cause the laminopathy restrictive dermopathy. J Invest Dermatol 2005; 125:913 - 9; http://dx.doi.org/10.1111/j.0022-202X.2005.23846.x; PMID: 16297189
- Navarro CL, Cadiñanos J, De Sandre-Giovannoli A, Bernard R, Courrier S, Boccaccio I, et al. Loss of ZMPSTE24 (FACE-1) causes autosomal recessive restrictive dermopathy and accumulation of Lamin A precursors. Hum Mol Genet 2005; 14:1503 - 13; http://dx.doi.org/10.1093/hmg/ddi159; PMID: 15843403
- Davies BS, Fong LG, Yang SH, Coffinier C, Young SG. The posttranslational processing of prelamin A and disease. Annu Rev Genomics Hum Genet 2009; 10:153 - 74; http://dx.doi.org/10.1146/annurev-genom-082908-150150; PMID: 19453251
- Fong LG, Ng JK, Meta M, Coté N, Yang SH, Stewart CL, et al. Heterozygosity for Lmna deficiency eliminates the progeria-like phenotypes in Zmpste24-deficient mice. Proc Natl Acad Sci USA 2004; 101:18111 - 6; http://dx.doi.org/10.1073/pnas.0408558102; PMID: 15608054
- Kubben N, Voncken JW, Konings G, van Weeghel M, van den Hoogenhof MM, Gijbels M, et al. Post-natal myogenic and adipogenic developmental: defects and metabolic impairment upon loss of A-type lamins. Nucleus 2011; 2:195 - 207; http://dx.doi.org/10.4161/nucl.2.3.15731; PMID: 21818413
- Dhe-Paganon S, Werner ED, Chi YI, Shoelson SE. Structure of the globular tail of nuclear lamin. J Biol Chem 2002; 277:17381 - 4; http://dx.doi.org/10.1074/jbc.C200038200; PMID: 11901143
- Krimm I, Ostlund C, Gilquin B, Couprie J, Hossenlopp P, Mornon JP, et al. The Ig-like structure of the C-terminal domain of lamin A/C, mutated in muscular dystrophies, cardiomyopathy, and partial lipodystrophy. Structure 2002; 10:811 - 23; http://dx.doi.org/10.1016/S0969-2126(02)00777-3; PMID: 12057196
- Loewinger L, McKeon F. Mutations in the nuclear lamin proteins resulting in their aberrant assembly in the cytoplasm. EMBO J 1988; 7:2301 - 9; PMID: 3056713
- Kapinos LE, Schumacher J, Mücke N, Machaidze G, Burkhard P, Aebi U, et al. Characterization of the head-to-tail overlap complexes formed by human lamin A, B1 and B2 “half-minilamin” dimers. J Mol Biol 2010; 396:719 - 31; http://dx.doi.org/10.1016/j.jmb.2009.12.001; PMID: 20004208
- Gerace L, Huber MD. Nuclear lamina at the crossroads of the cytoplasm and nucleus. J Struct Biol 2012; 177:24 - 31; http://dx.doi.org/10.1016/j.jsb.2011.11.007; PMID: 22126840
- Strelkov SV, Schumacher J, Burkhard P, Aebi U, Herrmann H. Crystal structure of the human lamin A coil 2B dimer: implications for the head-to-tail association of nuclear lamins. J Mol Biol 2004; 343:1067 - 80; http://dx.doi.org/10.1016/j.jmb.2004.08.093; PMID: 15476822
- Ho CY, Lammerding J. Lamins at a glance. J Cell Sci 2012; 125:2087 - 93; http://dx.doi.org/10.1242/jcs.087288; PMID: 22669459
- Sakaki M, Koike H, Takahashi N, Sasagawa N, Tomioka S, Arahata K, et al. Interaction between emerin and nuclear lamins. J Biochem 2001; 129:321 - 7; http://dx.doi.org/10.1093/oxfordjournals.jbchem.a002860; PMID: 11173535
- Lloyd DJ, Trembath RC, Shackleton S. A novel interaction between lamin A and SREBP1: implications for partial lipodystrophy and other laminopathies. Hum Mol Genet 2002; 11:769 - 77; http://dx.doi.org/10.1093/hmg/11.7.769; PMID: 11929849
- Kim JB, Spiegelman BM. ADD1/SREBP1 promotes adipocyte differentiation and gene expression linked to fatty acid metabolism. Genes Dev 1996; 10:1096 - 107; http://dx.doi.org/10.1101/gad.10.9.1096; PMID: 8654925
- Stierlé V, Couprie J, Ostlund C, Krimm I, Zinn-Justin S, Hossenlopp P, et al. The carboxyl-terminal region common to lamins A and C contains a DNA binding domain. Biochemistry 2003; 42:4819 - 28; http://dx.doi.org/10.1021/bi020704g; PMID: 12718522
- Sasseville AM, Langelier Y. In vitro interaction of the carboxy-terminal domain of lamin A with actin. FEBS Lett 1998; 425:485 - 9; http://dx.doi.org/10.1016/S0014-5793(98)00294-4; PMID: 9563518
- Simon DN, Zastrow MS, Wilson KL. Direct actin binding to A- and B-type lamin tails and actin filament bundling by the lamin A tail. Nucleus 2010; 1:264 - 72; http://dx.doi.org/10.4161/nucl.1.3.11799; PMID: 21327074
- Haque F, Mazzeo D, Patel JT, Smallwood DT, Ellis JA, Shanahan CM, et al. Mammalian SUN protein interaction networks at the inner nuclear membrane and their role in laminopathy disease processes. J Biol Chem 2010; 285:3487 - 98; http://dx.doi.org/10.1074/jbc.M109.071910; PMID: 19933576
- Hale CM, Shrestha AL, Khatau SB, Stewart-Hutchinson PJ, Hernandez L, Stewart CL, et al. Dysfunctional connections between the nucleus and the actin and microtubule networks in laminopathic models. Biophys J 2008; 95:5462 - 75; http://dx.doi.org/10.1529/biophysj.108.139428; PMID: 18790843
- Khatau SB, Hale CM, Stewart-Hutchinson PJ, Patel MS, Stewart CL, Searson PC, et al. A perinuclear actin cap regulates nuclear shape. Proc Natl Acad Sci USA 2009; 106:19017 - 22; http://dx.doi.org/10.1073/pnas.0908686106; PMID: 19850871
- Chen CY, Chi YH, Mutalif RA, Starost MF, Myers TG, Anderson SA, et al. Accumulation of the inner nuclear envelope protein Sun1 is pathogenic in progeric and dystrophic laminopathies. Cell 2012; 149:565 - 77; http://dx.doi.org/10.1016/j.cell.2012.01.059; PMID: 22541428
- Dechat T, Korbei B, Vaughan OA, Vlcek S, Hutchison CJ, Foisner R. Lamina-associated polypeptide 2alpha binds intranuclear A-type lamins. J Cell Sci 2000; 113:3473 - 84; PMID: 10984438
- Pekovic V, Harborth J, Broers JL, Ramaekers FC, van Engelen B, Lammens M, et al. Nucleoplasmic LAP2alpha-lamin A complexes are required to maintain a proliferative state in human fibroblasts. J Cell Biol 2007; 176:163 - 72; http://dx.doi.org/10.1083/jcb.200606139; PMID: 17227891
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970; 227:680 - 5; http://dx.doi.org/10.1038/227680a0; PMID: 5432063
- Thomas JO, Kornberg RD. An octamer of histones in chromatin and free in solution. Proc Natl Acad Sci USA 1975; 72:2626 - 30; http://dx.doi.org/10.1073/pnas.72.7.2626; PMID: 241077
- Matsudaira P. Sequence from picomole quantities of proteins electroblotted onto polyvinylidene difluoride membranes. J Biol Chem 1987; 262:10035 - 8; PMID: 3611052
- Testerink C, Dekker HL, Lim ZY, Johns MK, Holmes AB, Koster CG, et al. Isolation and identification of phosphatidic acid targets from plants. Plant J 2004; 39:527 - 36; http://dx.doi.org/10.1111/j.1365-313X.2004.02152.x; PMID: 15272872
- Shevchenko A, Wilm M, Vorm O, Mann M. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal Chem 1996; 68:850 - 8; http://dx.doi.org/10.1021/ac950914h; PMID: 8779443
- Schandorff S, Olsen JV, Bunkenborg J, Blagoev B, Zhang Y, Andersen JS, et al. A mass spectrometry-friendly database for cSNP identification. Nat Methods 2007; 4:465 - 6; http://dx.doi.org/10.1038/nmeth0607-465; PMID: 17538625