Abstract
Chromosome cohesion, mediated by the cohesin complex, is essential for the process of chromosome segregation. Mutations in cohesin and its regulators are associated with a group of human diseases known as the cohesinopathies. These diseases are characterized by defects in head, face, limb, and heart development, mental retardation, and poor growth. The developmental features of the diseases are not well explained by defects in chromosome segregation, but instead are consistent with changes in gene expression during embryogenesis. Thus a central question to understanding the cohesinopathies is how mutations in cohesin lead to changes in gene expression. One of the prevailing models is that cohesin binding to promoters and enhancers directly regulates transcription. I propose that in addition cohesin may influence gene expression via translational mechanisms. If true, cohesinopathies may be related in etiology to another group of human diseases known as ribosomopathies, diseases caused by defects in ribosome biogenesis. By considering this possibility we can more fully evaluate causes and treatments for the cohesinopathies.
Introduction
The cohesin complex is a 4 subunit ring structure that holds sister chromatids together from the time of DNA replication until the chromosomes divide upon cell division (). The complex binds to chromosomes in hundreds of locations along chromosome arms as well as centromeres where it is important for chromosome segregation. In 2004 came the first reports associating mutations in a cohesin loading factor, NIPBL, with human disease.Citation1-Citation3 In the past 8 years this finding has been followed with several more reports linking mutations in several different cohesin genes with human disease.Citation4 Despite the well-established function of the cohesin complex in chromosome segregation, the cohesinopathies intriguingly do not manifest with severe cellular defects in chromosome segregation. Instead, the cohesinopathies are developmental and growth disorders, characterized by defects in head, face, limb, gastrointestinal, and heart development and mental retardation.Citation4 This implies there are unexpected functions for the cohesin complex and has challenged the field to ask how defects in cohesin can cause these developmental abnormalities.
Figure 1. The cohesin ring is composed of 4 subunits, Smc1 (blue), Smc3 (red), Scc3 (yellow), and Mcd1/Rad21/Scc1 (green). The ring is loaded onto DNA by the Scc2/NIPBL-Scc4 complex (not depicted). The region of Smc3 in contact with Mcd1 is acetylated (ac) by Eco1 and deacetylated by Hos1. The table shows the genes mutated in various cohesinopathies. Modified from reference Citation39.

The prevailing hypothesis is that the association of cohesin with chromosome arms, and in particular with promoters and enhancers, regulates gene expression. Cohesinopathy mutations in some way compromise this association, leading to differential gene expression without compromising functions in chromosome segregation. This hypothesis can also be thought of as the “many targets” hypothesis wherein the binding of cohesin at many sites in the genome directly regulates transcription. While there is evidence that this type of mechanism is at work at some genes, the fact remains that many of the genes with altered expression in cohesin mutants are not associated with cohesin binding.
A second, non-exclusive hypothesis is the “smoking gun.” If cohesin binding at a few key loci regulates their expression, this could cause many indirect changes in gene expression. For instance, if cohesin binds to and regulates the c-Myc locus, which is a transcriptional activator of many genes, its reduced expression could affect c-Myc targets. There is evidence that cohesin binds to the c-Myc gene and promotes its expression.Citation5 Are other central regulators controlled by cohesin? While the major focus of the prevailing models relates to mechanisms that influence levels of mRNAs which in turn impact levels of proteins, mechanisms underlying translation itself may have a critical role. In this article I propose that cohesin binding could regulate loci that influence translation. Changes in translation may be able to account for a significant fraction of the gene expression changes associated with mutations in cohesin and provide a new paradigm for thinking about the cohesinopathies.
Ribosome Biogenesis
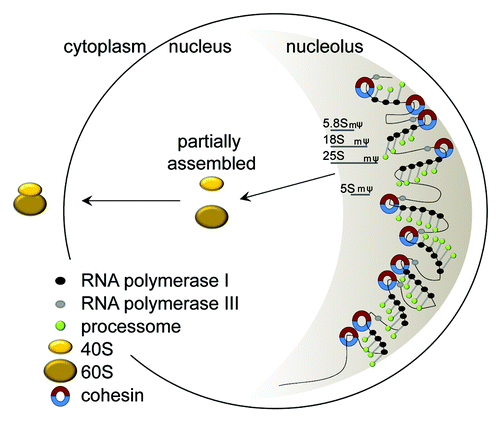
Ribosomes are the large protein-RNA machines in the cytoplasm that take an mRNA and translate it into a protein. Ribosomes are made up of 79 different proteins, including those in the small 40S subunit (RPSs) and those in the large 60S subunit (RPLs). Ribosomes also contain 4 different structural RNAs. Three of the RNAs are made by RNA polymerase I from a single RNA precursor, 2 of which are incorporated into the large subunit and 1 into the small subunit. The large subunit also contains an RNA made by RNA polymerase III. These RNAs are cotranscriptionally processed and modified by methylation and pseudouridylation in the nucleolus guided by a large cadre of proteins and snoRNAs (processome). The initial phases of 40S and 60S assembly also occur in the nucleolus. The production of ribosomes is tightly orchestrated and is the main energy requiring process in a proliferative cell ().
Figure 2. Assembly of ribosomes. rRNAs are made in the nucleolus, which is a single crescent shaped structure that fills about 30% of the budding yeast nucleus. The rDNA resides in the nucleolus. The rDNA in yeast consists of 150 repeats of a 9.1 kb sequence that generates the rRNAs that become the structural components of the ribosome. Only about half of the repeats are transcribed at any given time, and these tend to cluster together. RNA polymerase I (black circles) synthesizes a single 35S transcript which is processed and modified (pseudouridylated, ψ and methylated, m) by the processome (green circles) concurrent with transcription to generate 5.8S, 18S, and 25S. The 5S transcript is synthesized by RNA polymerase III (gray circles). These rRNAs are partially assembled with ribosomal proteins into precursors of the 40S and 60S subunits in the nucleus. Final assembly of the ribosome occurs in the cytoplasm (yellow ovals) after export. Cohesin (blue-red circles) associates with the rDNA in every genome examined. This cohesin could help to organize the active rDNA repeats for transcription. Mutations that affect the number of cohesive rings could have a negative impact on nucleolar form and function.
Mutations that impair ribosome biogenesis have long been known to confer developmental phenotypes. For example, “Minutes” are a class of mutants described in flies and mice that have delayed development, low fertility and viability, and small body size. The Minute phenotype in flies is caused by haploinsufficiency for any one of 64 different ribosomal proteins.Citation6 Minute flies have short bristles, probably due to the high rate of protein synthesis required in a single cell during a short developmental period. Demand for ribosomes in dividing cells is incredibly high; they are estimated to be produced at 2,000 per minute in yeast and 14,000 per minute in Hela cells. For this reason the genes encoding rRNAs are present in hundreds of copies in a genome. A reduction in the number of rDNA repeats gives rise to a different class of mutants in D. melanogaster called bobbed mutants. Similar to the Minutes, bobbed mutants have small bristles and developmental delay.
Recently it has emerged that several different human genetic diseases are associated with defects in ribosome biogenesis. This group of diseases is referred to as the ribosomopathies (). The genes mutated in these diseases include (1) RNA polymerase I and III subunits, (2) ribosomal protein subunits and (3) a modifier of the rRNA. One of the biggest surprises is the specificity of developmental defects that can be caused by mutations that affect different aspects of ribosome biogenesis. For instance, the main feature of Diamond Blackfan anemia is reduced production of red blood cells while the main features of Treacher Collins syndrome is defective craniofacial development.Citation7 However, one commonality may be that fast proliferating tissues are more sensitive to translational deficiencies.
Table 1. Ribosomopathies
Translational Reprogramming
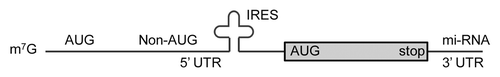
While a global translation deficiency can result from insufficient ribosomes, individual mRNAs may carry sequences that further influence their translation (). The variable use of these sequences is sometimes referred to as translational reprogramming, that is, using the pool of existing mRNAs but altering their translation. This is a strategy to rapidly change the proteome. For instance, upstream open reading frames (uORFs) are sequences in the 5′UTR of a mRNA where ribosomes can bind and may stall under normal conditions but not in starvation conditions. For some genes, alternate start codons may be used for translation initiation. Translation of an individual mRNA can be inhibited by microRNA binding in the 3′UTR.
Figure 3. mRNA sequences that can affect translation. Within the 5′UTR region, an mRNA might contain an AUG (uORF) that a ribosome will bind to and stall translation. Non-AUG codons and internal ribosome entry sites (IRES) are sometimes used to initiate translation. Under some conditions there may be readthrough of the normal stop codon. The non-AUG initiation and stop codon readthrough can change the peptide that is synthesized. An mi-RNA binding site in the 3′UTR can block translation.
Although cap dependent translation is most common, internal ribosome entry sites (IRES) can also be used for translation. IRES sequences were first discovered in viruses, but over 100 cellular eukaryotic genes have been reported to have an IRES that can be used to initiate translation. Viruses also have the best characterized examples of frameshifting and stop codon readthrough, but these alternative translation strategies are starting to pop up in association with eukaryotic mRNAs. In the human disease dyskeratosis congenita, mutations in dyskerin, which is the enzyme responsible for pseudouridylation of rRNA, are associated with increased rates of ribosomal frameshifting and reduced IRES usage without any major changes in the global efficiency of translation.Citation8,Citation9
Translational reprogramming occurs during development and in human disease. Changes in translational control are critical for cancer initiation and progressionCitation10,Citation11. The development of the axial skeleton in mice is dependent on RPL38; mutation of RPL38 leads to specific defects in translating homeobox mRNAs, with the outcome being homeotic transformations.Citation12 In yeast cells in which the RPS25a gene was deleted, cap independent (IRES) translation was reduced while cap dependent translation was normal, demonstrating that changes in a protein component of the ribosome can alter IRES usage.Citation13 These examples and others suggest translational reprogramming may in some instances be associated with specialized ribosomes with different translational properties, which could impact different classes of genes.Citation14
Cohesinopathies: Roberts Syndrome
Roberts syndrome (RBS) is caused by homozygous mutation of ESCO2, an acetyltransferase that acetylates the Smc3 subunit of the cohesin complex to “lock” the complex in a cohesive state. Most patients have no detectable ESCO2 protein. However, a few patients have a mutation in the active site of the enzyme. In this case a protein is made that lacks acetyltransferase activity, demonstrating that the disease can be attributed to the loss of this activity.Citation15 Animal models for Roberts syndrome show defects in cell proliferation and an increase in apoptosis.Citation16 Human cell lines are defective in DNA damage repair.Citation17 The most striking feature of chromosomes in RBS cells is heterochromatin puffing at the centromeres and nucleolar organizing regions in the genome, suggesting cohesion at these regions might be disturbed.Citation18 Interestingly, in one of the first publications identifying the cohesin complex in yeast in 1997, a picture of DNA from a cohesin mutant was shown and “puffy” rDNA is apparent.Citation19
We found that an RBS human cell line displays decreased translation.Citation20 RBS fibroblasts incorporate 35S-methionine into protein at reduced levels compared with either a normal control or the RBS cells complemented by ESCO2. Second, RBS fibroblasts have fewer actively translating ribosomes. Finally, the production of the structural rRNAs is reduced. Together, these findings suggest that one of the defects associated with RBS is a global reduction in translational efficiency.
Under starvation conditions, translation is downregulated to save energy. Under these conditions some mRNAs are preferentially translated, such as those that contain uORFs. This state may be akin in some ways to RBS. In a yeast strain containing the acetyltransferase mutation found in human RBS cells, the uORFs in the GCN4 promoter are bypassed and the protein is made at higher levels.Citation20 Gcn4 is often described as a starvation transcription factor, and it induces the expression of a group of genes. uORFs have been predicted to be present in approximately half of the gene promoters in the human genome.Citation21 We speculate that some of the differential gene expression associated with RBS may be due to this type of translational change.
How can we explain reduced translational efficiency in RBS? We have shown that the amount of rRNA that is produced in both yeast and human cells is reduced. Since this RNA can limit ribosome biogenesis, I speculate that this reduced level of rRNA contributes to defects in ribosome biogenesis. Cohesin binds at the rDNA in every species in which its binding has been mapped. Defects in cohesion at the rDNA may lead to defects in nucleolar organization and in fact, nucleolar morphology in scc2 (NIPBL) and eco1 (ESCO2) yeast mutants is aberrant.Citation22,Citation23 This in turn could lead to defects in nucleolar function, including transcription by RNA polymerase I, co-transcriptional processing and modification of the rRNAs, and assembly of ribosomes. It is even possible that the ribosomes produced have altered properties. The molecular details of this hypothesis remain to be tested.
Cohesinopathies: Cornelia de Lange Syndrome
Cornelia de Lange syndrome (CdLS) is caused by mutation in one copy of a cohesin gene, including Smc1 or Smc3, which are subunits of the cohesin ring, HDAC8, which is a deacetylase for the ring, and most commonly, NIPBL/Scc2.Citation4,Citation24 NIPBL is a loading factor for the cohesin complex. A related cohesinopathy is caused by mutation in Rad21, another subunit of the cohesin ring.Citation25 Gene expression profiling conducted in mouse and zebrafish models for these cohesinopathies as well as human cell lines reveals many small changes in gene expression.Citation16,Citation26,Citation27 However, the expression signature in human CdLS cells and a CdLS zebrafish model is distinct from that observed in models for Roberts syndrome,Citation16,Citation26 suggesting underlying molecular differences in these diseases.
While an smc1 mutant in yeast had decreased 35S-methionine incorporation, the decrease was half of that observed in the RBS background.Citation20 While there may be a mild global defect in translation, translational reprogramming may also contribute to CdLS. While most studies to date have focused on differential expression of mRNAs in the cohesinopathies, very little work has been done to examine how protein levels change. For instance, if cohesin mutants have reduced IRES usage, the production of proteins from IRES containing mRNAs could be reduced. Some important regulators that have experimentally validated IRESs in humans include RUNX1, BCL2, VEGF, FGF1, HSP70, eiF4G and the MYC genes.Citation28 Cohesin has also been suggested to influence promoter choice, which could influence the 5′UTR of the transcript and therefore translation.Citation29
Knockdown of ESCO2, Rad21, or Smc3 in zebrafish is associated with increased levels of p53, but knockdown of Nipbl is not.Citation5,Citation16,Citation30,Citation31 The elevation in p53 has been attributed to a variety of causes including DNA damage, but another potential explanation is nucleolar stress, which is associated with elevated levels of p53.Citation32 Since p53 is not uniformly triggered, there must be important molecular and cellular variation in the outcome arising from different mutations in cohesin associated genes. For instance, different mutations may compromise rDNA cohesion and nucleolar function to different degrees. This could contribute to different gene mutations causing diverse cohesinopathy syndromes.
In addition to a putative role in rDNA transcription, Scc2 could affect the transcription of other regions of the genome. Scc2 has been shown to bind at ribosomal protein genes, tRNA genes, and snoRNA genes in yeastCitation23 and cohesin binds at TFIIIC sites in mouse cells.Citation33 TFIIIC is a transcription factor for RNA polymerase III. tRNA genes, which are transcribed by RNA polymerase III, normally cluster in yeast, but are dispersed in yeast cohesin mutants.Citation22 Alterations in the expression of ribosomal protein genes, tRNA genes, or snoRNA genes could potentially affect translation. Recently, Treacher Collins syndrome, a developmental disorder characterized by abnormal craniofacial development, has been shown to be caused by mutations in subunits of RNA polymerase I and III.Citation34 Mutations that disturb the production of non-coding RNAs that are critical for translation could affect development. The relationship between translation and development remains to be explored in animal models of CdLS.
Summary and Perspective
Total loss of cohesion leads to precocious sister separation and chromosome missegregation. This state is not likely compatible with life. However, partial loss of cohesin function is associated with human disease. This partial loss of cohesin function may affect transcription and translation without having a catastrophic effect on chromosome segregation. While cohesin may regulate the expression of some genes directly, I propose that there may be substantial indirect regulation of genes due to differential expression of non-coding RNAs. These” smoking guns” may include the rRNA, tRNA, and snoRNA genes. I speculate that changes in translation may contribute to the cohesinopathies. Coupling chromosome associated processes to translation may provide the cell with a useful feedback loop to regulate proliferation.
If translational mechanisms contribute to the cohesinopathies, then the cohesinopathies may be viewed in some aspects as ribosomopathies.Citation7 In this case, one can view potential treatments from a different perspective, capitalizing on pharmaceutical methods that have been developed to modulate translation. For example, L-leucine has been shown to stimulate mammalian target of rapamycin (mTOR), a key node in translational pathways.Citation35,Citation36 L-leucine has a substantial rescue effect on development and anemia in animal models for the ribosomopathies 5q and Diamond Blackfan anemia.Citation37,Citation38 The more we understand about the molecular etiology of the cohesinopathies, the better positioned we will be to find effective therapies.
Acknowledgments
I would like to thank Robb Krumlauf and Baoshan Xu for critical reading.
References
- Gillis LA, McCallum J, Kaur M, DeScipio C, Yaeger D, Mariani A, et al. NIPBL mutational analysis in 120 individuals with Cornelia de Lange syndrome and evaluation of genotype-phenotype correlations. Am J Hum Genet 2004; 75:610 - 23; http://dx.doi.org/10.1086/424698; PMID: 15318302
- Krantz ID, McCallum J, DeScipio C, Kaur M, Gillis LA, Yaeger D, et al. Cornelia de Lange syndrome is caused by mutations in NIPBL, the human homolog of Drosophila melanogaster Nipped-B. Nat Genet 2004; 36:631 - 5; http://dx.doi.org/10.1038/ng1364; PMID: 15146186
- Tonkin ET, Wang TJ, Lisgo S, Bamshad MJ, Strachan T. NIPBL, encoding a homolog of fungal Scc2-type sister chromatid cohesion proteins and fly Nipped-B, is mutated in Cornelia de Lange syndrome. Nat Genet 2004; 36:636 - 41; http://dx.doi.org/10.1038/ng1363; PMID: 15146185
- Liu J, Krantz ID. Cohesin and human disease. Annu Rev Genomics Hum Genet 2008; 9:303 - 20; http://dx.doi.org/10.1146/annurev.genom.9.081307.164211; PMID: 18767966
- Rhodes JM, Bentley FK, Print CG, Dorsett D, Misulovin Z, Dickinson EJ, et al. Positive regulation of c-Myc by cohesin is direct, and evolutionarily conserved. Dev Biol 2010; 344:637 - 49; http://dx.doi.org/10.1016/j.ydbio.2010.05.493; PMID: 20553708
- Marygold SJ, Roote J, Reuter G, Lambertsson A, Ashburner M, Millburn GH, et al. The ribosomal protein genes and Minute loci of Drosophila melanogaster. Genome Biol 2007; 8:R216; http://dx.doi.org/10.1186/gb-2007-8-10-r216; PMID: 17927810
- Narla A, Ebert BL. Ribosomopathies: human disorders of ribosome dysfunction. Blood 2010; 115:3196 - 205; http://dx.doi.org/10.1182/blood-2009-10-178129; PMID: 20194897
- Yoon A, Peng G, Brandenburger Y, Zollo O, Xu W, Rego E, et al. Impaired control of IRES-mediated translation in X-linked dyskeratosis congenita. Science 2006; 312:902 - 6; http://dx.doi.org/10.1126/science.1123835; PMID: 16690864
- Jack K, Bellodi C, Landry DM, Niederer RO, Meskauskas A, Musalgaonkar S, et al. rRNA pseudouridylation defects affect ribosomal ligand binding and translational fidelity from yeast to human cells. Mol Cell 2011; 44:660 - 6; http://dx.doi.org/10.1016/j.molcel.2011.09.017; PMID: 22099312
- Stumpf CR, Ruggero D. The cancerous translation apparatus. Curr Opin Genet Dev 2011; 21:474 - 83; http://dx.doi.org/10.1016/j.gde.2011.03.007; PMID: 21543223
- Amsterdam A, Sadler KC, Lai K, Farrington S, Bronson RT, Lees JA, et al. Many ribosomal protein genes are cancer genes in zebrafish. PLoS Biol 2004; 2:E139; http://dx.doi.org/10.1371/journal.pbio.0020139; PMID: 15138505
- Kondrashov N, Pusic A, Stumpf CR, Shimizu K, Hsieh AC, Xue S, et al. Ribosome-mediated specificity in Hox mRNA translation and vertebrate tissue patterning. Cell 2011; 145:383 - 97; http://dx.doi.org/10.1016/j.cell.2011.03.028; PMID: 21529712
- Landry DM, Hertz MI, Thompson SR. RPS25 is essential for translation initiation by the Dicistroviridae and hepatitis C viral IRESs. Genes Dev 2009; 23:2753 - 64; http://dx.doi.org/10.1101/gad.1832209; PMID: 19952110
- Xue S, Barna M. Specialized ribosomes: a new frontier in gene regulation and organismal biology. Nat Rev Mol Cell Biol 2012; 13:355 - 69; http://dx.doi.org/10.1038/nrm3359; PMID: 22617470
- Gordillo M, Vega H, Trainer AH, Hou F, Sakai N, Luque R, et al. The molecular mechanism underlying Roberts syndrome involves loss of ESCO2 acetyltransferase activity. Hum Mol Genet 2008; 17:2172 - 80; http://dx.doi.org/10.1093/hmg/ddn116; PMID: 18411254
- Mönnich M, Kuriger Z, Print CG, Horsfield JA. A zebrafish model of Roberts syndrome reveals that Esco2 depletion interferes with development by disrupting the cell cycle. PLoS One 2011; 6:e20051; http://dx.doi.org/10.1371/journal.pone.0020051; PMID: 21637801
- van der Lelij P, Godthelp BC, van Zon W, van Gosliga D, Oostra AB, Steltenpool J, et al. The cellular phenotype of Roberts syndrome fibroblasts as revealed by ectopic expression of ESCO2. PLoS One 2009; 4:e6936; http://dx.doi.org/10.1371/journal.pone.0006936; PMID: 19738907
- Vega H, Waisfisz Q, Gordillo M, Sakai N, Yanagihara I, Yamada M, et al. Roberts syndrome is caused by mutations in ESCO2, a human homolog of yeast ECO1 that is essential for the establishment of sister chromatid cohesion. Nat Genet 2005; 37:468 - 70; http://dx.doi.org/10.1038/ng1548; PMID: 15821733
- Michaelis C, Ciosk R, Nasmyth K. Cohesins: chromosomal proteins that prevent premature separation of sister chromatids. Cell 1997; 91:35 - 45; http://dx.doi.org/10.1016/S0092-8674(01)80007-6; PMID: 9335333
- Bose T, Lee KK, Lu S, Xu B, Harris B, Slaughter B, et al. Cohesin proteins promote ribosomal RNA production and protein translation in yeast and human cells. PLoS Genet 2012; 8:e1002749; http://dx.doi.org/10.1371/journal.pgen.1002749; PMID: 22719263
- Calvo SE, Pagliarini DJ, Mootha VK. Upstream open reading frames cause widespread reduction of protein expression and are polymorphic among humans. Proc Natl Acad Sci U S A 2009; 106:7507 - 12; http://dx.doi.org/10.1073/pnas.0810916106; PMID: 19372376
- Gard S, Light W, Xiong B, Bose T, McNairn AJ, Harris B, et al. Cohesinopathy mutations disrupt the subnuclear organization of chromatin. J Cell Biol 2009; 187:455 - 62; http://dx.doi.org/10.1083/jcb.200906075; PMID: 19948494
- D’Ambrosio C, Schmidt CK, Katou Y, Kelly G, Itoh T, Shirahige K, et al. Identification of cis-acting sites for condensin loading onto budding yeast chromosomes. Genes Dev 2008; 22:2215 - 27; http://dx.doi.org/10.1101/gad.1675708; PMID: 18708580
- Deardorff MA, Bando M, Nakato R, Watrin E, Itoh T, Minamino M, et al. HDAC8 mutations in Cornelia de Lange syndrome affect the cohesin acetylation cycle. Nature 2012; 489:313 - 7; http://dx.doi.org/10.1038/nature11316; PMID: 22885700
- Deardorff MA, Wilde JJ, Albrecht M, Dickinson E, Tennstedt S, Braunholz D, et al. RAD21 mutations cause a human cohesinopathy. Am J Hum Genet 2012; 90:1014 - 27; http://dx.doi.org/10.1016/j.ajhg.2012.04.019; PMID: 22633399
- Liu J, Zhang Z, Bando M, Itoh T, Deardorff MA, Clark D, et al. Transcriptional dysregulation in NIPBL and cohesin mutant human cells. PLoS Biol 2009; 7:e1000119; http://dx.doi.org/10.1371/journal.pbio.1000119; PMID: 19468298
- Kawauchi S, Calof AL, Santos R, Lopez-Burks ME, Young CM, Hoang MP, et al. Multiple organ system defects and transcriptional dysregulation in the Nipbl(+/-) mouse, a model of Cornelia de Lange Syndrome. PLoS Genet 2009; 5:e1000650; http://dx.doi.org/10.1371/journal.pgen.1000650; PMID: 19763162
- Mokrejs M, Masek T, Vopálensky V, Hlubucek P, Delbos P, Pospísek M. IRESite--a tool for the examination of viral and cellular internal ribosome entry sites. Nucleic Acids Res 2010; 38:Database issue D131 - 6; http://dx.doi.org/10.1093/nar/gkp981; PMID: 19917642
- Monahan K, Rudnick ND, Kehayova PD, Pauli F, Newberry KM, Myers RM, et al. Role of CCCTC binding factor (CTCF) and cohesin in the generation of single-cell diversity of protocadherin-α gene expression. Proc Natl Acad Sci U S A 2012; 109:9125 - 30; http://dx.doi.org/10.1073/pnas.1205074109; PMID: 22550178
- Muto A, Calof AL, Lander AD, Schilling TF. Multifactorial origins of heart and gut defects in nipbl-deficient zebrafish, a model of Cornelia de Lange Syndrome. PLoS Biol 2011; 9:e1001181; http://dx.doi.org/10.1371/journal.pbio.1001181; PMID: 22039349
- Ghiselli G, Coffee N, Munnery CE, Koratkar R, Siracusa LD. The cohesin SMC3 is a target the for beta-catenin/TCF4 transactivation pathway. J Biol Chem 2003; 278:20259 - 67; http://dx.doi.org/10.1074/jbc.M209511200; PMID: 12651860
- Boulon S, Westman BJ, Hutten S, Boisvert FM, Lamond AI. The nucleolus under stress. Mol Cell 2010; 40:216 - 27; http://dx.doi.org/10.1016/j.molcel.2010.09.024; PMID: 20965417
- Carrière L, Graziani S, Alibert O, Ghavi-Helm Y, Boussouar F, Humbertclaude H, et al. Genomic binding of Pol III transcription machinery and relationship with TFIIS transcription factor distribution in mouse embryonic stem cells. Nucleic Acids Res 2012; 40:270 - 83; http://dx.doi.org/10.1093/nar/gkr737; PMID: 21911356
- Dauwerse JG, Dixon J, Seland S, Ruivenkamp CA, van Haeringen A, Hoefsloot LH, et al. Mutations in genes encoding subunits of RNA polymerases I and III cause Treacher Collins syndrome. Nat Genet 2011; 43:20 - 2; http://dx.doi.org/10.1038/ng.724; PMID: 21131976
- Bonfils G, Jaquenoud M, Bontron S, Ostrowicz C, Ungermann C, De Virgilio C. Leucyl-tRNA synthetase controls TORC1 via the EGO complex. Mol Cell 2012; 46:105 - 10; http://dx.doi.org/10.1016/j.molcel.2012.02.009; PMID: 22424774
- Han JM, Jeong SJ, Park MC, Kim G, Kwon NH, Kim HK, et al. Leucyl-tRNA synthetase is an intracellular leucine sensor for the mTORC1-signaling pathway. Cell 2012; 149:410 - 24; http://dx.doi.org/10.1016/j.cell.2012.02.044; PMID: 22424946
- Jaako P, Debnath S, Olsson K, Bryder D, Flygare J, Karlsson S. Dietary L-leucine improves the anemia in a mouse model for Diamond-Blackfan anemia. Blood 2012; 120:2225 - 8; http://dx.doi.org/10.1182/blood-2012-05-431437; PMID: 22791294
- Payne EM, Virgilio M, Narla A, Sun H, Levine M, Paw BH, et al. L-leucine improves the anemia and developmental defects associated with Diamond-Blackfan anemia and del(5q) MDS by activating the mTOR pathway. Blood 2012; 120:2214 - 24; http://dx.doi.org/10.1182/blood-2011-10-382986; PMID: 22734070
- Xiong B, Gerton JL. Regulators of the Cohesion Network. Annu Rev Biochem 2012; 79:131 - 53; http://dx.doi.org/10.1146/annurev-biochem-061708-092640; PMID: 20331362