Abstract
Mutations in the LMNA gene are associated with a spectrum of human dystrophic diseases termed the “nuclear laminopathies.” We recently found that the accumulation of the inner nuclear envelope proteins SUN1 is pathogenic in progeric and dystrophic laminopathies. This conclusion arose from the unexpected observation that the deletion of Sun1, instead of accelerating aging, actually ameliorated the progeric and dystrophic phenotypes in Lmna-deficient mice. In human cells, knocking down SUN1 corrected the nuclear aberrancies and the senescent tendencies of HGPS (Hutchinson-Gilford progeria syndrome) skin fibroblasts. Here we offer additional comments on the contributions of SUN1 and the process of normal protein turnover to cellular aging.
Keywords: :
Main Text
Nuclear lamin A is a component of the nuclear lamina associated with the nucleoplasmic surface of the inner nuclear membrane (INM).Citation1,Citation2 Mutations in the LMNA gene cause a spectrum of human dystrophic diseases, including cardiomyopathy, neurodystrophy, lipodystrophy and premature aging, collectively termed the “nuclear laminopathies.”Citation1,Citation3-Citation5 Since the 1980s, an increasing understanding of nuclear lamin functions has emerged;Citation6 however, questions remain regarding the causality between mutations in lamin A and various tissue-specific debilitating disorders.Citation3,Citation5,Citation7-Citation9
The SUN domain was first identified based on sequence alignment of Sad1 of S. pombe and UNC-84 of C. elegans.Citation10 Mammals have four SUN-domain proteins, SUN1, SUN2, SUN3 and SPAG4. SUN1 and SUN2 are in the INM in somatic cells, but they translocate to telomeres in the prophase of meiotic cells.Citation11,Citation12 The SUN-domain proteins interact with the C-terminal KASH domain of the Nesprin (also named Syne, synaptic nuclear envelope) proteins within the LINC (links the nucleoskeleton and cytoskeleton) complex, which provides a direct connection between the nuclear lamina and the cytoskeleton, thereby contributing to nuclear positioning and cellular rigidity.Citation13 Giant Nesprin1 and Nesprin2 (i.e., Syne1 and Syne2) isoforms localize to the outer nuclear membrane where they bridge the connection with the actin cytoskeleton via an N-terminal actin binding domain.
Because the SUN domain proteins are the only protein family currently known to connect the nucleoskeleton and cytoskeleton,14,Citation15 and because SUN1 is a direct interacting partner of lamin A, we hypothesized that SUN1 may play a role in human laminopathies. Our prediction was that a knock out of Sun1 would additively worsen the progeric phenotype of Lmna-deficient cells and animals. Unexpectedly, using human HGPS (Hutchinson-Gilford progeria syndrome) skin fibroblasts and two mouse models, Lmna−/− and LmnaΔ9 mice, that respectively represent dystrophic and progeric laminopathies, we foundCitation16 that a common explanation for these lamin-associated dysfunctions appear to be the over accumulation of the INM SUN1 protein. Accordingly, when we deleted the Sun1 geneCitation12 from Lmna−/− and LmnaΔ9 mice, the life span of the mice became significantly extended. Of note, a reversal of aging has also been observed in a separate progeria-like Zmpste24−/− mouse model (Zmpste24 is a metalloprotease of prelamin A). The pathologic phenotype of Zmpste24−/− mice, which arises from failed processing of prelamin A, was found to be alleviated by the additional knock out of p53 (i.e., Zmpste24−/−p53−/− mouse).Citation17 Increased proliferation of human HGPS cells was also seen when p53 was inactivated by the human papillomavirus (HPV) E6 protein.Citation18 However, these p53-dependent findings appear to be mechanistically distinct from Sun1 deletion-mediated increase in the longevity of Lmna-deficient mice which occurs in a p53 wild type setting.Citation12,Citation16
What are the explanations for the increased longevity of Sun1−/−Lmna−/− and Sun1−/−LmnaΔ9 mice? By analyzing Sun1 expression in mouse embryonic fibroblasts (MEFs), we found that Lmna−/− and LmnaΔ9, but not wild type, cells showed significant over accumulation of SUN1 protein in the nuclear envelope (NE) and the Golgi apparatus. This over accumulation arises from reduced protein turnover, not increased gene transcription. Moreover, the mislocation of INM protein SUN1 into the Golgi triggers nuclear envelope rupture, a phenotype that is commonly seen in Lmna−/− MEFs. Similar to the findings in Lmna−/− MEFs, increased SUN1 expression was also found in human HGPS cells, suggesting that a common pathogenic event in Lmna−/−, LmnaΔ9, and HGPS cells converges at SUN1 protein misaccumulation.
Because Lmna−/−, Lmna−/−Sun1−/−, LmnaΔ9 or LmnaΔ9Sun1−/− mice do not express wild type lamin A, a previous explanation for lamin A-associated dystrophic or progeric diseases was that they occur in these mice because of loss in lamin A function.Citation19,Citation20 On the other hand, our new SUN1 observations add another wrinkle to the aging question.Citation21 SUN1 mislocation and its over accumulation suggest that laminopathies may also in part be caused through an organelle storage disorder-like mechanism. Thus a part of the pathological impetus of laminopathies may arise, like other age-dependent degenerative diseases such as lysosome storage diseases (e.g., Fabry, Tay-Sachs, Gaucher, Niemann-Pick, Pompe and Krabbe) and ER storage diseases (e.g., cystic fibrosis, a1-antitrypsin deficiency, hereditary hypoparathyroidism, and procollagen type I, II, and IV deficiency), by the “over stuffing” of misaccumulated material in subcellular organelles (e.g., lysosomes, ER, Golgi or nuclear envelope) that elicits stress signals unfavorable to normal cellular physiology.Citation22-Citation26
A feature common to the above metabolic disorders is that the affected individuals appear normal at birth, but manifest progressive symptoms as they age. Unlike early passaged Lmna-deficient MEFs, in multiply passaged human HGPS fibroblasts, a major accumulation of SUN1 in the Golgi was not seen.Citation16 This could be because the over accumulation of SUN1 is potently toxic in HGPS cells; hence, all such late passaged cells are eliminated due to non-viability. On the other hand, although there was no Golgi over accumulation, multiply-passaged HGPS cells do show significant over accumulation of SUN1 in the NE. If SUN1-Golgi over accumulated HGPS cells are eliminated while SUN1-NE over accumulated counterparts persist, this would suggest that the latter mislocation creates a milder, more tolerated phenotype and could represent a form of “NE storage disease.” Indeed, as frequently noted, HGPS patients are born normal, but quickly develop age-associated changes including alopecia, atherosclerosis, osteolysis and severe lipodystrophy between 12 and 18 mo;Citation27 these changes become more severe as the individuals age.
An intriguing question from our work is why does SUN1 over accumulate in the NE and Golgi? A full answer awaits better characterization of SUN1 sequence determinants for localization in and trafficking to the NE, Golgi and ER.Citation28 Nevertheless, we envision that the following processes may be relevant. In anterograde transport, proteins synthesized in the ER exit in transport vesicles that bud from the ER and congregate at the ER-Golgi-intermediate compartment (ERGIC).Citation24 By vesicles or tubules they are delivered to the cis-Golgi and then move (by vesicular transport or cisternal maturation) through the Golgi cisternae to the trans-Golgi network (TGN). Secretory proteins that fail to exit the ER result in protein accumulation and pathological ER storage. Conversely, retrograde (Golgi-to-ER) transport retrieves ER residents and other constituents that cycle between these two compartments.Citation29 Furthermore, because the NE is composed of two lipid membranes that are continuous with the ER, one view is that INM proteins synthesized in the ER are transported along the rough ER, through the nuclear pore complex to arrive at the inner nuclear membrane.Citation1,Citation3 SUN1 accumulation in both the NE and the Golgi represents two opposite destinations of ER retrograde and anterograde trafficking (). Although yet unknown, SUN1 may undergo post-translational modifications through its lamin A-dependent interaction in the NE. In the absence of lamin A, SUN1 may not be modified properly and thus cannot achieve normal turnover; and this could explain its proclivity for over accumulation. Possibly, over accumulated SUN1 may aggregate into a conformation that favors ER-to-Golgi anterograde transport into the Golgi. In the case of HGPS, which heterozygously expresses one wild type allele of lamin A and one mutant allele of lamin A, LAΔ50, the single wild type copy of lamin A may produce enough functional lamin A protein to sufficiently direct the appropriate localization of SUN1 to the NE.
Figure 1. The cause and reversal of laminopathies. In normal cell, SUN1 (red dots) is located in the inner nuclear membrane and interacts with the nucleoskeleton (via nuclear lamin A) and the cytoskeletal actin (via Syne1 and Syne2) and microtubules in supporting nuclear integrity. Loss of functional lamin A (LAΔ, either Lmna−/− or LmnaΔ9) is reported to result in accumulated SUN1 in the NE and Golgi (through anterograde transport), leading to nuclear morphology aberrations and cellular senescence. The removal of over accumulated SUN1 by genetic deletion or RNA silencing ameliorates nuclear aberrancies and prolongs longevity in mice. An alternative route to reversing the cellular phenotype of progeria is through activating autophagy using rapamycin. Rapamycin inhibits mTOR, activates autophagy, and thereby enhances the degradation of progerin (LAΔ50) in HGPS cells. The removal of abnormally accumulated progerin, as well as other protein moieties, is postulated to abolish nuclear blebbing and alleviate the onset of cellular senescence. ERGIC, ER-Golgi-intermediate compartment; TGN, trans-Golgi network; Syne, synaptic nuclear envelope; mTOR, mammalian target of rapamycin.
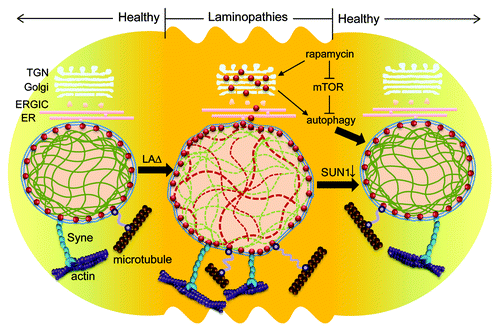
Why do other INM proteins not over accumulate? Perhaps they do. HGPS cells show characteristic NPC clustering,Citation30 Emerin mislocalizationCitation31 and LAP2 loss,Citation32 although SUN2 localization seems to be normal.Citation8,Citation16 What governs (mis)localization is not fully understood, but we hypothesize that the INM proteins may distribute non-randomly and may occupy discrete “territories” in the inner nuclear membrane similar to the concept of “chromosome territories.”Citation33 Thus, the INM proteins may migrate to designated loci of the INM for various nuclear functions; and lamin A may be one of several determinants that govern these movements. In the absence of functional lamin A, these INM proteins may disseminate freely in various context-dependent manner.
What these new findings mean for therapeutic treatment of HGPS-like diseases remains to be verified. Currently, one line of thought is that HGPS arises from a dominant mutant form of prelamin A protein (named LAΔ50 or progerin) that contains a CaaX motif which can be durably farnesylated. Farnesylated progerin is considered to be pathogenic, and thus farnesyltransferase inhibitors (FTIs) that were originally developed to block the farnesylation of Ras oncogene for the treatment of cancer has been applied with the goal of treating HGPS. Using a FTI, R115777 (Tipifarnib; also known as Zarnestra), Mallampalli, et al. demonstrated that FTI treatment of cells reversed the abnormal nuclear morphology associated with progerin;Citation34 on the other hand other investigators have not always replicated the same outcome.Citation35 If the mislocation and misaccumulation in cells of INM proteins such as SUN1 are further contributors to the pathology of HGPS-like diseases, then FTI treatments are unlikely to correct these additional causes. Interestingly, treatment of HGPS cells with rapamycin, an mTOR inhibitor and a strong immunosuppressant that is also a potent inducer of the autophagy pathway, was found to alleviate in part the pathologies of HGPS cells such as nuclear blebbing and cellular senescence ().Citation36 This result makes mechanistic sense if rapamycin treatment enhances cellular degradation of over accumulated and mislocated nuclear envelope proteins like LAΔ50 and SUN1 through its induction of autophagy (). Interestingly, rapamycin treatment of wild type mice has been shown to prolong their life span by 14%,Citation37 suggesting that physiological aging and its consequences may also be influenced by age-dependent inefficiencies in normal protein degradation/turnover.
The salient observation from our new study is that the elimination of an INM protein, SUN1, ameliorates cellular and organ pathologies of premature aging in mice. A correlative interpretation of our work is that aging (whether premature or physiological) is a consequence of the improper disposal of accumulated cellular proteins that occur with extreme robustness in the Lmna-deficient mice. If aging is a result of abnormal protein turnover, then treatments for premature aging HGPS-like diseases should consider alleviating pathogenic upstream (i.e., treating progerin) as well as downstream (i.e., SUN1 misaccumulation) events. Without addressing downstream protein misaccumulation, therapeutic resolution of progeria or EDMD pathologies are unlikely to be achieved.
| Abbreviations: | ||
| SUN | = | Sad1-UNC-84 homology |
| HGPS | = | Hutchinson-Gilford progeria syndrome |
| EDMD | = | Emery-Dreifuss muscular dystrophy |
Acknowledgments
Our work was supported by NIAID intramural funds, the NHRI, Taiwan (NHRI 01A1-CSPP13–014), and NSC, Taiwan (NSC 98–2320-B-400–009-MY3).
References
- Burke B. The nuclear envelope: filling in gaps. Nat Cell Biol 2001; 3:E273 - 4; http://dx.doi.org/10.1038/ncb1201-e273; PMID: 11781582
- Gruenbaum Y, Margalit A, Goldman RD, Shumaker DK, Wilson KL. The nuclear lamina comes of age. Nat Rev Mol Cell Biol 2005; 6:21 - 31; http://dx.doi.org/10.1038/nrm1550; PMID: 15688064
- Burke B, Stewart CL. Life at the edge: the nuclear envelope and human disease. Nat Rev Mol Cell Biol 2002; 3:575 - 85; http://dx.doi.org/10.1038/nrm879; PMID: 12154369
- Burke B, Mounkes LC, Stewart CL. The nuclear envelope in muscular dystrophy and cardiovascular diseases. Traffic 2001; 2:675 - 83; http://dx.doi.org/10.1034/j.1600-0854.2001.21001.x; PMID: 11576443
- Worman HJ, Courvalin JC. How do mutations in lamins A and C cause disease?. J Clin Invest 2004; 113:349 - 51; PMID: 14755330
- Gerace L, Blobel G. The nuclear envelope lamina is reversibly depolymerized during mitosis. Cell 1980; 19:277 - 87; http://dx.doi.org/10.1016/0092-8674(80)90409-2; PMID: 7357605
- Burtner CR, Kennedy BK. Progeria syndromes and ageing: what is the connection?. Nat Rev Mol Cell Biol 2010; 11:567 - 78; http://dx.doi.org/10.1038/nrm2944; PMID: 20651707
- Haque F, Mazzeo D, Patel JT, Smallwood DT, Ellis JA, Shanahan CM, et al. Mammalian SUN protein interaction networks at the inner nuclear membrane and their role in laminopathy disease processes. J Biol Chem 2010; 285:3487 - 98; http://dx.doi.org/10.1074/jbc.M109.071910; PMID: 19933576
- Chi YH, Chen ZJ, Jeang KT. The nuclear envelopathies and human diseases. J Biomed Sci 2009; 16:96; http://dx.doi.org/10.1186/1423-0127-16-96; PMID: 19849840
- Malone CJ, Fixsen WD, Horvitz HR, Han M. UNC-84 localizes to the nuclear envelope and is required for nuclear migration and anchoring during C. elegans development. Development 1999; 126:3171 - 81; PMID: 10375507
- Ding X, Xu R, Yu J, Xu T, Zhuang Y, Han M. SUN1 is required for telomere attachment to nuclear envelope and gametogenesis in mice. Dev Cell 2007; 12:863 - 72; http://dx.doi.org/10.1016/j.devcel.2007.03.018; PMID: 17543860
- Chi YH, Cheng LI, Myers T, Ward JM, Williams E, Su Q, et al. Requirement for Sun1 in the expression of meiotic reproductive genes and piRNA. Development 2009; 136:965 - 73; http://dx.doi.org/10.1242/dev.029868; PMID: 19211677
- Haque F, Lloyd DJ, Smallwood DT, Dent CL, Shanahan CM, Fry AM, et al. SUN1 interacts with nuclear lamin A and cytoplasmic nesprins to provide a physical connection between the nuclear lamina and the cytoskeleton. Mol Cell Biol 2006; 26:3738 - 51; http://dx.doi.org/10.1128/MCB.26.10.3738-3751.2006; PMID: 16648470
- Razafsky D, Hodzic D. Bringing KASH under the SUN: the many faces of nucleo-cytoskeletal connections. J Cell Biol 2009; 186:461 - 72; http://dx.doi.org/10.1083/jcb.200906068; PMID: 19687252
- Padmakumar VC, Libotte T, Lu W, Zaim H, Abraham S, Noegel AA, et al. The inner nuclear membrane protein Sun1 mediates the anchorage of Nesprin-2 to the nuclear envelope. J Cell Sci 2005; 118:3419 - 30; http://dx.doi.org/10.1242/jcs.02471; PMID: 16079285
- Chen CY, Chi YH, Mutalif RA, Starost MF, Myers TG, Anderson SA, et al. Accumulation of the inner nuclear envelope protein Sun1 is pathogenic in progeric and dystrophic laminopathies. Cell 2012; 149:565 - 77; http://dx.doi.org/10.1016/j.cell.2012.01.059; PMID: 22541428
- Varela I, Cadiñanos J, Pendás AM, Gutiérrez-Fernández A, Folgueras AR, Sánchez LM, et al. Accelerated ageing in mice deficient in Zmpste24 protease is linked to p53 signalling activation. Nature 2005; 437:564 - 8; http://dx.doi.org/10.1038/nature04019; PMID: 16079796
- Kudlow BA, Stanfel MN, Burtner CR, Johnston ED, Kennedy BK. Suppression of proliferative defects associated with processing-defective lamin A mutants by hTERT or inactivation of p53. Mol Biol Cell 2008; 19:5238 - 48; http://dx.doi.org/10.1091/mbc.E08-05-0492; PMID: 18843043
- Mounkes LC, Kozlov S, Hernandez L, Sullivan T, Stewart CL. A progeroid syndrome in mice is caused by defects in A-type lamins. Nature 2003; 423:298 - 301; http://dx.doi.org/10.1038/nature01631; PMID: 12748643
- Sullivan T, Escalante-Alcalde D, Bhatt H, Anver M, Bhat N, Nagashima K, et al. Loss of A-type lamin expression compromises nuclear envelope integrity leading to muscular dystrophy. J Cell Biol 1999; 147:913 - 20; http://dx.doi.org/10.1083/jcb.147.5.913; PMID: 10579712
- Liu B, Jin DY, Zhou Z. From loss to gain: role for SUN1 in laminopathies. Cell Biosci 2012; 2:21; http://dx.doi.org/10.1186/2045-3701-2-21; PMID: 22709970
- Filocamo M, Morrone A. Lysosomal storage disorders: molecular basis and laboratory testing. Hum Genomics 2011; 5:156 - 69; http://dx.doi.org/10.1186/1479-7364-5-3-156; PMID: 21504867
- Luheshi LM, Crowther DC, Dobson CM. Protein misfolding and disease: from the test tube to the organism. Curr Opin Chem Biol 2008; 12:25 - 31; http://dx.doi.org/10.1016/j.cbpa.2008.02.011; PMID: 18295611
- Rutishauser J, Spiess M. Endoplasmic reticulum storage diseases. Swiss Med Wkly 2002; 132:211 - 22; PMID: 12087487
- Crews L, Masliah E. Molecular mechanisms of neurodegeneration in Alzheimer’s disease. Hum Mol Genet 2010; 19:R1 R12 - 20; http://dx.doi.org/10.1093/hmg/ddq160; PMID: 20413653
- Kim I, Xu W, Reed JC. Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nat Rev Drug Discov 2008; 7:1013 - 30; http://dx.doi.org/10.1038/nrd2755; PMID: 19043451
- Merideth MA, Gordon LB, Clauss S, Sachdev V, Smith AC, Perry MB, et al. Phenotype and course of Hutchinson-Gilford progeria syndrome. N Engl J Med 2008; 358:592 - 604; http://dx.doi.org/10.1056/NEJMoa0706898; PMID: 18256394
- Turgay Y, Ungricht R, Rothballer A, Kiss A, Csucs G, Horvath P, et al. A classical NLS and the SUN domain contribute to the targeting of SUN2 to the inner nuclear membrane. EMBO J 2010; 29:2262 - 75; http://dx.doi.org/10.1038/emboj.2010.119; PMID: 20551905
- Lorente-Rodríguez A, Barlowe C. Entry and exit mechanisms at the cis-face of the Golgi complex. Cold Spring Harb Perspect Biol 2011; 3:a005207; http://dx.doi.org/10.1101/cshperspect.a005207; PMID: 21482742
- Liu Q, Pante N, Misteli T, Elsagga M, Crisp M, Hodzic D, et al. Functional association of Sun1 with nuclear pore complexes. J Cell Biol 2007; 178:785 - 98; http://dx.doi.org/10.1083/jcb.200704108; PMID: 17724119
- Vaughan A, Alvarez-Reyes M, Bridger JM, Broers JL, Ramaekers FC, Wehnert M, et al, Whitfield WGF. Both emerin and lamin C depend on lamin A for localization at the nuclear envelope. J Cell Sci 2001; 114:2577 - 90; PMID: 11683386
- Scaffidi P, Misteli T. Reversal of the cellular phenotype in the premature aging disease Hutchinson-Gilford progeria syndrome. Nat Med 2005; 11:440 - 5; http://dx.doi.org/10.1038/nm1204; PMID: 15750600
- Misteli T. Concepts in nuclear architecture. Bioessays 2005; 27:477 - 87; http://dx.doi.org/10.1002/bies.20226; PMID: 15832379
- Mallampalli MP, Huyer G, Bendale P, Gelb MH, Michaelis S. Inhibiting farnesylation reverses the nuclear morphology defect in a HeLa cell model for Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A 2005; 102:14416 - 21; http://dx.doi.org/10.1073/pnas.0503712102; PMID: 16186497
- Verstraeten VL, Peckham LA, Olive M, Capell BC, Collins FS, Nabel EG, et al. Protein farnesylation inhibitors cause donut-shaped cell nuclei attributable to a centrosome separation defect. Proc Natl Acad Sci U S A 2011; 108:4997 - 5002; http://dx.doi.org/10.1073/pnas.1019532108; PMID: 21383178
- Cao K, Graziotto JJ, Blair CD, Mazzulli JR, Erdos MR, Krainc D, et al. Rapamycin reverses cellular phenotypes and enhances mutant protein clearance in Hutchinson-Gilford progeria syndrome cells. Sci Transl Med 2011; 3:89ra58; http://dx.doi.org/10.1126/scitranslmed.3002346; PMID: 21715679
- Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 2009; 460:392 - 5; PMID: 19587680