Abstract
The nuclear pore complex (NPC) mediates macromolecular exchange between nucleus and cytoplasm. It is a regulated channel whose functional properties are modulated in response to the physiological status of the cell. Identifying the factors responsible for regulating NPC activity is crucial to understand how intracellular signaling cues are integrated at the level of this channel to control nucleocytoplasmic trafficking. For proteins lacking active translocation signals the NPC acts as a molecular sieve limiting passage across the nuclear envelope (NE) to proteins with a MW below ~40 kD. Here, we investigate how this permeability barrier is altered in paradigms of cell death and cell survival, i.e., apoptosis induction via staurosporine, and enhanced viability via overexpression of Bcl-2. We monitor dynamic changes of the NPC’s size-exclusion limit for passive diffusion by confocal time-lapse microscopy of cells undergoing apoptosis, and use different diffusion markers to determine how Bcl-2 expression affects steady-state NE permeability. We show that staurosporine triggers an immediate and gradual leakiness of the NE preceding the appearance of apoptotic hallmarks. Bcl-2 expression leads to a constitutive increase in NE permeability, and its localization at the NE is sufficient for the effect, evincing a functional role for Bcl-2 at the nuclear membrane. In both settings, NPC leakiness correlates with reduced Ca2+ in internal stores, as demonstrated by fluorometric measurements of ER/NE Ca2+ levels. By comparing two cellular models with opposite outcome these data pinpoint ER/NE Ca2+ as a general and physiologically relevant regulator of the permeability barrier function of the NPC.
Introduction
In eukaryotic cells the nuclear envelope (NE) provides a physical barrier that secludes and organizes genomic material within the nucleus. Molecular traffic across the NE occurs exclusively via nuclear pore complexes (NPCs), multimeric macromolecular channels spanning both lipid layers of the NE.
The NPC acts as diffusion barrier for inert molecules with a MW larger than ≈40 kD and facilitates the translocation of much larger proteins up to a flux rate of 1,000/s.Citation1 This activity is dynamically regulated in response to physiological or pathological signaling cues.Citation2,Citation3 Functional control of the NPC may occur via alterations in NPC composition,Citation4 by post-translational modification of nuclear pore proteins (Nups),Citation5,Citation6 and by modulating the dimensions of the channel itself.Citation7 These structural changes impinge on transport capacity and selectivity of the NPC as well as on its size-exclusion limit for passive translocation. Thus, one important layer of regulation of nucleo-cytoplasmic trafficking resides at the NPC itself.Citation8
Active cell proliferation and programmed cell death are examples for cellular states known to induce modifications of NPC structure and function. Oncogene-transformed, hyperproliferating cells display larger NPC diameters than their resting counterparts.Citation9,Citation10 In mitotic cells, the level of Nup96 is downregulated to allow efficient G1/S transition.Citation11 In apoptotic cells, NPC dismantling and breakdown of the NE permeability barrier are the consequence of caspase-mediated proteolysis of a subset of nucleoporins (Nups).Citation12-Citation16 Yet another study has evidenced that calpains, Ca2+-activated proteases, cleave Nups in neuronal cells undergoing excitotoxic death resulting in nuclear accumulation of the cytoplasmic protein GAPDH.Citation17 Disruption of NPC components is not the only mechanism of NE permeabilization, since nuclear leakiness has been observed also in the absence of Nup proteolysis in apoptotic and virus-infected cells.Citation12,Citation17,Citation18 Also the proapoptotic Bcl-2 family proteins Bax and Bak were proposed to affect nucleo-cytoplasmic protein partitioning: histone H1 and nucleophosmin were shown to redistribute to the cytoplasm as a consequence of Bax/Bak overexpression independently of caspase activity.Citation19 This result highlights yet another link between the apoptosis machinery and nucleo-cytoplasmic trafficking.
Ca2+ functions as second messenger in the cellular response to very diverse endogenous and exogenous signals ranging from noxious environmental insults to growth stimulatory factors. Numerous studies have proposed a role for Ca2+ in the regulation of NPC structure and function. The underlying mechanisms are not fully clear yet (reviewed inCitation20-Citation22). The NE harbours functional inositol(1,4,5)-trisphosphate receptors (InsP3Rs), as well as ryanodine receptors (RyRs) and nicotinic acid adenine dinucleotide phosphate receptors (NAADPRs). In response to their cognate agonistic signals these receptors can induce [Ca2+] gradients in the lumen of the NE, which is continuous to the lumen of the endoplasmic reticulum (ER),Citation23 or in the cytosol, or in both compartments. Depletion of ER/NE Ca2+ stores was shown to inhibit passage of inert molecules and transport cargo in some studiesCitation24,Citation25 but not in others.Citation26,Citation27 Alterations in NPC topology in response to Ca2+ have been observed by (immuno-)electron and scanning force microscopy. “Close” vs. “open” conformations of the nucleoplasmic basket structure could be distinguished underpinning the role of Ca2+ in structural rearrangements of the NPC.Citation28-Citation31 Intriguingly, Bax and Bak, initially characterized as key players in apoptotic cytochrome C release from mitochondria, were later involved in maintaining ER [Ca2+]. Cells lacking both Bax and Bak were shown to have a drastically reduced ER [Ca2+] and fail to mobilize Ca2+ from the ER to mitochondria.Citation32,Citation33 These results suggest that Bcl-2 family proteins might impinge on the NPC via modulation of ER/NE Ca2+ levels, in line with the abovementioned findings on Bax/Bak-dependent apoptotic nuclear protein redistribution.
Prompted by these considerations, we asked whether changes in ER/NE [Ca2+] could provide a common denominator for functional modulation of the NPC under conditions as diverse as cell death and cell survival. We addressed this question by investigating first the NE permeability barrier in cells treated to undergo apoptosis by staurosporine (STS). This broad spectrum kinase inhibitor is a well-established and efficient apoptosis inducer known to elicit intracellular [Ca2+] elevation via release from internal stores.Citation32,Citation34 We then compared this death model with overexpression of Bcl-2, a paradigm for enhanced cell survival and proliferation (reviewed in ref. Citation35). Bcl-2 is known to affect ER [Ca2+] through direct interaction with InsP3Rs.Citation36,Citation37
Our results provide evidence for a major role of ER/NE [Ca2+] in modulating the NE permeability barrier independently of cellular fate and identify Bcl-2 as a regulator of the size exclusion limit of the NE for passive diffusion.
Results
Staurosporine induces a pre-apoptotic, caspase-independent leakiness of the NE
We first investigated how STS treatment affects the permeability barrier function of the NE. To this end, we performed single-cell, confocal time-lapse recordings of HeLa cells expressing a reporter for passive passage across the NE, a tetrameric mCherry fusion protein (4xCherry) (; Fig. S1A and B). Images were taken approx. every 8 min, and a minimum of 13 cells was recorded in each experiment. The nuclear influx of 4xCherry was quantified over time as described in “Materials and Methods” and in Figure S1D. To monitor apoptosis progression the cells were stained with low concentrations of the vital DNA dye Hoechst 33342. This enabled to visualize nuclear condensation as a hallmark of caspase-dependent apoptotic execution (Fig. S1C). To account for the asynchronicity of the apoptotic process, data from each time series were split into two parts and displayed on separate x-axes. The left portion of the x-axis shows traces from the start of the experiment up to time points immediately preceding nuclear condensation (pre-apoptotic). On the right x-axis, traces were aligned with respect to the time of nuclear condensation (time point 0). This arrangement properly visualizes changes in nuclear permeability occurring at nuclear execution.
Figure 1. STS, but not TRAIL, induces a pre-apoptotic gradual increase in nuclear permeability coincident with depletion of ER/NE Ca2+. (A) Time course of nuclear entry of 4xCherry in HeLa cells treated with either STS (open rectangles) or TRAIL (open circles). A region of interest corresponding to the area of the nucleus was obtained by manual segmentation (see Fig. S1D). The mean intensities measured in the 4xCherry channel are plotted over time on two separate x-axes. On the left x-axis, time points from the start of image acquisition until the onset of nuclear condensation are displayed. On the right x-axis, time points immediately preceding and following nuclear condensation are shown. Here, traces were aligned with respect to the time point of nuclear condensation (time 0, dashed blue line). Red arrows indicate the time points corresponding to the frames shown in (B). Green arrows refer to the time points of Ca2+ measurement shown in (C) and (D). Data points are the average of at least 13 cells. Error bars show the SEM (B) Confocal images of HeLa cells expressing the nuclear permeability marker 4xCherry treated with STS or TRAIL. The time points refer to the time course shown in (A) (red arrows). (C) Characteristic traces of Fluo-4 fluorescence in HeLa cells treated to undergo apoptosis. HeLa cells were left untreated (black), or treated for 60 min with either STS (light gray) or TRAIL (dark gray), and loaded with the Ca2+ indicator Fluo-4. TG (5μm) was added at the indicated time point (arrow) to release Ca2+ from internal stores. (D) Comparison of Ca2+ content in internal stores after treatment with STS or TRAIL for 60 min. The difference between basal and peak fluorescence was calculated. Bars indicate the amount of Ca2+ mobilization in treated cells as percentage of control, untreated cells. Experiments were performed in triplicates. *** p < 0.001.
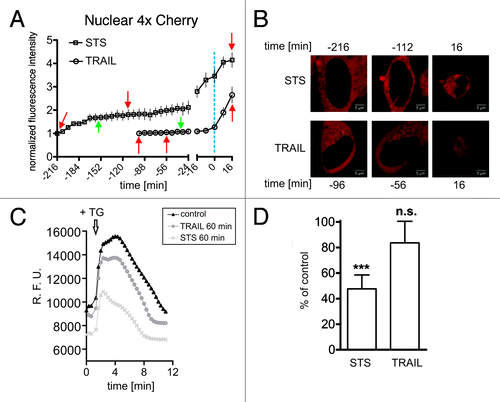
STS-treated cells died on average 3.5 h after drug addition, when 80% displayed apoptotic nuclei (not shown). Traces of nuclear 4xCherry fluorescence displayed a characteristic early-onset and gradual increase in signal intensity indicating that nuclei became partially accessible to the permeability marker shortly after drug addition (, left x-axis, open rectangles). At later time points immediately preceding chromatin condensation, a steep increase in nuclear fluorescence was observed, suggesting a rapid collapse of the NE permeability barrier (, right x-axis, open rectangles). To determine whether this behavior is generally associated with apoptotic cell demise we treated cells with TRAIL, a death receptor ligand triggering cell death via the extrinsic pathway.Citation38 Cells underwent apoptosis quite rapidly as expected (~1.5 h) but in contrast to STS treatment nuclei efficiently excluded 4xCherry until chromatin condensation ensued (, open circles, left x-axis). In the final phase, the NE permeability barrier collapsed in these cells similarly to STS-treated cells (, right x-axis). These data suggest that the observed pre-apoptotic increase in nuclear accessibility is a distinctive feature of STS-induced cell death. To address the question about a potential involvement of Ca2+ we determined ER/NE [Ca2+] levels at selected time points after treatment with both drugs. We used Fluo-4 as a Ca2+-specific indicator, in combination with thapsigargin (TG), an inhibitor of the ER Ca2+-ATPase.Citation39 TG induces the discharging of ER/NE Ca2+ resulting in an abrupt increase of its cytosolic concentration which can be detected with Fluo-4. Thus, TG-induced Ca2+ transients are an established measure of ER/NE Ca2+ content. At 60 min after STS addition we observed a 50% reduction of ER Ca2+ levels, contrarily to cells stimulated for the same time period with the death-receptor ligand TRAIL. The latter had no significant effect on ER/NE [Ca2+] (). These results suggest that the early increase in the size exclusion limit of the NE for passive diffusion observed under STS treatment is associated with the depletion of ER/NE [Ca2+] levels induced by this drug.
Early NE leakiness precedes apoptotic cleavage of nucleoporin Nup153 and is not dependent on caspase activity
According to previous studies, collapse of the nucleocytoplasmic permeability barrier in apoptosis is brought about by the caspase-mediated cleavage of Nups.Citation14 To temporally correlate the permeability alterations of the NE observed in STS- and TRAIL-treated cells with NPC proteolysis we monitored a fluorescently labeled fusion of Nup153 over time. Nup153 is a component of the NPC located at the nuclear basket and is efficiently cleaved by executioner caspases.Citation12,Citation16 In control experiments we verified that the Nup153-GFP fusion protein is processed upon treatment of HeLa cells with STS or TRAIL in a caspase-dependent fashion (Fig. S2). In our confocal time series of apoptotic cells we then quantified the average intensity of the Nup153-GFP signal at the nuclear rim by manual segmentation, taking care to avoid regions in which the NE formed convolutions, as seen quite frequently in STS-treated cells. Such convolutions appear as out-of-focus regions of the nuclear rim signal (Fig. S1D). Both STS and TRAIL treated cells showed a clear and abrupt reduction of Nup153-GFP fluorescence at the nuclear rim at the onset of chromatin condensation. This effect was abrogated in the presence of the pan-caspase inhibitor zVAD and by inactivation of the caspase cleavage site in Nup153 at amino acid position 349 (, right x-axes). In the time window preceding chromatin condensation, Nup153-GFP traces were quite similar for STS and TRAIL treatment and both were not affected by zVAD (, left axes). According to these data, loss of Nup153-GFP signal from the nuclear rim is an indicator of caspase-dependent dismantling of the NPC which occurs concomitantly to chromatin condensation. We then investigated the effect of zVAD on the kinetics of nuclear 4xCherry influx. Caspase-inhibition prevented the final, abrupt collapse of the permeability barrier in both apoptosis models, but had no effect on the early-onset, moderate increase in nuclear accessibility observed in cells treated with STS (). In these cells caspase-3-like activity was not detectable until 2h after stimulation, when partial nuclear entry of 4xCherry was already established. This assay also confirmed that zVAD was highly effective in blocking caspase activation (). In sum, these results support the existence of an early, caspase-independent leakiness of the NE that precedes the apoptotic dismantling of the NPC triggered by STS.
Figure 2. Caspase-dependent cleavage of Nup153-GFP coincides with chromatin condensation in TRAIL and STS-induced apoptosis. HeLa cells expressing Nup153-GFP (green line) or its caspase-uncleavable mutant Nup153-D349N-GFP (red line) were treated with STS or TRAIL. The former were additionally preincubated with the pan-caspase inhibitor zVAD (black line). Regions of interest corresponding to the nuclear rim were defined interactively taking care to omit convoluted regions (see also Material and Methods and Fig. S1D). Normalized Nup153-GFP signal intensities are plotted over time as in . The dashed blue line indicates the time point of nuclear condensation. Data points are the average of at least 15 cells. Error bars show the SEM.
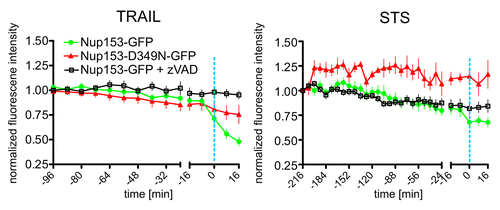
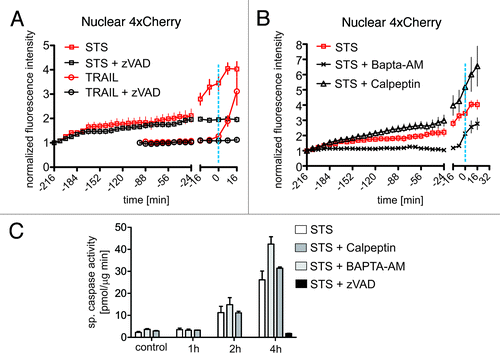
Figure 3. STS-induced early nuclear leakiness is mediated by Ca2+ but neither by caspases nor calpains. (A) Hela cells were treated with STS (open rectangles) and TRAIL (open circles) in the absence (red lines) or presence (black lines) of the pan-caspase inhibitor zVAD. Nuclear 4xCherry intensities were extracted from confocal time series and plotted over time as in . The dashed blue line indicates the onset of nuclear condensation. (B) Hela cells were treated with STS alone (red open rectangles), or in presence of the calpain inhibitor calpeptin (black open triangles), or in presence of the Ca2+ chelator BAPTA-AM (black crosses). Nuclear 4xCherry intensities were extracted from confocal time series and plotted over time as in . The dashed blue line indicates the onset of nuclear condensation. (C) Hela cells were treated with STS either in absence (white bars) or presence of the following inhibitors: calpeptin (0.5 μM, dark gray bars), BAPTA-AM (10 μM, light gray bars), and zVAD (20 μM, black bar). The inhibitors were added 30 min prior to STS treatment. Untreated cells served as control. Caspase activity was measured fluorometrically in cell lysates at the indicated time points.
Early pre-apoptotic permeabilization of the nuclear envelope depends on Ca2+, but not on calpain activation
The NPC has been shown to be the target of non-caspase proteolytic systems, in particular of the Ca2+-dependent protease family of calpains. In primary cortical neurons, Ca2+ overload caused by exposure to excitotoxic glutamate concentrations activates calpains resulting in the degradation of several Nups and the concomitant loss of proper nucleo-cytoplasmic partitioning of reporter molecules.Citation17 To investigate the potential involvement of calpains in STS-mediated nuclear permeabilization in HeLa cells we performed live-cell microscopy experiments in the presence of the cell-permeable calpain inhibitor calpeptin. The efficacy of the inhibitor was confirmed using an in vitro calpain activity assay (Fig. S3). Differently from neuronal excitotoxicity, calpeptin did not protect from NE permeability changes induced by STS in HeLa cells both on the early and late time scale (). Early nuclear leakiness, however, was efficiently prevented in the presence of BAPTA-AM, a cell permeable, high-affinity Ca2+ chelator that removes intracellular free [Ca2+] also in internal stores.Citation40 In the presence of BAPTA-AM mobilization of free [Ca2+] from the ER/NE is blocked. This compound did not inhibit the collapse of the NE permeability barrier occurring at nuclear condensation (). In addition, up to 2 h after treatment with STS caspase-3-like activity was not significantly affected either by calpeptin or by BAPTA-AM (). This lack of correlation between caspase activation and the early nuclear redistribution of 4xCherry in response to the Ca2+ chelator further supports the existence of a caspase-independent effect of STS at the NE which requires mobilization of Ca2+ from internal stores. At the final stage of chromatin condensation, the NE permeability barrier collapses, a process dominated by the effect of executioner caspases and evidently not dependent either on Ca2+ or on calpains.
Exogenous expression of Bcl-2 at the nuclear membrane increases NE permeability
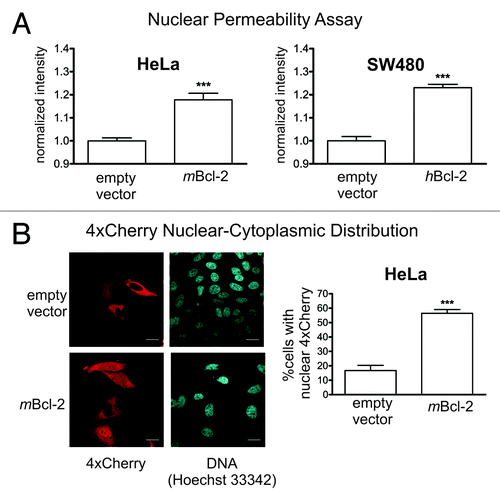
Bcl-2 is an antiapoptotic protein with potent pro-survival activity whose expression leads to a reduction in ER/NE Ca2+ levels.Citation41 Bcl-2 family proteins Bax and Bak have been suggested to increase nuclear permeabilityCitation19 as part of their proapoptotic activity. Their antiapoptotic cousin Bcl-2 has not been investigated so far. Therefore we sought to determine whether Bcl-2 expression might impinge on NE permeability and whether this would involve ER/NE [Ca2+]. First, we compared HeLa cells stably expressing wildtype murine Bcl-2 (HeLa-mbcl-2), and SW480 cells expressing the human counterpart (SW480-hbcl-2) (Fig. S4A) with the respective control cell lines using our nuclear permeability assay (NPA).Citation42,Citation43 Cells were partially permeabilized with digitonin, incubated with fluorescently labeled 70 kD dextran, and imaged by confocal microscopy. Passage of dextran across the NE was quantified by measuring the average fluorescence signal intensity in cell nuclei. In both HeLa and SW480 cells, Bcl-2 expression led to a marked increase in dextran nuclear entry as compared with control (). This finding was confirmed with transient transfections of HeLa-mBcl-2 cells using the permeability marker 4xCherry. Cells with a homogenously distributed fluorescence signal were scored positive for nuclear 4xCherry. Bcl-2 expression resulted in a significantly higher percentage of these cells as compared with control (). Finally, we used bead loading to introduce fluorescent 70 kD dextran into Bcl-2 overexpressing HeLa cells, and obtained again similar results (Fig. S4B). Altogether these data demonstrate that overexpression of Bcl-2 leads to an increase in the size exclusion limit of the NE for passive diffusion, a finding quite unexpected from previous studies and from our own data linking nuclear leakiness to apoptotic cell death.
Figure 4. Bcl-2 overexpression leads to increased nuclear envelope permeability. (A) Nuclear entry of 70 kD fluorescent dextran was assessed in HeLa and SW480 cells stably expressing murine and human bcl-2, respectively, using the Nuclear Permeability Assay. Cells transfected with the empty vector served as controls. In each experiment, the average nuclear fluorescence intensity was normalized to the respective control measurement. A total of at least 800 nuclei were evaluated semi-automatically. *** p < 0.001. (B) HeLa cells stably expressing murine Bcl-2 and the corresponding empty vector were transiently transfected with a construct encoding the permeability marker 4xCherry. Left panel, cell cultures were inspected microscopically for the presence of 4xCherry in the nucleus. A representative set of confocal images is shown. Right panel, quantification of microscopic images. Only cells exhibiting a homogenous distribution of the marker protein between nucleus and cytoplasm were scored as positive. The experiment was performed twice counting at least 150 cells per repeat. Scale bar: 10 μm. *** p < 0.001.
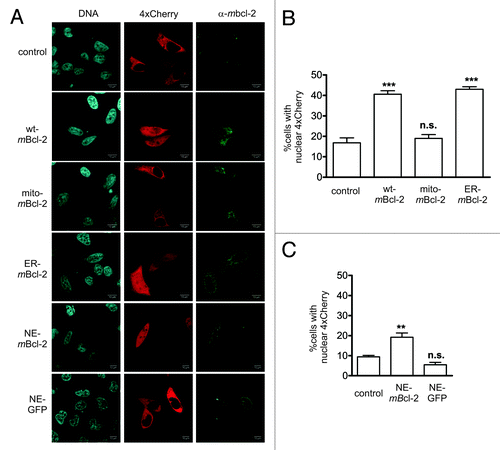
Bcl-2 is known to insert into all intracellular membranes including those of mitochondria, endoplasmic reticulum and nucleus (own unpublished observations and ref. Citation44). To examine whether the effect of Bcl-2 on the nuclear permeability barrier depends on the specific subcellular localization of this protein we compared nuclear entry of 4xCherry in cells expressing full length Bcl-2, and Bcl-2 variants targeted either to the endoplasmic reticulum via fusion to the C-terminus of cytochrome b5 (ER-mBcl2Citation45), or to the mitochondria (mito-mBcl2Citation46) or to the NE (NE-mBcl2, this study). The latter Bcl-2 variant was generated by replacing the C-terminal transmembrane domain of Bcl-2 with the KASH domain of human Nesprin-2, a protein tethering the actin cytoskeleton to the nuclear membraneCitation47 (Fig. S5A). At high expression levels NE-mBcl2 showed a tendency to mislocalize to the ER (not shown). We therefore expressed the corresponding gene under the control of a tetracycline-regulatable promoter in HeLa Tet Off cells. At low expression levels we could achieve an exclusive localization of NE-Bcl2 at the nuclear rim, as judged by its colocalization with Nesprin-2 in confocal images (Fig. S5B). HeLa cells were transiently transfected with plasmid vectors encoding wildtype, mitochondrial, and ER targeted Bcl-2. NE-targeted Bcl-2 was expressed in HeLa Tet Off cells in the presence of doxycyclin. NE-targeted GFP served as control. Nuclear permeability was assessed by contrasfection of 4xCherry and counting cells with equal distribution of the permeability marker between nucleus and cytoplasm. Bcl-2 localization was verified via immunocytochemistry. An increase in NE permeability comparable to that caused by wildtype Bcl-2 was obtained only in cells in which Bcl-2 localized at the ER and the NE but not at the mitochondria. We observed a 2-fold increase with respect to control cells expressing only the permeability marker (). These data indicate that localization of Bcl-2 at the NE is sufficient to mediate its activity on the nuclear permeability barrier.
Figure 5. Bcl-2 localization at the nuclear membrane is sufficient to increase the size exclusion limit of the NE. (A) Confocal images of HeLa cells transiently expressing the permeability marker 4xCherry alone (control) and in combination with either wildtype murine bcl-2 (wt-mBcl-2) or bcl-2 variants targeted to the mitochondria (mito-mBcl-2) and the endoplasmic reticulum (ER-mBcl-2). Nuclear envelope targeted Bcl-2 (NE-mBcl-2) was expressed in HeLa Tet Off cells in the presence of 1 μg/ml doxycyclin to avoid strong overexpression and mislocalization. Nuclear envelope targeted GFP (NE-GFP) served as control. Bcl-2 localization was determined by immunocytochemistry using antibodies specific for murine Bcl-2. The NE-Bcl2 protein was not detectable at the nuclear rim under fixation conditions required to preserve 4xCherry localization. NE-Bcl-2 expressing cells were nevertheless readily identifiable by a punctuate staining pattern. Proper localization of NE-Bcl2 was verified in parallel transfection experiments using digitonin as a permeabilizing agent (see Figure S5 and Material and Methods). Red: 4xCherry; green: mBcl-2; blue: DNA (Hoechst 33342). (B) and (C) Quantitation of microscopic images obtained from HeLa cells (B) and from HeLa Tet Off cells (C). Cells expressing mBcl-2 and its variants were inspected for nuclear 4xCherry localization as described in . At least 150 cells per experimental condition were counted. ** p < 0.01. In HeLa Tet off cells, expression of NE-targeted mBcl-2 resulted in a 2-fold increase in the number of cells displaying nuclear 4xCherry localization with respect to control cells. For wt mBcl-2 this increase was 2.4-fold.
Bcl-2 effects at the NE are mediated by Ca2+
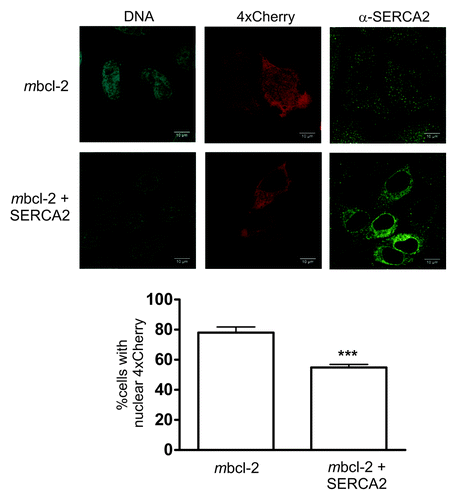
The effect of Bcl-2 as regulator of ER Ca2+ levels is well documented (reviewed in ref. Citation48). Based on our observation in STS-treated HeLa cells suggesting that Ca2+ is involved in controlling the size exclusion limit of the NE, and having detected that nuclear membrane-associated Bcl-2 might increase this limit, we inferred a role for Ca2+ as downstream mediator of Bcl-2 at the NE. To address this question, we first determined ER/NE Ca2+ levels in Bcl-2-expressing cells using Fluo-4 and TG. Hela cells stably expressing wildtype Bcl-2 showed a marked reduction in TG-induced Ca2+ release from internal stores as compared with control cells, in line with previous studiesCitation41 (, left panel, left bar). We also recorded capacitative Ca2+ influx, a secondary Ca2+ response ensuing in cells with depleted internal stores when Ca2+ is added back to the culture medium.Citation49 In Bcl-2 overexpressing cells, this response was shown to be downregulated as adaptation to the long-term reduction of Ca2+ in their internal stores.Citation50 Readdition of Ca2+ (1 mM) to HeLa cells previously exposed to TG evoked a steep increase in fluorophore signal indicative of Ca2+ capacitative influx. This effect was significantly diminished in Bcl-2 overexpressing cells (, right panel, left bar). These data confirm that Bcl-2 overexpression leads to altered Ca2+ levels in the ER/NE supporting our assumption of a role of Ca2+ in the Bcl-2-dependent increase in NE permeability in our experimental model. We then sought to mimic the Ca2+-mediated, Bcl-2 dependent effect on nuclear permeability by manipulating Ca2+ levels in the ER/NE independently of Bcl-2. To this end, we cultured HeLa cells for 18 h in medium at low Ca2+ concentration (0.1 mM), a treatment leading to an adaptive reduction of Ca2+ levels in the ER/NE.Citation50 These cells displayed a marked reduction of TG-induced Ca2+ release as well as a lowered capacitative Ca2+ influx as compared with controls, similarly to Bcl-2 overexpressing cells (, both panels, right bars). Analysis of nucleo-cytoplasmic 4xCherry distribution after adaptation to low calcium conditions indicated a significant increase in the number of cells with nuclear 4xCherry, although not to the same extent as following Bcl-2 overexpression (). Finally, we restored high Ca2+ levels in the ER/NE of Bcl2-expressing Hela cells by coexpressing the ER Ca2+-ATPase SERCA2. Correction of ER/NE [Ca2+] by SERCA2 overexpression has been demonstrated in Bax/Bak double knockout cells, which also display a decreased [Ca2+] ER.Citation51 In line with our working hypothesis, SERCA2 effectively counteracted the Bcl-2 induced increase in nuclear permeability further implicating ER/NE Ca2+ levels in the Bcl-2-dependent regulation of NE permeability ().
Figure 6. Adaptation to low Ca2+ conditions reduces ER Ca2+ levels similarly to Bcl-2 expression, and mimics the Bcl-2 dependent increase in NE permeability. (A) Typical traces of Fluo-4 fluorescence in HeLa cells stably expressing Bcl-2 (light gray), and in control cells transfected with empty vector. The latter were grown either in normal medium (black) or incubated for 18h in low Ca2+ medium (0.1 mM, dark gray). Internal stores were depleted by TG addition (5 μm, indicated by the first arrow). After recovery, extracellular Ca2+ was added back (1 mM, second arrow) by replacing the medium. A second phase of Ca2+ mobilization is observed (capacitative Ca2+ influx). Data points are the average of 5 cells. Error bars show the SEM (B) Quantification of ER Ca2+ levels (left panel) and capacitative Ca2+ influx (right panel). The difference between basal and peak fluorescence values in the first and second phase of Ca2+ elevation are expressed as percent of the values determined in control (empty vector) cells. Experiments were performed in triplicates. * p < 0.05; *** p < 0.001. (C) HeLa control cells (empty vector) were incubated for 6h in low Ca2+ medium (0.1 mM) or in control medium (1 mM Ca2+) prior to transfection with a construct encoding the permeability marker 4xCherry. After overnight incubation, cell cultures were inspected microscopically for the presence of 4xCherry in the nucleus. Only those cells exhibiting at least a homogenous distribution of the marker between nucleus and cytoplasm were scored as positive. A total of at least 150 cells were scored. * p < 0.05.
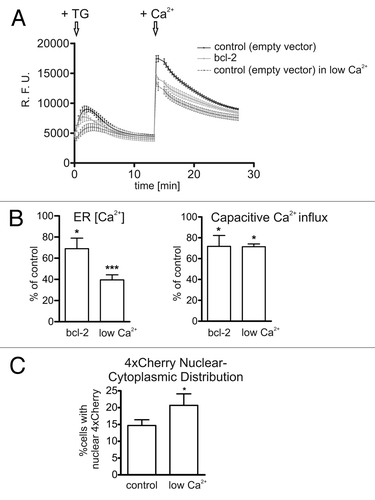
Figure 7. Replenishment of ER/NE Ca2+ levels via SERCA2 expression restores the NE permeability barrier. (A) Representative confocal images of HeLa cells transfected with a construct encoding 4xCherry alone or in combination with an expression plasmid for SERCA2. (B) Quantification of cells displaying nuclear 4xCherry localization. At least 100 cells were counted per experiment. Only cells coexpressing SERCA2 and 4xCherry were included in the evaluation. The analysis was performed as described in .*** p < 0.001.
In sum, these data show that Bcl-2 overexpression lowers the size exclusion limit of the nuclear envelope by altering Ca2+ homeostasis at the ER/NE, and that localization of the protein at the nuclear membrane is sufficient for the effect.
Discussion
The identification of general mechanisms that govern NPC activity in response to different extra- and intracellular signaling cues is of prime importance for understanding the NPC’s recognized involvement in basic nuclear functions, not only nucleo-cytoplasmic trafficking but also chromatin organization and gene expression.Citation52,Citation53 The present work focuses on the NPC’s activity as molecular sieve and investigates how this functional parameter is affected by a drug treatment triggering cell death, vs. a condition of increased cell survival and proliferation. In both settings we detect an increase in the size-exclusion limit of the NPC and show data indicating Ca2+ as a common mediator of the effect.
In STS-treated cells, we detect an early, pre-apoptotic leakiness of the NE concomitantly to a decrease in NE/ER Ca2+ levels. The effect is efficiently suppressed by a Ca2+ chelator and mimicked by artificial downregulation of [Ca2+] in internal stores. It is well established that Ca2+ release channels, such as IP3Rs and RyRs, carry multiple phosphorylation sites and that phosphorylation status modulates channel activity. To date at least 12 kinases are known to directly phosphorylate the IP3Rs.Citation54 Treatment with STS, a pan-kinase inhibitor with broad substrate specificityCitation55 may thus directly interfere with Ca2+ homeostasis by altering Ca2+ channel activity. The association of the IP3R inhibitory protein, IRBIT, to IP3Rs is also regulated by phosphorylation.Citation56 Dephosphorylated IRBIT, which might predominate in kinase-inhibited cells, cannot bind to IP3R. As a consequence, the receptor becomes sensitive to IP3 concentrations that exist in cells under resting conditions, which might result in a lower threshold for Ca2+ release from the NE/ER.
Ca2+ leakage from the ER of apoptotic cells has also been shown to result from caspase-3 dependent cleavage of IP3Rs, generating a constitutively leaky “channel-only” domain.Citation57 The latter however, represents a late apoptotic event downstream of caspase activationCitation58 which is difficult to reconcile with the early-stage nuclear permeabilization observed here, occurring almost immediately after exposure to STS and prior to the caspase-mediated cleavage of Nups. We can also rule out the involvement of calpain-mediated NPC proteolysis in our system, contrarily to what has been reported in neurons challenged with high-doses of glutamate to trigger excitotoxic cell death.
Our data do not support hyperphosphorylation of Nup62 as being involved in the STS-dependent early nuclear leakiness. Nup62, a heavily O-glycosylated Nup of the central channel was shown to be excessively phosphorylated in meningo-virus infected cells displaying loss of NE permeability function. In fact, in that study, STS was used to block virus-mediated permeabilization of the NE.Citation18 A more likely candidate for mediating nuclear leakiness in our experimental model is gp210, a large, N-glycosylated integral nuclear membrane protein associated with the NPC via its short C-terminus exposed toward the cytoplasm. The bulk of the protein extends into the lumen of the NE. gp210, whose cisternal domain possesses five EF hand calcium-binding motives, has been proposed to function as Ca2+ sensor within the NE, and to mediate Ca2+-dependent structural changes of the NPC.Citation24 Moreover, the C-terminal cytoplasmic tail of gp210 is phosphorylated in a cell-cycle dependent manner.Citation59 gp210 is required for nuclear envelope breakdown at the onset of mitosis and is possibly involved in early NPC destabilization, functions which depend upon phosphorylation of the C-terminus.Citation60 Thus, gp210 could potentially respond both to alterations in NE Ca2+ levels and to the activity of cytosolic kinases. Whether both signaling cues converge onto gp210 and how this affects NPC structure is not known. Interestingly, stable exogenous expression of Bcl-2 in R6 rat fibroblasts which are devoid of the nucleoporin gp210 does not elicit an increase in basal nuclear permeability (data not shown) further supporting a role of gp210 in transducing Ca2+ signals to the NPC.
So far, the impairment of the permeability barrier function of the NPC has been mostly associated with stress conditions leading to cell demise. Thus, the finding that exogenous Bcl-2 expression, a condition leading to increased survival and stress resistance also increases basal nuclear permeability was unexpected. By showing that these cells possess a reduced steady-state luminal ER [Ca2+], we provide a possible explanation to this seemingly surprising observation. We propose that both, STS treatment as well as Bcl-2 overexpression increase NE permeability via lowering luminal NE/ER [Ca2+]. Bcl-2 has been shown to localize at the nuclear membrane where it could directly interact with IP3Rs.Citation37,Citation61-Citation63 Importantly, we show that targeting Bcl-2 at the nuclear membrane is sufficient for its permeabilizing activity. Direct binding of Bcl-2 to all three IP3R isoforms has been described.Citation37,Citation64 Recently these interactions have been shown to enhance the sensitivity of the IP3Rs to IP3 resulting in a decreased steady-state [Ca2+] at the ER.Citation37 We therefore predict that Bcl-2-IP3R interaction at the nuclear membrane will result in a similar reduction of [Ca2+] in the nuclear cisterna. This correlation between ER/NE Ca2+ levels and NE permeability is in agreement with numerous studies on Ca2+-mediated structural rearrangements of the NPC.
Our results cannot exclude the existence of further mechanisms of NE permeabilization unrelated to the NPC. High levels of Bcl-2 could destabilize the NE. Alternatively, the marked cytoskeletal deformations following STS treatmentCitation65 might trigger limited and controlled disruption of the NE. In parvovirus-infected cells such transient NE disruptions were reported to mediate nuclear delivery of the virus.Citation66 NE deformations involving changes in the phosphorylation status of lamin A/C have been also implicated in SV-40 infection of non-dividing cells.Citation67
The observation that in STS-treated cells, early nuclear permeabilization can be prevented by BAPTA without affecting final apoptotic execution raises the question about the role of early leakiness of the NE in the apoptotic program. In the presence of BAPTA, STS-treated cells activate caspases () and undergo apoptosis. However, we could not identify nuclei with a morphology corresponding to stage 3 nuclear apoptosis (nuclear collapse/ disassembly)Citation68 in our confocal movies. In the absence of BAPTA, about 20% of the cells reach this stage within the time of observation (see Fig. S6B). This finding on the one hand could reflect a requirement for Ca2+ for completing apoptotic chromatin fragmentation, as already proposed.Citation69 On the other hand, it argues against NE dilation being a proapoptotic signal per se. While in the context of apoptosis, early nuclear leakiness might facilitate nuclear disassembly and influence the final apoptotic morphology similarly to what has been reported for the cleavage of B-type lamins,Citation70 it most likely won’t interfere with basic cellular functions in a healthy cell. Our observation that signal-dependent nuclear transport remains fully functional during the first phase of STS-mediated nuclear permeabilization corroborates this interpretation (data not shown). Our data showing that in tumor cells such as Bcl-2 expressing HeLa and SW480, more than 60% of the cell population shows equal nucleo-cytoplasmic distribution of an inert permeability marker indicate that large variations in the size-exclusion limit of the NE are well tolerated and compatible with cell survival and proliferation. Large-scale structural rearrangements of the NPC are known to occur in connection with the passage of very large cargo, such as mRNP particlesCitation71 and transient increases in the uptake of transport substrates were observed upon stimulation with growth factorsCitation72 or during the cell cycle.Citation10 In bcl-2 overexpressing cells, we observe a constitutive alteration of nuclear membrane permeability which we interpret as an adaptive response to lowered steady-state [Ca2+] in the lumen of the NE. Whether this persistent enhanced accessibility of the nuclear compartment can be established only in the presence of a general death suppressor such as Bcl-2, or whether high NE permeability may promote oncogenic functions of Bcl-2 associated e.g., with cell cycle regulation or DNA repairCitation73 are challenging questions for the future.
Materials and Methods
Plasmid constructs
p4xCherry was created by consecutively inserting four copies of the cDNA sequence encoding the red fluorescent GFP analog mCherryCitation74 into pcDNA3.1 (Invitrogen). Removal of the internal start codons was confirmed by DNA sequence analysis. pRSET-B-mCherry was kindly provided by Roger Tsien (University of California, La Jolla) and used as template for PCR amplification.
Expression constructs encoding murine bcl-2 proteins targeted to different subcellular compartments were a kind gift of Prof Christoph Borner (University of Freiburg): pcDNA3-mbcl2 (full length murine bcl-2), pcDNA3-mbcl2-cytb5 (ER-targeted murine bcl-2) and pcDNA3-mbcl2-RK/Chbcl2XL (mitochondria-targeted murine bcl-2).
The inducible expression construct for NE-targeted murine bcl-2 (NE-Bcl-2) was designed in analogy to a previously described NE-targeted GFP protein.Citation75 The KASH domain of human Nesprin-2 (aa 6833–6883) was amplified from pEGFP-C1-Tm-Nesprin-2 (kind gift of Dr Akis Karakesisoglou, University of Durham) by PCR and introduced into the EcoRI and BamHI sites of pTRE MCS (Clontech) resulting in pTRE-KASH. pTRE-KASH-mBcl-2 was obtained by inserting a mbcl-2 fragment from pcDNA3-mbcl2 lacking the transmembrane domain of bcl-2 into pTRE-KASH. The control plasmid pTRE-GFP-KASH was constructed by insertion of the EGFP coding sequence into pTRE-KASH via PCR.
The pSERCA2 expression construct was kindly provided by Luca Scorrano (University of Padova, Italy). pNup153-GFP was a kind gift of Jan Ellenberg (EMBL Heidelberg). pNup153-D349N-GFP was generated by PCR mutagenesis using the following primers: 153D349NFwd 5′-GTG GGA TAG ATA TCA CAA ATT TTC AGG CCA AAA GAG AAA AG-3′ and 153D439NRev 5′-CTT TTC TCT TTT GGC CTG AAA ATT TGT GAT ATC TAT CCC AC-3′.
Cell culture and transfections
HeLa cervix carcinoma cells stably expressing murine bcl-2 (HeLa-mbcl-2) and SW480 colon carcinoma cells stably expressing human Bcl-2 (SW480-hbcl-2) and the respective empty vector control cell lines were a kind gift of Prof. Christoph Borner (University of Freiburg).
HeLa Tet Off cells expressing the tetracycline controlled transactivator (Clontech) were kindly provided by Dr. Thomas Meergans (University of Konstanz).
All cell lines were maintained in DMEM supplemented with 100 U/ml penicillin, 100 ∝g/ml streptomycin, 2 mM l-glutamine (Invitrogen) and 10% fetal calf serum (Sigma). Cells were cultivated at 37°C with 5% CO2 in a humidified atmosphere. For live cell microscopy, cells were seeded in glass-bottom dishes (ibidi) and transfected with Effectene (Quiagen) according to the manufacturer’s instructions 24 h prior to the experiment.
HeLa Tet Off cells were transfected using the calcium phosphate co-precipitation method. Cells were used for further experiments ~24 h after transfection.
For bead loading experiments, cells were seeded in MatTek glass bottom culture dishes with a glass inset of ~1 cm2 and grown to a density of ~60% prior to the experiment.
Live cell imaging
Imaging experiments were performed in culture medium devoid of Phenol Red to reduce background fluorescence. After exchanging the medium, 150 ∝g/ml Hoechst 33342 was added for labeling nuclear DNA, and the cells were incubated at the microscope stage at 37°C and 5% CO2 for 1–2 h before starting image acquisition. Confocal time-lapse series were recorded at a Zeiss LSM510 Meta confocal microscope equipped with a stage-top incubator and an objective heater using a 63 × /1.4 NA Plan-Apochromat objective lens (Carl Zeiss Microimaging). Laser settings were optimized to ensure cell viability for the whole duration of the experiments. As an additional control for phototoxicity, it was verified that imaging conditions were compatible with cell division (Fig. S6A). Cell nuclei were tracked and focused using an autofocus macro kindly provided by J. Ellenberg.Citation76 For each experiment, 10–13 cells were imaged at a 5 × zoom in a time interval of about eight minutes. If necessary, a delay between acquisition rounds was introduced to keep this time frame constant. At each position, a z-stack consisting of three sections at a spacing of 0.6 μm was recorded. After the first imaging cycle, the acquisition routine was paused and the apoptotic inducer STS (Sigma Aldrich, 0.5 μm) or lz-TRAIL (kind gift of H. Walczak, 300 ng/ml) was added. Inhibitors were added 30 min prior to STS addition at the following concentrations: zVAD-fmk (20 μM), calpeptin (5 μM) and BAPTA-AM (10 μM). For bleaching correction, time courses of untreated cells were recorded under identical conditions.
Quantification of confocal time series
Image analysis was performed using the open source software Image J. Image series were visually inspected and for each time point one z-section showing the nuclear rim in focus in the Nup153-GFP channel was selected. The images were combined into new stacks for quantification. This procedure was necessary because cells alter their morphology during the apoptotic process.
For the same reason, automatic segmentation of the cell nucleus in the Hoechst channel was not applicable. Therefore, all three channels were segmented interactively. Nuclear condensation and NE permeabilization were measured in the Hoechst and the 4xCherry channel, respectively. To this end, a ROI corresponding to the inner boundary of the nucleus was first defined in the Nup153-GFP channel and copied into the Hoechst and the 4xCherry images. For quantifying NE integrity, signal intensities were measured in the Nup153-GFP channel in a second ROI including the nuclear rim (Fig. S1D). Folded regions of the NE were omitted. Intensity values were background substracted, corrected for bleaching and normalized. The time point of nuclear condensation in each time series was defined as an increase in Hoechst fluorescence by a factor of at least 1.07 occurring within two consecutive frames. Cells go through apoptosis asynchronously. To better visualize the abrupt changes in nuclear permeability and NE integrity occurring at the final stage of nuclear execution, intensity values for these two channels were plotted on two separate time-axis. In the first part of the time course intensity values were displayed from the start of image acquisition up to the time point immediately preceding nuclear condensation. In the second part, traces were aligned with respect to the time point of chromatin condensation as determined in the Hoechst channel.
Assessment of nuclear permeability
Nuclear Permeability Assay (NPA) was performed as described previously.Citation43 Briefly, cells were seeded onto glass coverslips ~20 h before the start of the experiment. Cell membranes were selectively permeabilized with digitonin (24 μg/ml). The semi-permeabilized cells were incubated with the fluorescent permeability marker 70 kDa Texas Red-labeled dextran (Sigma) at a concentration of 20 mg/ ml and then imaged at the confocal microscope (Zeiss LSM510 Meta). Images were quantitated semi-automatically by a custom-made software extracting the average nuclear fluorescence intensity. Fluorescence intensity in the nucleus is a measure of nuclear penetration of 70 kDa dextran and thus an indicator of passive permeability of the nuclear envelope.
For the experiment shown in Figure S3B cells were loaded with 70 kDa Texas Red-labeled dextran using glass beads.Citation77,Citation78 Briefly, glass beads with a diameter of ~100 ∝m (Sigma) were treated overnight with 5 M NaOH, washed with ethanol and dried by evaporation. The culture medium was removed and 20 μl dextran solution was added to the glass inset of MatTek culture dishes (20 mg/ml 70 kDa Texas Red-labeled dextran (Sigma) in PBS). Subsequently, a monolayer of clean glass beads was added into the dish cavity and the dish was quickly shaken. Beads were then removed by washing with PBS, medium added back and the cells incubated for 45 min at 37°C. Confocal images were taken and evaluated as described above.
In cells expressing Bcl-2 and its mutants, nuclear permeability was assessed by measuring the percentage of cells displaying nuclear localization of 4xCherry. Images were taken at fixed time points after transfection of 4xCherry and Bcl-2 expression contructs by confocal fluorescence microscopy. Only cells showing at least the same 4xCherry signal in the nucleus as in the cytosol were scored as having a nuclear 4xCherry localization.
Immunocytochemistry
Immunocytochemical detection of mbcl-2 was performed using antibodies specific for murine bcl-2 (mouse monoclonal anti-bcl-2 clone 10C4, Santa Cruz), at a dilution of 1:500 in 10% NGS/PBS. Cells were shortly washed with 5 mM MgCl2/PBS, permeabilised with 0.3% digitonin/PBS for 5 min on ice and rinsed again in 5 mM MgCl2/PBS. Fixation was performed with 4% PFA/PBS for 10 min at room temperature followed by washing in PBS. Reactive aldehyde groups were blocked with 50 mM NH4Cl for 10 min. Unspecific binding sites were saturated with 1% BSA/PBS for 30 min. Incubation with the 10C4 anti-bcl-2 antibody was overnight at 4°C. The secondary antibody conjugated to Alexa488 (Molecular Probes) was diluted 1:400 in 10% NGS/PBS and incubated for 1 h. Excessive washing steps between incubations removed unbound antibodies. DNA was stained with 200 ng/ml Hoechst 33342 for 10 min.
For co-detection of 4xCherry and Bcl-2, cells were fixed overnight at 4°C in 4% PFA/PBS to prevent loss of the permeability marker during permeabilization. After fixation cells were permeabilized for 5 min with 0.1% Triton X-100/PBS at room temperature, rinsed in PBS and incubated in 50 mM NH4Cl/PBS for 10 min. Subsequently, cells were treated with cold acetone at -20°C for 8 min, rinsed in PBS and incubated in 1% BSA/PBS for 30 min. Antibody incubations and Hoechst staining of DNA were performed as described above.
Immunostaining of Bcl-2 at the nuclear membrane required a permeabilization step prior to fixation and was therefore not compatible with simultaneous detection of 4xCherry. In this case, the protocol used for co-detection of 4xCherry and Bcl-2 resulted in a punctuate staining pattern that nevertheless allowed for the identification of cotransfected cells and image quantification. The correct localization of NE-Bcl-2 was confirmed in parallel transfections using digitonin permeabilization.
For co-detection of 4xCherry and SERCA2, cells were fixed overnight at 4°C in 4% PFA/PBS and processed as decribed above with omission of the acetone treatment. The SERCA2 antibody (IID8, Alexis) was diluted 1:750 in 1% BSA/PBS.
Preparation of whole cell extracts and immunoblot analysis
For the preparation of cell lysates, HeLa cell cultures were placed on ice, and protease inhibitors (Complete Mix; Roche Applied Science) and dithiothreitol (1mM) were added directly to the growth media. The cells were then gently scraped off the dish with a rubber policeman, washed in ice-cold PBS, resuspended in 95°C lysis buffer (50 mM Tris/HCl, pH 8.0, 0.5% SDS, 1 mM dithiothreitol), and heated at 95°C for 10 min. The cell debris was removed by centrifugation at 20,000 × g for 10 min. For the detection of Nup-153, SDS-PAGE was performed according to Thomas and Kornberg,Citation79 while for other proteins it was according to Laemmli.Citation80 Proteins were blotted onto nitrocellullose using a wet blot chamber (Bio-Rad Trans-Blot Cell) or a Semidry Blotter (Biometra), and filters incubated in TNT buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 0.05% Tween 20) with 5% milk powder at room temperature for 1 h. Incubation with primary antibodies at 4°C was in TNT with milk overnight. Filter washings were in TNT alone. Incubations with horseradish peroxidase-coupled secondary antibodies (1:2000, Dako) were in TNT with milk at room temperature for 1 h. The filter strip with the biotinylated molecular weight marker (Bio-Rad) was incubated separately with horseradish peroxidase-coupled avidin for 30 min at room temperature. Immunoblots were visualized with a chemiluminescent image analyzer (LAS-1000; Fujifilm). Murine and human Bcl-2 were detected using the antibody N19 (Santa Cruz) at a dilution of 1:2000. The anti-actin antibody (Chemicon) was used at 1:50000 dilution, the anti-Nup-153 / GFP antibody (Progen, clone nup7A8) at 1:50 dilution, and the anti-DsRed antibody (Clontech) at 1:500 dilution.
Fluorometric determination of protease activity, and of ER/NE [Ca2+]
For measurement of caspase activity, a total of 3 × 105 HeLa cells were seeded in six-well plates 24 h prior to the experiment and treated with STS (0.5 μm). To block caspase activation zVAD-fmk (20 μM) was added 30 min prior apoptotic stimulation At the indicated time points, the cells were placed on ice and after the addition of protease inhibitors, they were gently scraped off the dish and collected by centrifugation. Cells were lysed in 25 mM HEPES, pH 7.5, 5 mM MgCl2, 1 mM EGTA, and 0.5% Triton X-100, and the cleavage of DEVD-7-amino-4-trifluoromethyl coumarin (afc) (40 μM) was monitored fluorometrically in reaction buffer (50 mM HEPES, pH 7.5, 10 mM dithiothreitol, 1% sucrose, 0.1% CHAPS {3-[(3-cholamidopropyl)-dimethylammonio]-2-hydroxy-1-propanesulfonate)} over a period of 20 min at 37°C, with a λex (excitation wavelength) value of 390 nm and a λem (emission wavelength) value of 505 nm. The activity was calibrated by using afc standard solutions. Measurements were run in triplicate.
Calpain activity was measured using a Calpain Activity Assay Kit (Abcam, ab65308) according to the manufacturer’s instructions. For apoptosis induction, HeLa cells were treated with 0.5 μM STS for 2h. To block calpain activity, 5 μM calpeptin (Merck) was added 30 min prior to STS treatment. Cell lysates were prepared from 2 × 106 cells. Protein concentrations were determined using BCA (Pierce Biotechnology). Lysates were spiked with 10 ∝M ∝l active calpain. After addition of the calpain substrate (Ac-LLY-afc), samples were transferred to a black UV-light permeable 96-well plate and incubated at 37°C for 60 min. Fluorescence emission of afc was detected as above.
For Ca2+ measurements, cells were cultured in 96 multiwell plates for 24 h and washed carefully with Ca2+-buffer (140 mM NaCl, 5 mM KCl, 1mM CaCl2, 1 mM NaH2PO4, 1mM MgSO4, 5.5 mM Glucose, 20 mM Hepes pH 7.4). Cells were loaded with the [Ca2+] indicator Fluo-4 (Invitrogen) by incubation in 150 ∝l loading buffer (Ca2+-buffer containing Fluo-4 and pluronic acid at final concentrations of 4 ∝M and 0.08% respectively) for 60 min at RT in the dark. After two washings with Ca2+-buffer containing 1mM probenecid, cells were incubated for 30 min at RT for complete de-esterification of intracellular Fluo-4. Immediately before measurement, the buffer was replaced with fresh Ca2+-buffer with probenecid. Fluorescence emission was detected with a microplate fluorescence reader (Genios Plus, Tecan) equipped with appropriate filters (λex = 488 nM, λem = 510–570 nM). After measurement of basal fluorescence, 50 ∝l of buffer solution were replaced with 50 μl of thapsigargin (TG) containing Ca2+-buffer (final TG concentration 5 μM). TG induces release of Ca2+ from internal stores. After Fluo-4 signal intensities had recovered to the basal level (~10 min) the medium was replaced with Ca2+-buffer (1 mM Ca2+) to measure the capacitative Ca2+ influx.
To measure ER/NE Ca2+ and capacitative Ca2+ influx in cells adapted to low Ca2+ conditions, cells were seeded in 96 multiwell plates two days prior to the experiment. After 24 h the growth medium was exchanged with low-Ca2+-buffer (Ca2+-buffer containing 0.1 mM Ca2+). The cells were adapted to low Ca2+ conditions for 24 h. Fluorometric measurements were performed as described above using low-Ca2+-buffer for loading the cells with Fluo-4 and subsequent washing steps. Capacitative Ca2+ influx was measured after readdition of Ca2+. Control measurements using Ca2+-buffer (1 mM) were conducted in parallel.
| Abbreviations: | ||
| NPC | = | nuclear pore complex |
| NE | = | nuclear envelope |
| Nup | = | nuclear pore protein |
| ER | = | endoplasmic reticulum |
| STS | = | staurosporine |
| TG | = | thapsigargin |
Additional material
Download Zip (836.2 KB)Acknowledgments
We thank Christoph Borner (University of Freiburg) for the Bcl-2 expression plasmids and cell lines, Luca Scorrano (University of Padova) for pSERCA2, Jan Ellenberg (EMBL, Heidelberg) for pNup153-GFP, Akis Karakesisoglou (University of Durham) for pEGFP-C1-Tm-Nesprin-2, Henning Walczak (Imperial College London) for lz-TRAIL and Thomas Meergans for HeLa Tet-Off cells. We are grateful to Felix Schönenberger for support in image analysis, and to Anja Holtz for helpful discussions and sequence analysis of the 4xCherry expression plasmid.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here: http://www.landesbioscience.com/journals/nucleus/article/21982/
References
- Ribbeck K, Görlich D. Kinetic analysis of translocation through nuclear pore complexes. EMBO J 2001; 20:1320 - 30; http://dx.doi.org/10.1093/emboj/20.6.1320; PMID: 11250898
- Wälde S, Kehlenbach RH. The Part and the Whole: functions of nucleoporins in nucleocytoplasmic transport. Trends Cell Biol 2010; 20:461 - 9; http://dx.doi.org/10.1016/j.tcb.2010.05.001; PMID: 20627572
- Wente SR, Rout MP. The nuclear pore complex and nuclear transport. Cold Spring Harb Perspect Biol 2010; 2:a000562; http://dx.doi.org/10.1101/cshperspect.a000562; PMID: 20630994
- Satterly N, Tsai PL, van Deursen J, Nussenzveig DR, Wang Y, Faria PA, et al. Influenza virus targets the mRNA export machinery and the nuclear pore complex. Proc Natl Acad Sci U S A 2007; 104:1853 - 8; http://dx.doi.org/10.1073/pnas.0610977104; PMID: 17267598
- Kosako H, Yamaguchi N, Aranami C, Ushiyama M, Kose S, Imamoto N, et al. Phosphoproteomics reveals new ERK MAP kinase targets and links ERK to nucleoporin-mediated nuclear transport. Nat Struct Mol Biol 2009; 16:1026 - 35; http://dx.doi.org/10.1038/nsmb.1656; PMID: 19767751
- Makhnevych T, Lusk CP, Anderson AM, Aitchison JD, Wozniak RW. Cell cycle regulated transport controlled by alterations in the nuclear pore complex. Cell 2003; 115:813 - 23; http://dx.doi.org/10.1016/S0092-8674(03)00986-3; PMID: 14697200
- Allen TD, Rutherford SA, Bennion GR, Wiese C, Riepert S, Kiseleva E, et al. Three-dimensional surface structure analysis of the nucleus. Methods Cell Biol 1998; 53:125 - 38; http://dx.doi.org/10.1016/S0091-679X(08)60877-8; PMID: 9348507
- Terry LJ, Shows EB, Wente SR. Crossing the nuclear envelope: hierarchical regulation of nucleocytoplasmic transport. Science 2007; 318:1412 - 6; http://dx.doi.org/10.1126/science.1142204; PMID: 18048681
- Feldherr CM, Akin D. The permeability of the nuclear envelope in dividing and nondividing cell cultures. J Cell Biol 1990; 111:1 - 8; http://dx.doi.org/10.1083/jcb.111.1.1; PMID: 2365731
- Feldherr CM, Akin D. Signal-mediated nuclear transport in proliferating and growth-arrested BALB/c 3T3 cells. J Cell Biol 1991; 115:933 - 9; http://dx.doi.org/10.1083/jcb.115.4.933; PMID: 1955463
- Chakraborty P, Wang Y, Wei JH, van Deursen J, Yu H, Malureanu L, et al. Nucleoporin levels regulate cell cycle progression and phase-specific gene expression. Dev Cell 2008; 15:657 - 67; http://dx.doi.org/10.1016/j.devcel.2008.08.020; PMID: 19000832
- Ferrando-May E, Cordes V, Biller I, Görlich D, Mirkovic J, Nicotera P. Caspases mediate nucleoporin cleavage but not early redistribution of transport factors and mRNA in apoptosis. Cell Death Differ 2001; 8:495 - 505; http://dx.doi.org/10.1038/sj.cdd.4400837; PMID: 11423910
- Patre M, Tabbert A, Hermann D, Walczak H, Rackwitz HR, Cordes VC, et al. Caspases target only two architectural components within the core structure of the nuclear pore complex. J Biol Chem 2006; 281:1296 - 304; http://dx.doi.org/10.1074/jbc.M511717200; PMID: 16286466
- Faleiro L, Lazebnik Y. Caspases disrupt the nuclear-cytoplasmic barrier. J Cell Biol 2000; 151:951 - 9; http://dx.doi.org/10.1083/jcb.151.5.951; PMID: 11085998
- Kihlmark M, Imreh G, Hallberg E. Sequential degradation of proteins from the nuclear envelope during apoptosis. J Cell Sci 2001; 114:3643 - 53; PMID: 11707516
- Buendia B, Santa-Maria A, Courvalin JC. Caspase-dependent proteolysis of integral and peripheral proteins of nuclear membranes and nuclear pore complex proteins during apoptosis. J Cell Sci 1999; 112:1743 - 53; PMID: 10318766
- Bano D, Dinsdale D, Cabrera-Socorro A, Maida S, Lambacher N, McColl B, et al. Alteration of the nuclear pore complex in Ca(2+)-mediated cell death. Cell Death Differ 2010; 17:119 - 33; http://dx.doi.org/10.1038/cdd.2009.112; PMID: 19713973
- Bardina MV, Lidsky PV, Sheval EV, Fominykh KV, van Kuppeveld FJ, Polyakov VY, et al. Mengovirus-induced rearrangement of the nuclear pore complex: hijacking cellular phosphorylation machinery. J Virol 2009; 83:3150 - 61; http://dx.doi.org/10.1128/JVI.01456-08; PMID: 19144712
- Lindenboim L, Blacher E, Borner C, Stein R. Regulation of stress-induced nuclear protein redistribution: a new function of Bax and Bak uncoupled from Bcl-x(L). Cell Death Differ 2010; 17:346 - 59; http://dx.doi.org/10.1038/cdd.2009.145; PMID: 19816507
- Bootman MD, Fearnley C, Smyrnias I, MacDonald F, Roderick HL. An update on nuclear calcium signalling. J Cell Sci 2009; 122:2337 - 50; http://dx.doi.org/10.1242/jcs.028100; PMID: 19571113
- Sarma A, Yang W. Calcium regulation of nucleocytoplasmic transport. Protein Cell 2011; 2:291 - 302; http://dx.doi.org/10.1007/s13238-011-1038-x; PMID: 21528351
- Gerasimenko O, Gerasimenko J. New aspects of nuclear calcium signalling. J Cell Sci 2004; 117:3087 - 94; http://dx.doi.org/10.1242/jcs.01295; PMID: 15226390
- Wu X, Bers DM. Sarcoplasmic reticulum and nuclear envelope are one highly interconnected Ca2+ store throughout cardiac myocyte. Circ Res 2006; 99:283 - 91; http://dx.doi.org/10.1161/01.RES.0000233386.02708.72; PMID: 16794184
- Greber UF, Gerace L. Depletion of calcium from the lumen of endoplasmic reticulum reversibly inhibits passive diffusion and signal-mediated transport into the nucleus. J Cell Biol 1995; 128:5 - 14; http://dx.doi.org/10.1083/jcb.128.1.5; PMID: 7822421
- Stehno-Bittel L, Perez-Terzic C, Clapham DE. Diffusion across the nuclear envelope inhibited by depletion of the nuclear Ca2+ store. Science 1995; 270:1835 - 8; http://dx.doi.org/10.1126/science.270.5243.1835; PMID: 8525380
- Strübing C, Clapham DE. Active nuclear import and export is independent of lumenal Ca2+ stores in intact mammalian cells. J Gen Physiol 1999; 113:239 - 48; http://dx.doi.org/10.1085/jgp.113.2.239; PMID: 9925822
- Wei X, Henke VG, Strübing C, Brown EB, Clapham DE. Real-time imaging of nuclear permeation by EGFP in single intact cells. Biophys J 2003; 84:1317 - 27; http://dx.doi.org/10.1016/S0006-3495(03)74947-9; PMID: 12547812
- Paulillo SM, Powers MA, Ullman KS, Fahrenkrog B. Changes in nucleoporin domain topology in response to chemical effectors. J Mol Biol 2006; 363:39 - 50; http://dx.doi.org/10.1016/j.jmb.2006.08.021; PMID: 16962132
- Stoffler D, Schwarz-Herion K, Aebi U, Fahrenkrog B. Getting across the nuclear pore complex: new insights into nucleocytoplasmic transport. Can J Physiol Pharmacol 2006; 84:499 - 507; http://dx.doi.org/10.1139/y06-001; PMID: 16902595
- Stoffler D, Fahrenkrog B, Aebi U. The nuclear pore complex: from molecular architecture to functional dynamics. Curr Opin Cell Biol 1999; 11:391 - 401; http://dx.doi.org/10.1016/S0955-0674(99)80055-6; PMID: 10395558
- Perez-Terzic C, Pyle J, Jaconi M, Stehno-Bittel L, Clapham DE. Conformational states of the nuclear pore complex induced by depletion of nuclear Ca2+ stores. Science 1996; 273:1875 - 7; http://dx.doi.org/10.1126/science.273.5283.1875; PMID: 8791595
- Nutt LK, Chandra J, Pataer A, Fang B, Roth JA, Swisher SG, et al. Bax-mediated Ca2+ mobilization promotes cytochrome c release during apoptosis. J Biol Chem 2002; 277:20301 - 8; http://dx.doi.org/10.1074/jbc.M201604200; PMID: 11909872
- Oakes SA, Scorrano L, Opferman JT, Bassik MC, Nishino M, Pozzan T, et al. Proapoptotic BAX and BAK regulate the type 1 inositol trisphosphate receptor and calcium leak from the endoplasmic reticulum. Proc Natl Acad Sci U S A 2005; 102:105 - 10; http://dx.doi.org/10.1073/pnas.0408352102; PMID: 15613488
- Kruman I, Guo Q, Mattson MP. Calcium and reactive oxygen species mediate staurosporine-induced mitochondrial dysfunction and apoptosis in PC12 cells. J Neurosci Res 1998; 51:293 - 308; http://dx.doi.org/10.1002/(SICI)1097-4547(19980201)51:3<293::AID-JNR3>3.0.CO;2-B; PMID: 9486765
- Yip KW, Reed JC. Bcl-2 family proteins and cancer. Oncogene 2008; 27:6398 - 406; http://dx.doi.org/10.1038/onc.2008.307; PMID: 18955968
- Pinton P, Rizzuto R. Bcl-2 and Ca2+ homeostasis in the endoplasmic reticulum. Cell Death Differ 2006; 13:1409 - 18; http://dx.doi.org/10.1038/sj.cdd.4401960; PMID: 16729032
- Eckenrode EF, Yang J, Velmurugan GV, Foskett JK, White C. Apoptosis protection by Mcl-1 and Bcl-2 modulation of inositol 1,4,5-trisphosphate receptor-dependent Ca2+ signaling. J Biol Chem 2010; 285:13678 - 84; http://dx.doi.org/10.1074/jbc.M109.096040; PMID: 20189983
- Walczak H, Degli-Esposti MA, Johnson RS, Smolak PJ, Waugh JY, Boiani N, et al. TRAIL-R2: a novel apoptosis-mediating receptor for TRAIL. EMBO J 1997; 16:5386 - 97; http://dx.doi.org/10.1093/emboj/16.17.5386; PMID: 9311998
- Thastrup O, Cullen PJ, Drøbak BK, Hanley MR, Dawson AP. Thapsigargin, a tumor promoter, discharges intracellular Ca2+ stores by specific inhibition of the endoplasmic reticulum Ca2(+)-ATPase. Proc Natl Acad Sci U S A 1990; 87:2466 - 70; http://dx.doi.org/10.1073/pnas.87.7.2466; PMID: 2138778
- Preston SF, Berlin RD. An intracellular calcium store regulates protein synthesis in HeLa cells, but it is not the hormone-sensitive store. Cell Calcium 1992; 13:303 - 12; http://dx.doi.org/10.1016/0143-4160(92)90065-Z; PMID: 1377981
- Foyouzi-Youssefi R, Arnaudeau S, Borner C, Kelley WL, Tschopp J, Lew DP, et al. Bcl-2 decreases the free Ca2+ concentration within the endoplasmic reticulum. Proc Natl Acad Sci U S A 2000; 97:5723 - 8; http://dx.doi.org/10.1073/pnas.97.11.5723; PMID: 10823933
- Roehrig S, Tabbert A, Ferrando-May E. In vitro measurement of nuclear permeability changes in apoptosis. Anal Biochem 2003; 318:244 - 53; http://dx.doi.org/10.1016/S0003-2697(03)00242-2; PMID: 12814628
- Grote P, Ferrando-May E. In vitro assay for the quantitation of apoptosis-induced alterations of nuclear envelope permeability. Nat Protoc 2006; 1:3034 - 40; http://dx.doi.org/10.1038/nprot.2006.460; PMID: 17406565
- Schinzel A, Kaufmann T, Borner C. Bcl-2 family members: integrators of survival and death signals in physiology and pathology [corrected]. [corrected] Biochim Biophys Acta 2004; 1644:95 - 105; http://dx.doi.org/10.1016/j.bbamcr.2003.09.006; PMID: 14996494
- Häcki J, Egger L, Monney L, Conus S, Rossé T, Fellay I, et al. Apoptotic crosstalk between the endoplasmic reticulum and mitochondria controlled by Bcl-2. Oncogene 2000; 19:2286 - 95; http://dx.doi.org/10.1038/sj.onc.1203592; PMID: 10822379
- Kaufmann T, Schlipf S, Sanz J, Neubert K, Stein R, Borner C. Characterization of the signal that directs Bcl-x(L), but not Bcl-2, to the mitochondrial outer membrane. J Cell Biol 2003; 160:53 - 64; http://dx.doi.org/10.1083/jcb.200210084; PMID: 12515824
- Padmakumar VC, Libotte T, Lu W, Zaim H, Abraham S, Noegel AA, et al. The inner nuclear membrane protein Sun1 mediates the anchorage of Nesprin-2 to the nuclear envelope. J Cell Sci 2005; 118:3419 - 30; http://dx.doi.org/10.1242/jcs.02471; PMID: 16079285
- Rong Y, Distelhorst CW. Bcl-2 protein family members: versatile regulators of calcium signaling in cell survival and apoptosis. Annu Rev Physiol 2008; 70:73 - 91; http://dx.doi.org/10.1146/annurev.physiol.70.021507.105852; PMID: 17680735
- Hofer AM, Fasolato C, Pozzan T. Capacitative Ca2+ entry is closely linked to the filling state of internal Ca2+ stores: a study using simultaneous measurements of ICRAC and intraluminal [Ca2+]. [Ca2+] J Cell Biol 1998; 140:325 - 34; http://dx.doi.org/10.1083/jcb.140.2.325; PMID: 9442108
- Pinton P, Ferrari D, Magalhães P, Schulze-Osthoff K, Di Virgilio F, Pozzan T, et al. Reduced loading of intracellular Ca(2+) stores and downregulation of capacitative Ca(2+) influx in Bcl-2-overexpressing cells. J Cell Biol 2000; 148:857 - 62; http://dx.doi.org/10.1083/jcb.148.5.857; PMID: 10704437
- Scorrano L, Oakes SA, Opferman JT, Cheng EH, Sorcinelli MD, Pozzan T, et al. BAX and BAK regulation of endoplasmic reticulum Ca2+: a control point for apoptosis. Science 2003; 300:135 - 9; http://dx.doi.org/10.1126/science.1081208; PMID: 12624178
- Tran EJ, Wente SR. Dynamic nuclear pore complexes: life on the edge. Cell 2006; 125:1041 - 53; http://dx.doi.org/10.1016/j.cell.2006.05.027; PMID: 16777596
- Xylourgidis N, Fornerod M. Acting out of character: regulatory roles of nuclear pore complex proteins. Dev Cell 2009; 17:617 - 25; http://dx.doi.org/10.1016/j.devcel.2009.10.015; PMID: 19922867
- Vanderheyden V, Devogelaere B, Missiaen L, De Smedt H, Bultynck G, Parys JB. Regulation of inositol 1,4,5-trisphosphate-induced Ca2+ release by reversible phosphorylation and dephosphorylation. Biochim Biophys Acta 2009; 1793:959 - 70; http://dx.doi.org/10.1016/j.bbamcr.2008.12.003; PMID: 19133301
- Karaman MW, Herrgard S, Treiber DK, Gallant P, Atteridge CE, Campbell BT, et al. A quantitative analysis of kinase inhibitor selectivity. Nat Biotechnol 2008; 26:127 - 32; http://dx.doi.org/10.1038/nbt1358; PMID: 18183025
- Devogelaere B, Beullens M, Sammels E, Derua R, Waelkens E, van Lint J, et al. Protein phosphatase-1 is a novel regulator of the interaction between IRBIT and the inositol 1,4,5-trisphosphate receptor. Biochem J 2007; 407:303 - 11; http://dx.doi.org/10.1042/BJ20070361; PMID: 17635105
- Nakayama T, Hattori M, Uchida K, Nakamura T, Tateishi Y, Bannai H, et al. The regulatory domain of the inositol 1,4,5-trisphosphate receptor is necessary to keep the channel domain closed: possible physiological significance of specific cleavage by caspase 3. Biochem J 2004; 377:299 - 307; http://dx.doi.org/10.1042/BJ20030599; PMID: 12968951
- Assefa Z, Bultynck G, Szlufcik K, Nadif Kasri N, Vermassen E, Goris J, et al. Caspase-3-induced truncation of type 1 inositol trisphosphate receptor accelerates apoptotic cell death and induces inositol trisphosphate-independent calcium release during apoptosis. J Biol Chem 2004; 279:43227 - 36; http://dx.doi.org/10.1074/jbc.M403872200; PMID: 15284241
- Favreau C, Worman HJ, Wozniak RW, Frappier T, Courvalin JC. Cell cycle-dependent phosphorylation of nucleoporins and nuclear pore membrane protein Gp210. Biochemistry 1996; 35:8035 - 44; http://dx.doi.org/10.1021/bi9600660; PMID: 8672508
- Galy V, Antonin W, Jaedicke A, Sachse M, Santarella R, Haselmann U, et al. A role for gp210 in mitotic nuclear-envelope breakdown. J Cell Sci 2008; 121:317 - 28; http://dx.doi.org/10.1242/jcs.022525; PMID: 18216332
- Hoetelmans R, van Slooten HJ, Keijzer R, Erkeland S, van de Velde CJ, Dierendonck JH. Bcl-2 and Bax proteins are present in interphase nuclei of mammalian cells. Cell Death Differ 2000; 7:384 - 92; http://dx.doi.org/10.1038/sj.cdd.4400664; PMID: 10773823
- Givol I, Tsarfaty I, Resau J, Rulong S, da Silva PP, Nasioulas G, et al. Bcl-2 expressed using a retroviral vector is localized primarily in the nuclear membrane and the endoplasmic reticulum of chicken embryo fibroblasts. Cell Growth Differ 1994; 5:419 - 29; PMID: 8043516
- Monaghan P, Robertson D, Amos TA, Dyer MJ, Mason DY, Greaves MF. Ultrastructural localization of bcl-2 protein. J Histochem Cytochem 1992; 40:1819 - 25; http://dx.doi.org/10.1177/40.12.1453000; PMID: 1453000
- Chen R, Valencia I, Zhong F, McColl KS, Roderick HL, Bootman MD, et al. Bcl-2 functionally interacts with inositol 1,4,5-trisphosphate receptors to regulate calcium release from the ER in response to inositol 1,4,5-trisphosphate. J Cell Biol 2004; 166:193 - 203; http://dx.doi.org/10.1083/jcb.200309146; PMID: 15263017
- Pelling AE, Veraitch FS, Chu CP, Mason C, Horton MA. Mechanical dynamics of single cells during early apoptosis. Cell Motil Cytoskeleton 2009; 66:409 - 22; http://dx.doi.org/10.1002/cm.20391; PMID: 19492400
- Cohen S, Marr AK, Garcin P, Panté N. Nuclear envelope disruption involving host caspases plays a role in the parvovirus replication cycle. J Virol 2011; 85:4863 - 74; http://dx.doi.org/10.1128/JVI.01999-10; PMID: 21367902
- Butin-Israeli V, Ben-nun-Shaul O, Kopatz I, Adam SA, Shimi T, Goldman RD, et al. Simian virus 40 induces lamin A/C fluctuations and nuclear envelope deformation during cell entry. Nucleus 2011; 2:320 - 30; http://dx.doi.org/10.4161/nucl.2.4.16371; PMID: 21941111
- Toné S, Sugimoto K, Tanda K, Suda T, Uehira K, Kanouchi H, et al. Three distinct stages of apoptotic nuclear condensation revealed by time-lapse imaging, biochemical and electron microscopy analysis of cell-free apoptosis. Exp Cell Res 2007; 313:3635 - 44; http://dx.doi.org/10.1016/j.yexcr.2007.06.018; PMID: 17643424
- Oshimi Y, Miyazaki S. Fas antigen-mediated DNA fragmentation and apoptotic morphologic changes are regulated by elevated cytosolic Ca2+ level. J Immunol 1995; 154:599 - 609; PMID: 7529280
- Rao L, Perez D, White E. Lamin proteolysis facilitates nuclear events during apoptosis. J Cell Biol 1996; 135:1441 - 55; http://dx.doi.org/10.1083/jcb.135.6.1441; PMID: 8978814
- Kiseleva E, Goldberg MW, Daneholt B, Allen TD. RNP export is mediated by structural reorganization of the nuclear pore basket. J Mol Biol 1996; 260:304 - 11; http://dx.doi.org/10.1006/jmbi.1996.0401; PMID: 8757794
- Feldherr CM, Akin D. Regulation of nuclear transport in proliferating and quiescent cells. Exp Cell Res 1993; 205:179 - 86; http://dx.doi.org/10.1006/excr.1993.1073; PMID: 8453991
- Green DR, McKinnon PJ. A survivor hits the breaks. Mol Cell 2008; 29:411 - 2; http://dx.doi.org/10.1016/j.molcel.2008.02.003; PMID: 18313378
- Shaner NC, Campbell RE, Steinbach PA, Giepmans BN, Palmer AE, Tsien RY. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat Biotechnol 2004; 22:1567 - 72; http://dx.doi.org/10.1038/nbt1037; PMID: 15558047
- Crisp M, Liu Q, Roux K, Rattner JB, Shanahan C, Burke B, et al. Coupling of the nucleus and cytoplasm: role of the LINC complex. J Cell Biol 2006; 172:41 - 53; http://dx.doi.org/10.1083/jcb.200509124; PMID: 16380439
- Rabut G, Ellenberg J. Automatic real-time three-dimensional cell tracking by fluorescence microscopy. J Microsc 2004; 216:131 - 7; http://dx.doi.org/10.1111/j.0022-2720.2004.01404.x; PMID: 15516224
- Grote P, Ferrando-May E. Quantitative measurement of nuclear permeability changes in apoptosis. Nat Protoc 2006; 1:3034 - 40; http://dx.doi.org/10.1038/nprot.2006.460; PMID: 17406565
- Mcneil PL. Incorporation of macromolecules into living cells. Methods Cell Biol 1989; 29:153 - 73; http://dx.doi.org/10.1016/S0091-679X(08)60193-4; PMID: 2643758
- Manders EM, Kimura H, Cook PR. Direct imaging of DNA in living cells reveals the dynamics of chromosome formation. J Cell Biol 1999; 144:813 - 21; http://dx.doi.org/10.1083/jcb.144.5.813; PMID: 10085283
- Thomas JO, Kornberg RD. An octamer of histones in chromatin and free in solution. Proc Natl Acad Sci U S A 1975; 72:2626 - 30; http://dx.doi.org/10.1073/pnas.72.7.2626; PMID: 241077
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970; 227:680 - 5; http://dx.doi.org/10.1038/227680a0; PMID: 5432063