Abstract
Over 300 mutations in the LMNA gene, encoding A-type lamins, are associated with 15 human degenerative disorders and premature aging syndromes. Although genomic instability seems to contribute to the pathophysiology of some laminopathies, there is limited information about what mutations cause genomic instability and by which molecular mechanisms. Mouse embryonic fibroblasts depleted of A-type lamins or expressing mutants lacking exons 8–11 (LmnaΔ8–11/Δ8–11) exhibit alterations in telomere biology and DNA repair caused by cathepsin L-mediated degradation of 53BP1 and reduced expression of BRCA1 and RAD51. Thus, a region encompassing exons 8–11 seems essential for genome integrity. Given that deletion of lamin A exon 9 in the mouse (LmnaΔ9/Δ9) results in a progeria phenotype, we tested if this domain is important for genome integrity. LmnaΔ9/Δ9 MEFs exhibit telomere shortening and heterochromatin alterations but do not activate cathepsin L-mediated degradation of 53BP1 and maintain expression of BRCA1 and RAD51. Accordingly, LmnaΔ9/Δ9 MEFs do not present genomic instability, and expression of mutant lamin A Δexon9 in lamin-depleted cells restores DNA repair factors levels and partially rescues nuclear abnormalities. These data reveal that the domain encoded by exon 9 is important to maintain telomere homeostasis and heterochromatin structure but does not play a role in DNA repair, thus pointing to other exons in the lamin A tail as responsible for the genomic instability phenotype in LmnaΔ8–11/Δ8–11 mice. Our study also suggests that the levels of DNA repair factors 53BP1, BRCA1 and RAD51 could potentially serve as biomarkers to identify laminopathies that present with genomic instability.
Introduction
A-type lamins (lamins A and C) are type-V intermediate filaments that form the nuclear lamina, a proteinaceous meshwork underlying the inner nuclear membrane, which also extends throughout the nucleoplasm. They are major contributors to nuclear shape and architecture.Citation1-Citation3 Lamins A and C arise as different mature proteins through alternative splicing of the LMNA gene and post-translational modifications. Lamin A is synthesized as a prelamin A precursor that undergoes farnesylation and carboxymethylation at the CAAX motif at the carboxyl terminus, followed by proteolytic cleavage of 15 amino acids, including the modified terminal cysteine. A-type lamins have been implicated in many essential nuclear processes, including DNA replication and repair, gene transcription and silencing, positioning of nuclear pore complexes, chromatin remodeling and nuclear envelope breakdown and reassembly during mitosis.Citation4-Citation6
Laminopathies are diseases caused by mutations in genes encoding proteins of the nuclear lamina.Citation6,Citation7 In particular, over 300 mutations have been identified in the LMNA gene that are associated with a variety of human diseases,Citation6 ranging from muscular and adipose tissue dystrophies to premature aging syndromes such as Hutchinson-Gilford progeria syndrome (HGPS).Citation8,Citation9 Although it is clear that A-type lamins are critical for nuclear function, the molecular mechanisms affected by the different mutations in the A-type lamins and how they contribute to the broad variety of diseases remain ill defined.
Mouse models of laminopathies have been instrumental to understand the molecular basis of these diseases.Citation6,Citation10,Citation11 The LmnaΔ8–11/Δ8–11 mouse model is one of the best-characterized.Citation12 Originally generated as a knockout of A-type lamins, a recent study showed that it is not a complete knockoutCitation13 and that low levels of a truncated form of lamin A are produced lacking the domain encoded for by exons 8–11 of the LMNA gene. These mice develop abnormalities that resemble human Emery-Dreifuss muscular dystrophy and die at 5‒6 weeks of age with severe growth retardation and cardiomyopathy. In addition, several mouse models of progeria have been generated to study HGPS. Most HGPS cases are caused by a splicing defect in exon 11 of the LMNA gene. A substitution of C to T at nucleotide 1824 (G608G) introduces a cryptic donor splice site that results in the deletion of 50 amino acids near the carboxyl-terminus of prelamin A. This altered form of lamin A, known as progerin, is toxic to the cells. Mice homozygous for this mutation (G609G in mice) exhibit growth retardation and premature death at around 100 days.Citation14 Another premature aging mouse, Zmpste24 knockout,Citation15 lacks the metalloprotease that cleaves the farnesylated tail of prelamin A leading to accumulation of farnesylated prelamin A and absence of mature lamin A. These mice die at five months and exhibit growth retardation, skeletal defects, cardiomyopathy, muscular dystrophy, lipodystrophy, and a progeroid phenotype. A different mouse model was generated by introducing a point mutation in exon 9 of the LMNA gene that caused mRNA splicing defects and deletion of exon 9 (herein LmnaΔ9/Δ9), which is accompanied by low levels of mutant lamin A/C transcripts and proteins.Citation16 The LmnaΔ9/Δ9 mouse shares many tissues and molecular pathologies characteristic of progeria, with severe growth retardation and death within 3‒4 weeks, but with no evidence of muscular dystrophy.Citation17 Thus, these mice present a phenotype that is significantly different from LmnaΔ8–11/Δ8–11 mice.
At the cellular level, common defects in the laminopathies include ultrastructural defects of the nuclear envelope, loss of heterochromatin from the nuclear periphery, alterations in epigenetic marks characteristic of constitutive heterochromatin and increased genomic instability.Citation18-Citation21 Our previous studies demonstrated that LmnaΔ8–11/Δ8–11 MEFs present alterations in telomere structure, length and function, as well as on the stability of key factors in DNA repair.Citation22-Citation25 In particular, these mutant MEFs activate cathepsin L-mediated degradation of 53BP1 and downregulate transcription of BRCA1 and RAD51 genes. As a consequence, LmnaΔ8–11/Δ8–11 MEFs exhibit defects in the two main mechanisms of DNA double-strand break (DSB) repair: non-homologous end-joining (NHEJ) and homologous recombination (HR).
Here, we used MEFs from LmnaΔ9/Δ9 mice to determine if the domain of A-type lamins encoded for by exon 9 is important for the maintenance of genome stability. We find that LmnaΔ9/Δ9 MEFs present with telomere and chromatin defects but not genomic instability. Interestingly, expression of mutant lamin A Δexon9 protein is able to maintain normal levels of the DNA repair factors 53BP1, BRCA1 and RAD51, as well as rescue DNA repair defects in A-type lamins-depleted cells. Our study suggests that the domain encoded for by exon 9 is not critical for DNA repair, and that the levels of DNA repair factors such as 53BP1, BRCA1 and RAD51 have the potential to serve as biomarkers to identify laminopathy patients with genomic instability.
Results
Telomere shortening in LmnaΔ9/Δ9 mice
We previously showed that LmnaΔ8–11/Δ8–11 MEFs exhibit telomere shortening and complete telomere losses.Citation22,Citation24 Here, we determined if LmnaΔ9/Δ9 MEFs are compromised in their ability to maintain telomere length homeostasis. Since previous studies showed that lamin A/C transcripts carrying the Δexon9 mutation are not stable,Citation16 we first confirmed the global decrease of mutant lamin A/C proteins in MEFs isolated from these mice (Fig. S1).
Next, we measured telomere length by telomere restriction fragment (TRF) analysis and quantitative fluorescence in situ hybridization (Q-FISH) analysis (). LmnaΔ9/Δ9 MEFs show a clear faster migration of telomeres by TRF compared with wild-type, indicative of shorter telomere tracks. These results were confirmed by Q-FISH assays, which show an average telomere length approximately 6 Kb shorter in LmnaΔ9/Δ9 MEFs than in wild-type MEFs (). In addition, a 50‒75% decrease in the percentage of long telomeres (>65 Kb) was observed in LmnaΔ9/Δ9 MEFs. Statistical analysis shows that these differences in telomere length between genotypes are significant (*P = 0.0181). These data demonstrate that low expression of the lamin A ∆exon9 form leads to defects in the maintenance of telomere length homeostasis, recapitulating the defects observed in LmnaΔ8–11/Δ8–11 MEFs and in MEFs depleted of A-type lamins via lentiviral transduction with specific shRNAs.Citation22,Citation24 Thus, alterations in A-type lamins, via depletion or expression of mutant lamins, result in faster telomere attrition, which if recapitulated in human cells, could limit cell proliferation over time.
Figure 1.Telomere shortening and heterochromatin defects in LmnaΔ9/Δ9 MEFs. (A) Telomere restriction fragment (TRF) analysis shows faster migration of telomeres and thus telomere shortening in LmnaΔ9/Δ9 MEFs. (B) Three independent Q-FISH analyses of telomere length in wild-type and LmnaΔ9/Δ9 MEFs. Telomere length of at least 20 metaphases was measured per experiment. Note the decreased average telomere length in LmnaΔ9/Δ9 fibroblasts. (C) ChIP analysis performed on wild-type and LmnaΔ9/Δ9 MEFs with antibodies recognizing H3K9me3 and H4K20me3. Immunoprecipitated DNA was dot blotted and hybridized to a telomere probe, stripped and rehybridized to a centromere probe (major satellite). (D) Quantitation of immunoprecipitated telomeric DNA after normalization to input signals with H3K9me3 (3 experiments) or H4K20me3 (4 experiments). Bars represent standard deviation. (E) Western blots to monitor global levels of H3K9me3 and H4K20me3 in wild-type, LmnaΔ8–11/Δ8–11 and LmnaΔ9/Δ9 MEFs. β-Tubulin was used as loading control. (F) Immunofluorescence performed in wild-type and LmnaΔ9/Δ9 MEFs with antibodies recognizing H3K9me3 and H4K20me3 (green). DAPI is shown in blue. Note the diffuse and irregular distribution of heterochromatin domains in the mutant cells. *P, represents value of statistical significance; P ≤ 0.05 is considered significant.
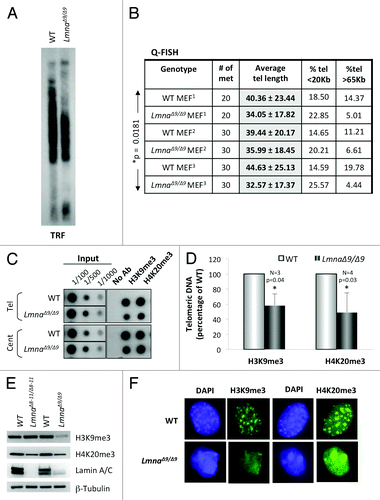
Telomeric and pericentric heterochromatin structure is altered in LmnaΔ9/Δ9 fibroblasts
Telomeric and pericentric domains have epigenetic modifications characteristic of heterochromatin, including trimethylation of histone H3 at lysine 9 (H3K9me3) and of histone H4 at lysine 20 (H4K20me3).Citation26,Citation27 We previously found that expression of Δexon8–11 mutant lamins affects the epigenetic status of telomeric and pericentric chromatin. In particular, decreased levels of H4K20me3 at both domains were observed in LmnaΔ8–11/Δ8–11 fibroblasts.Citation24 To determine if MEFs from the LmnaΔ9/Δ9 mouse model exhibit alterations in the structure of telomeric heterochromatin we performed chromatin immunoprecipitation (ChIP) assays with antibodies recognizing H3K9me3 and H4K20me3 marks (). We found a 40% decrease in the levels of both heterochromatic marks at telomeres in LmnaΔ9/Δ9 cells (). Collectively, these data support a role for A-type lamins in the maintenance of telomeric chromatin structure, with specific mutations in A-type lamins having differential impacts on the epigenetic status of telomeres.
Alterations of telomeric chromatin structure are often phenocopied by pericentric heterochromatin. Such is the case of LmnaΔ8–11/Δ8–11 MEFs, which show reduced levels of H4K20me3 at pericentric domains. To determine the levels of H3K9me3 and H4K20me3 at pericentric chromatin in LmnaΔ9/Δ9 fibroblasts, the membranes hybridized with a telomeric probe were stripped and rehybridized with a probe recognizing major satellite sequences. Interestingly, while the levels of H3K9me3 at pericentric chromatin were also reduced in the mutant cells (albeit to a lower extent than at telomeric chromatin), the levels of H4K20me3 were maintained in the mutant cells at these domains (). Our results indicate specificity with respect to epigenetic changes at telomeric vs. pericentric chromatin in LmnaΔ9/Δ9 MEFs. Furthermore, comparing global levels of heterochromatic marks between LmnaΔ8–11/Δ8–11 and LmnaΔ9/Δ9 MEFs by western revealed a global decrease of H4K20me3 in LmnaΔ8–11/Δ8–11 MEFs and a global decrease in both H3K9me3 and H4K20me3 in LmnaΔ9/Δ9 MEFs ().
Mutations in the LMNA gene and reduced lamin expression are associated with loss of heterochromatin from the nuclear periphery.Citation28 In mouse cells, heterochromatic domains, especially pericentric regions, are easily visualized by DAPI staining during interphase. In normal fibroblasts, pericentric domains appear as DAPI-positive clusters called chromocenters that are enriched in H3K9me3 and H4K20me3 marks. We performed immunofluorescence studies to test if expression of ∆exon9 lamin A mutant protein affects the structure and distribution of heterochromatin domains within the nucleus. We found that in mutant cells, DAPI staining is distributed throughout the nucleoplasm, exhibiting only a few areas of compacted pericentric chromatin (). Labeling with antibodies recognizing H3K9me3 and H4K20me3 mirrored the DAPI distribution, such that the heterochromatic marks in mutant cells were either distributed more diffusely throughout the nucleus or forming a few large aggregates. Overall, these studies indicate that low expression of the lamin A ∆exon9 mutant protein leads to a decrease in heterochromatic marks at telomeres and an overall disorganization of pericentric chromatin domains. These results support a role for the domain encoded by exon 9 of the LMNA gene in the nuclear compartmentalization of heterochromatin.
Fibroblasts from LmnaΔ9/Δ9 mice do not exhibit profound genomic instability
Alterations of telomere biology and defects in DNA repair can lead to chromosome end-to-end fusions and other chromosomal aberrations that have serious detrimental effects for the integrity of the genome. LmnaΔ8–11/Δ8–11 MEFs feature defective DNA repair pathways and increased basal DNA damage levels, as well as increased chromosomal aberrations.Citation19,Citation24 To determine whether the domain encoded by exon 9 is important for the maintenance of genomic stability, we tested if LmnaΔ9/Δ9 MEFs phenocopy these defects. FISH analysis performed with a telomere probe shows that mutant MEFs have only a modest increase in single telomere loss (STL) (). In addition, we did not find evidence of increased frequency of chromosome end-to-end fusions or chromosome and chromatid breaks in LmnaΔ9/Δ9 MEFs. Only an increase in the percentage of metaphases with complex aberrations (recombination figures, double minute chromosomes and aneuploidy) was noted, although at a low frequency (). To confirm that the LmnaΔ9/Δ9 fibroblasts were able to maintain chromosome stability, we monitored the presence of γH2AX foci. We did not find any evidence of basal levels of unrepaired DNA damage in these cells (). In addition, neutral comet assays performed to compare DNA repair kinetics between genotypes did not show any defects in the ability of LmnaΔ9/Δ9 fibroblasts to repair DNA DSBs resulting from ionizing radiation (). In summary, we did not find a profound increase in genomic instability in the LmnaΔ9/Δ9 mutant cells when compared with wild-type, thus revealing profound differences in the ability to maintain genome integrity between the LmnaΔ9/Δ9 and LmnaΔ8–11/Δ8–11 models.
Figure 2.Degree of genomic instability in LmnaΔ9/Δ9 MEFs. (A) Genomic instability was monitored in wild-type and LmnaΔ9/Δ9 MEFs by counting percentage of metaphase spreads showing single telomere losses (STL), chromosome end-to-end fusions, chromosome and chromatid breaks, and other complex aberrations. Images show examples of chromosomal aberrations. Note that LmnaΔ9/Δ9 fibroblasts do not show a marked increase in genomic instability. (B) Quantitation of percentage of cells presenting with basal levels of DNA damage, as assessed by the presence of >5 γH2AX foci by immunofluorescence. n, total number of cells analyzed. (C) Neutral comet assays were performed in wild-type and LmnaΔ9/Δ9 MEFs at different times (0‒150 min) post-irradiation with 8 Gy, to compare kinetics of DNA DSB repair. Average “olive moment,” a measure or unrepaired DNA damage, was calculated in 3 independent experiments with 25‒30 measurements per condition. Bars represent standard deviation. (D) Quantitation of CTSL, 53BP1, BRCA1, and RAD51 transcripts levels by qRT-PCR in wild-type and LmnaΔ9/Δ9 MEFs. (E) Western blots to monitor levels of 53BP1, BRCA1, RAD51, and CTSL proteins in MEFs from LmnaΔ8–11/Δ8–11, LmnaΔ9/Δ9 and their corresponding wild-type littermates. β-Tubulin was used as loading control.
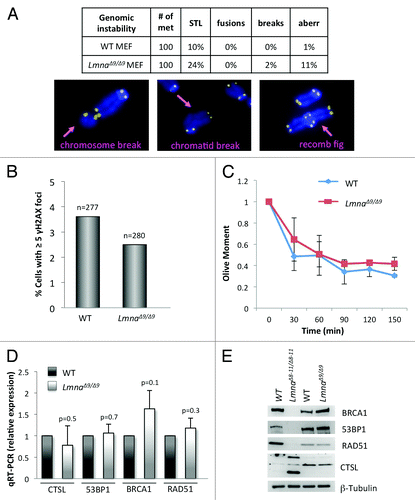
To gain insight into putative molecular mechanisms responsible for these differences, we compared the levels of key factors in the DNA damage response (DDR) regulated by A-type lamins. We previously showed that LmnaΔ8–11/Δ8–11 MEFs exhibit significant genomic instability, in part due to activation of CTSL-mediated degradation of 53BP1 protein, and reduced expression of BRCA1 and RAD51 genes. We tested whether LmnaΔ9/Δ9 fibroblasts are deficient in these DNA repair factors. As shown in , LmnaΔ9/Δ9 fibroblasts maintain nearly normal levels of CTSL, 53BP1, BRCA1, and RAD51 transcripts. In addition, comparing the levels of DNA repair factors between wild-type, LmnaΔ8–11/Δ8–11 and LmnaΔ9/Δ9 fibroblasts confirmed the upregulation of CTSL and decreased levels of 53BP1, BRCA1, and RAD51 in LmnaΔ8–11/Δ8–11, with no profound differences in the levels of any of these proteins between wild-type and LmnaΔ9/Δ9 fibroblasts (). Our results indicate that low expression of the mutant lamin A ∆exon9 protein does not lead to destabilization of 53BP1, loss of BRCA1 and RAD51 or profound genomic instability. This mutation leads, however, to telomere length changes and defects in heterochromatin structure.
Expression of lamin A ∆exon9 protein rescues 53BP1, BRCA1 and RAD51 levels in lamin-depleted cells
Our data suggest that a mutant lamin A protein lacking the domain encoded by exon 9 is able to maintain the levels of DNA repair factors 53BP1, BRCA1, and RAD51. To test this model directly, we generated lamin-deficient cells via lentiviral transduction of wild-type MEFs with a specific shRNA (shLmna) and shRNA control (shLuc). As shown in , depletion of lamins A/C results in increased expression of CTSL mRNA and protein, with the consequent destabilization of 53BP1 protein, as well as a marked reduction in BRCA1 and RAD51 mRNA and protein, as previously reported.Citation25 Next, we performed retroviral transduction of lamin-depleted cells with an empty vector control (EV), wild-type lamin A (LA) or the mutant lamin A ∆exon9 (∆Ex9LA) construct. Interestingly, expression of both proteins reduced the global levels of CTSL and rescued the levels of 53BP1, BRCA1, and RAD51 proteins (). Monitoring the levels of transcripts of these DNA repair factors revealed that ∆Ex9LA protein expression counteracts the transcriptional activation of CTSL and the repression of BRCA1 and RAD51 observed in lamin-depleted cells (). Furthermore, previous studies had shown that loss of A-type lamins leads to degradation of Rb family members pRb and p107 with no effect on p130.Citation25,Citation29,Citation30 Interestingly, reconstitution of a variety of disease-associated lamin mutants into lamin-deficient cells rescued Rb family members and the ability of these cells to respond to p16ink4a-induced growth arrest.Citation30 Here, we show that reconstitution of ∆Ex9LA protein expression also rescues the levels of pRb and p107 proteins in lamins-depleted cells. These results demonstrate that the domain encoded by exon 9 in the LMNA gene is not necessary to maintain transcription and stabilization of DNA repair factors and cell cycle proteins regulated by A-type lamins.
Figure 3. Effect of expression of ∆Ex9LA protein on DNA repair factors levels. (A) Wild-type MEFs were lentivirally transduced with shRNA control (shLuc) or shRNA specific for depletion of A-type lamins (shLmna). Subsequently, both types of cells were retrovirally transduced with an empty vector control (EV), and lamins-depleted cells were transduced with wild-type lamin A (LA) or with ∆Ex9LA mutant protein. Western blots were performed to monitor levels of proteins regulated by lamins (53BP1, BRCA1, RAD51, CTSL, and Rb family members). β-Tubulin was used as loading control. Note the marked depletion of lamins A/C by shLmna, and the reconstitution of wild-type and mutant lamin A (showing lower Mr). (B) Quantitation of CTSL, 53BP1, BRCA1 and RAD51 transcripts levels by qRT-PCR in lamins-proficient and -deficient cells in which wild-type LA or ∆Ex9LA mutant proteins were reconstituted. Graph shows average of 3 independent experiments (each conducted in triplicate), and bars represent standard deviation. P, represents value of statistical significance. When P ≤ 0.05, differences are considered significant and labeled with asterisk (*). (C) Western blots were performed in lamins-proficient (shLuc+EV), lamins-deficient cells (shaman+EV) and lamins-deficient cells expressing the G608G mutant form of lamin A. The levels of DNA repair factors and Rb family members regulated by lamins were detected.
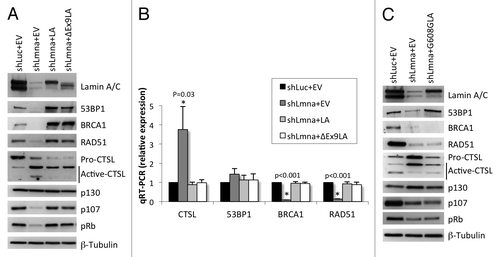
To determine if lamin mutants associated with genomic instability are able to rescue the levels of DNA repair factors, we reconstituted the mutant lamin A protein that is expressed in HGPS patients (G608G) (). Intriguingly, we found that after passaging the cells for approximately two weeks, G608GLA mutant protein was able to rescue to certain extent the levels of CTSL and 53BP1. In contrast, cells expressing G608GLA exhibit as low levels of BRCA1, RAD51, pRb and p107 proteins as lamin-deficient cells. These results support the notion that specific mutations in A-type lamins affect the expression and stability of different proteins, thus affecting differentially DNA repair mechanisms, genomic stability and cell cycle regulation.
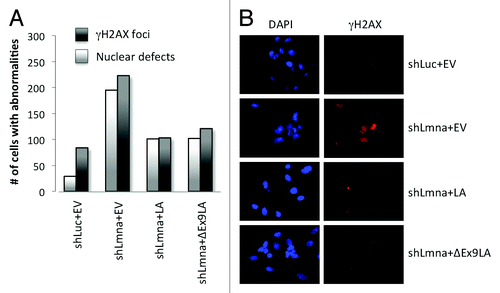
Altogether, our data indicate that cells expressing ∆Ex9LA mutant protein are able to maintain the levels of key factors in the maintenance of genomic stability. Supporting this conclusion, expression of ∆Ex9LA mutant protein reduced the basal levels of unrepaired DNA damage in lamin-depleted cells, as monitored by γH2AX foci (). In addition, ∆Ex9LA mutant protein reduced the extent of nuclear morphological abnormalities, as monitored by DAPI (), to a similar extent as wild-type lamin A protein. In summary, we show that the domain encoded for by exon 9 in the LMNA gene affects telomere homeostasis and heterochromatin structure, without having a major role in the maintenance of DNA repair and genome stability.
Figure 4. Expression of ∆Ex9LA partially rescues phenotypes of lamins-deficient cells. (A) Quantitation of cells presenting with >5 γH2AX foci, indicating basal levels of unrepaired DNA damage, as well as cells exhibiting nuclear morphological abnormalities by DAPI staining. Cells analyzed include MEFs transduced with control shRNA and empty vector (shLuc+EV); MEFs transduced with shRNA for lamins depletion and empty vector (shLmna+EV); as well as lamins-deficient MEFs in which LA or ∆Ex9LA proteins were reconstituted (shLmna+LA and shLmna+∆Ex9LA). Three hundred cells were analyzed per condition. Note how ∆Ex9LA expression partially rescues both phenotypes. (B) Images from the immunofluorescence study with γH2AX antibody and DAPI staining in (A).
Discussion
The broad range of diseases associated with either mutations in the LMNA gene or changes in the expression of A-type lamins has drawn much attention toward elucidating the functions of these structural nuclear proteins. Several lines of evidence indicate that lamin-related diseases are associated with increased genomic instability.Citation19,Citation24,Citation31 The notion that defects in the maintenance of telomeres and in DNA repair contribute to the pathophysiology of these diseases has been gaining momentum. However, the data supporting this notion remains scarce. Our previous studies showed that LmnaΔ8–11/Δ8–11 fibroblasts exhibit defects in telomere structure, length and function, increased chromosome and chromatid breaks, a higher degree of unrepaired DNA damage and an overall increase in genomic instability. The activation of CTSL-mediated degradation of 53BP1 and the transcriptional downregulation of BRCA1 and RAD51 genes are mechanisms underlying the genomic instability in these cells. These results support the notion that increased genomic instability contributes to the phenotype of some lamin-related diseases. The present study on LmnaΔ9/Δ9 mice shows, however, that increased genomic instability is not a phenotype shared by all lamin-related diseases. In contrast, alterations of telomere biology and heterochromatin defects are common to many models of laminopathies, although if and how these alterations contribute to the pathophysiology of these diseases remains unknown. Furthermore, our data begins to provide information about domains in A-type lamins that are important for maintenance of DNA repair factors, DNA repair mechanisms and overall genome integrity. In particular, we conclude that the globular tail domain encoded for by exons 8‒11 contains features that are critical for the maintenance of DNA repair. Within this C-terminal domain, the region of the protein encoded for by exon 9 does not seem to contribute to the genomic instability phenotype. This region, however, plays a key role in the regulation of transcription factors that modulate the deposition of a functional extracellular matrix.Citation17 Given that a number of mutations in lamin A exon 9 have been linked to diseases such as HGPS (R527C) and muscular dystrophy (L530P), it is tempting to speculate that these mutations could lead to defects in different signaling pathways without affecting genomic stability. Future studies with different disease-associated point mutations or more deletions in this domain will be necessary to delineate the specific region in the globular tail of A-type lamins that is responsible for the genomic instability phenotype.
Defects in chromatin structure
Fibroblasts from HGPS patients exhibit reduced levels of the heterochromatic marks HP1, H3K9me3 and H3K27me3, while featuring increased levels of H4K20me3 at pericentric chromatin domains.Citation18,Citation21 The defects in HP1 and H3K9me3 have been linked to a reduction of NURD subunits, specifically RBBP4, RBBP7 and HDAC1, which interact with lamin A.Citation32 The authors found that the loss of NURD subunits in HGPS cells was dependent upon the presence of progerin. It remains to be determined if expression of other mutant lamins impact on these chromatin-regulating factors. Interestingly, LmnaΔ8–11/Δ8–11 fibroblasts do not show decreased H3K9me3 at heterochromatic domains. Thus, this mutant form of lamins might not affect the expression of NURD factors. In contrast, we found a marked decrease in H4K20me3 levels at these domains.Citation24 The concomitant destabilization of Rb family members in LmnaΔ8–11/Δ8–11 fibroblastsCitation29 could be responsible for this defect.Citation27 In the present study we show that LmnaΔ9/Δ9 fibroblasts exhibit yet a different pattern of defects in histone marks. LmnaΔ9/Δ9 fibroblasts have decreased global levels of both H3K9me3 and H4K20me3, which are recapitulated in telomeric domains. Interestingly, pericentric chromatin exhibit decreased levels of H3K9me3, while maintaining H4K20me3 levels. Understanding the molecular mechanisms that allow LmnaΔ9/Δ9 fibroblasts to maintain H4K20me3 levels at pericentric but not telomeric chromatin will require further investigation. Of note, in addition to the defects in histone marks, we observed alterations in the structure of pericentric heterochromatin in LmnaΔ9/Δ9 cells. Altogether, these studies indicate that A-type lamins play a key role in the maintenance of heterochromatin domains, with different alterations in these proteins impacting on specific chromatin-modifying activities.
Defects in telomere biology
Alterations of telomere biology have been associated with lamin-related diseases. However, the available data shows clear differences between the different models studied. While HGPS fibroblasts undergo faster telomere attrition during proliferation, Zmpste24−/− cells maintain telomere length homeostasis.Citation15 In LmnaΔ8–11/Δ8–11, we observed both telomere shortening and telomere deletions. Interestingly, LmnaΔ9/Δ9 fibroblasts show telomere attrition but not a significant increase in STL.
Maintenance of telomere length and stability is a complex process that requires proper accessibility of telomerase to replicating telomeres and assembly of the shelterin complex after replication. The shelterin complex facilitates the formation of a T-loop structure at the end of the chromosome, which prevents NHEJ or HR events involving telomeres. We envision that the expression of lamin A ∆exon9 mutant proteins might hinder the accessibility of telomerase, while expression of lamin A ∆exon8–11 could affect both telomerase accessibility and telomere replication and/or repair, leading to terminal deletions. These differences suggest that specific mutations in the LMNA gene impact differentially on telomere maintenance mechanisms. However, we find it unlikely that the telomere defects play a major role in the phenotype of mouse models of laminopathies, given that in most cases the defects are modest. Other processes such as defects in DNA repair, cell cycle regulation and signaling pathways that condition the extracellular environment could possibly have a major contribution to the pathophysiology of these diseases.
Genomic instability in the laminopathies
Increased genomic instability has been observed in LmnaΔ8–11/Δ8–11 fibroblasts. In contrast, no evidence of increased genomic instability was found in LmnaΔ9/Δ9 fibroblasts. A striking difference between these genotypes is their ability to stabilize 53BP1 and maintain expression of BRCA1, and RAD51. We found that the levels of 53BP1, BRCA1 and RAD51 are markedly reduced in LmnaΔ8–11/Δ8–11 and lamin-depleted fibroblasts but not in LmnaΔ9/Δ9 fibroblasts. Overall, these studies provide new insights about how different mutations in lamin A impact specific molecular mechanisms responsible for the maintenance of genome integrity.
The DNA repair factors 53BP1, BRCA1 and RAD51 are recruited to sites of DNA DSBs and play a key role in the repair of breaks by NHEJ (53BP1) and HR (BRCA1 and RAD51). We previously reported that the decrease in 53BP1, BRCA1 and RAD51 levels observed in LmnaΔ8–11/Δ8–11 fibroblasts could explain the increased genomic instability observed in these cells. In support of this notion, LmnaΔ9/Δ9 cells, which maintain the levels of all these proteins, are able to maintain genome integrity.
The molecular mechanisms by which specific mutations in A-type lamins impact on the expression of CTSL, BRCA1 and RAD51 remain unknown. However, it is clear that transcription of these genes is affected by specific lamins mutations. It is plausible that the localization of these genes in the 3D nuclear space is affected by the mutant lamins, as shown for a number of genes.Citation33 Alternatively, the effect of lamins mutations in chromatin structure could alter the epigenetic status of these genes, resulting in deregulated transcription. Another possibility is a direct association of lamins with these genes to regulate transcriptional activation or repression. Additional studies need to be performed to identify the mechanisms responsible for these transcriptional changes.
Importantly, this study suggests that levels of 53BP1, BRCA1 and RAD51 could represent biomarkers to screen patients with different laminopathies and identify those patients that present with genomic instability. Future studies along this line could represent important advances for therapy.
Materials and Methods
Cell culture
Wild-type, LmnaΔ8–11/Δ8–11 and LmnaΔ9/Δ9 MEFs were generated in the laboratory of Stewart CL. Cells were maintained in DMEM supplemented with 10% FBS, antibiotics and antimycotics.
Ionizing radiation
For IRIF formation, cells were irradiated with 8 Gy and fixed and processed for immunofluorescence 1 h post-irradiation. For comet assays, cells were irradiated with 8 Gy and collected at different times post-irradiation.
Telomere length measurement
TRF
Cells were prepared in agarose plugs and TRF analysis was performed as described.Citation34
Quantitative FISH
Metaphase stage chromosomal spreads were prepared and hybridized as described.Citation35 Fluorescent images were taken using a Nikon 90i upright microscope and the intensity of telomere fluorescence was analyzed using the TFL-Telo program (gift from Lansdorp P, Vancouver, Canada). Images and fluorescence telomere values were obtained from at least 20 metaphases in all cases.
Chromatin immunoprecipitation
ChIP analyses were performed as describedCitation36 with modifications. We used 4 × 106 MEFs per condition and diluted 200 μL of lysate 1:10 in dilution buffer. Samples were pre-cleared with 25% slurry of Protein A/G PLUS-Agarose IP Reagent (sc-2003, Santa Cruz) that was blocked with E. coli genomic DNA and BSA. Chromatin was immunoprecipitated using anti-H3K9me3 and anti-H4K20me3 antibodies. Immunoprecipated DNA was slot-blotted onto a Hybond N+ membrane and hybridized to a telomeric probe (gift from de Lange T, Rockefeller University, NY) or a major satellite probe. The signal was quantitated using the ImageQuant software (Molecular Dynamics). Serial dilutions of the unbound fraction from the no antibody control were processed for inputs. We calculated the amount of telomeric DNA immunoprecipitated relative to the signal of the corresponding inputs. The ChIP values are represented as a percentage of the total input telomeric DNA, therefore correcting for the difference in the number of telomere repeats.
Immunofluorescence
Cells growing in coverslips were processed directly for IF or irradiated with 8 Gy and processed 1 h post-irradiation. Cells were fixed in 3.7% formaldehyde + 0.2% Triton-X 100 in PBS for 10 min at RT and blocked for 1 h at 37 °C in 10% serum in PBS. Incubations with antibodies were performed for 1 h at 37 °C. Washes were performed in PBS and slides were counterstained with DAPI in Vectashield (Vector). Microscopy and photo capture was performed on either a Nikon Eclipse 90i microscope using 60× or 100× oil objective lenses (NA 1.4 and 1.45, respectively) with a Photometrics Cool Snap ES2 digital camera and MetaMorph (Version 7.1.2.0) or a Leica DM5000 B microscope using 63× or 100× oil objective lenses (NA 1.4 and 1.3, respectively) with a Leica DFC350FX digital camera and the Leica Application Suite (Version 4.1.0).
Fluorescence in situ hybridization
FISH was performed on metaphase spreads. Cells were arrested in mitosis by treating with colcemid for 4 h and prepared for FISH by hypotonic swelling in 0.56% KCl, followed by fixation in 3:1 methanol:acetic acid. Cell suspensions were dropped onto slides and FISH was performed using a Cy3-telomeric PNA probe, and DNA counterstained using DAPI. Images were taken using a Nikon 90i upright microscope.
Comet assays
Neutral comet assays were performed using CometSlide assay kits (Trevigen). Cells were irradiated with 8 Gy and incubated at 37 °C for different periods of time (0‒150 min) to allow for DNA damage repair. Cells were embedded in agarose, lysed and subjected to neutral electrophoresis. Before image analysis, cells were stained with ethidium bromide and visualized under a fluorescence microscope. Single-cell electrophoresis results in a comet-shaped distribution of DNA. The comet head contains high molecular weight and intact DNA, and the tail contains the leading ends of migrating fragments. Olive comet moment was calculated by multiplying the percentage of DNA in the tail by the displacement between the means of the head and tail distributions. We utilized the program CometScore™ Version 1.5 (TriTek) to calculate Olive Comet Moment. A total of 25‒30 comets were analyzed per condition in each experiment.
Immunoblotting
Cells were lysed in RIPA buffer (150 mM NaCl, 50 mM Tris-HCl pH 7.4, 1% NP-40, 0.2% SDS, 0.25% sodium deoxycholate and 1 mM EDTA) containing HALT protease and phosphatase inhibitor cocktail (Pierce). Lysates were sheared using 10 passes through a 26-gauge needle followed by 10 passes through a 30-gauge needle. Sixty to 80 µg of total protein were separated by SDS-PAGE on a 4‒15% Criterion TGX Gel (Bio-Rad) and transferred to a nitrocellulose membrane using the Trans-Blot Turbo system (Bio-Rad). Membranes were developed using standard ECL technique and used Thermo Pierce ECL (Fisher) or Millipore Super ECL (EMD Millipore) for BRCA1 detection. A list of antibodies used is provided in the Supplemental Material.
Quantitative reverse-transcription PCR
cDNA was generated by reverse transcription of 1 μg total RNA using the GeneAmp® RNA PCR kit (Applied Biosystems, CA). qRT-PCR was performed using the 7900HT Fast Real-Time PCR system (Applied Biosystems) with the Taqman® Universal PCR Master Mix. Reactions were performed in triplicate and target gene and endogenous controls were amplified in the same plate. Relative quantitative measurements of target genes were determined by comparing the cycle thresholds.
Viral transduction
Retro- and lenti-viral transductions were performed as described.Citation24 Briefly, 293T cells were transfected with viral packaging (pUMVC3 or pHR’8.2ΔR) and envelope plasmids (p-CMV-VSV-G) along with the vector containing the cDNA or shRNA of interest. After 48 h, virus-containing media was harvested to infect target cells (MEFs). Retroviral transductions were performed as two 4‒6 h infections on sequential days and lentiviral as one 4 h infection. Cells were allowed to recover for 48 h, and selected with the appropriate drugs. Viral envelope and packaging plasmids were gifts from Sheila Stewart (Washington University). shRNAs were obtained from Sigma-Aldrich, and LA and ∆Ex9LA expression plasmids were generated in Stewart C’s laboratory.
Statistical analysis
With the exception of the ChIP experiments, a standard 2-sided, unpaired t-test was used to test for significance. Because the telomeric DNA content in the ChIP assays was always normalized to the DNA content of the wild-type cells, the mean of the ΔEx9 Homoz was tested using the null hypotheis that the mean equals 100 with degrees of freedom equal to the number of repeats minus 1. Results were considered significant at the 0.05 level.
| Abbreviations: | ||
| CTSL | = | cathepsin L |
| DSBs | = | double strand breaks |
| HR | = | homologous recombination |
| NHEJ | = | non-homologous end joining |
| IRIF | = | ionizing radiation induced foci |
Additional material
Download Zip (337 KB)Acknowledgments
We thank Caceres J for technical assistance. Research in the laboratory of Gonzalo S was supported by NIGMS Grant RO1 GM094513–01, DOD BCRP Idea Award BC110089, RDA from Siteman Cancer Center and Presidential Research Award from St Louis University. Gonzalez-Suarez I was recipient of a postdoctoral fellowship from AHA.
Author Contributions
Das A and Grotsky DA performed most of the experiments and contributed to the design of the project. Neumann MA, Kreienkamp R, Gonzalez-Suarez I and Redwood AB contributed with some experiments. Stewart CL provided MEFs from the LmnaΔ9/Δ9 mouse model, and LA and ∆Ex9LA expression plasmids. Kennedy BK provided G608GLA expression plasmid. Gonzalo S supervised the project and prepared the manuscript.
Disclosure of Potential Conflicts of Interest
No potential conflict of interest was disclosed.
Supplemental Materials
Supplemental materials may be found here:
www.landesbioscience.com/journals/nucleus/article/26873
References
- Broers JL, Ramaekers FC, Bonne G, Yaou RB, Hutchison CJ. Nuclear lamins: laminopathies and their role in premature ageing. Physiol Rev 2006; 86:967 - 1008; http://dx.doi.org/10.1152/physrev.00047.2005; PMID: 16816143
- Taddei A, Hediger F, Neumann FR, Gasser SM. The function of nuclear architecture: a genetic approach. Annu Rev Genet 2004; 38:305 - 45; http://dx.doi.org/10.1146/annurev.genet.37.110801.142705; PMID: 15568979
- Dechat T, Pfleghaar K, Sengupta K, Shimi T, Shumaker DK, Solimando L, Goldman RD. Nuclear lamins: major factors in the structural organization and function of the nucleus and chromatin. Genes Dev 2008; 22:832 - 53; http://dx.doi.org/10.1101/gad.1652708; PMID: 18381888
- Goldman RD, Gruenbaum Y, Moir RD, Shumaker DK, Spann TP. Nuclear lamins: building blocks of nuclear architecture. Genes Dev 2002; 16:533 - 47; http://dx.doi.org/10.1101/gad.960502; PMID: 11877373
- Gruenbaum Y, Margalit A, Goldman RD, Shumaker DK, Wilson KL. The nuclear lamina comes of age. Nat Rev Mol Cell Biol 2005; 6:21 - 31; http://dx.doi.org/10.1038/nrm1550; PMID: 15688064
- Schreiber KH, Kennedy BK. When lamins go bad: nuclear structure and disease. Cell 2013; 152:1365 - 75; http://dx.doi.org/10.1016/j.cell.2013.02.015; PMID: 23498943
- Burke B, Stewart CL. Life at the edge: the nuclear envelope and human disease. Nat Rev Mol Cell Biol 2002; 3:575 - 85; http://dx.doi.org/10.1038/nrm879; PMID: 12154369
- De Sandre-Giovannoli A, Bernard R, Cau P, Navarro C, Amiel J, Boccaccio I, Lyonnet S, Stewart CL, Munnich A, Le Merrer M, et al. Lamin a truncation in Hutchinson-Gilford progeria. Science 2003; 300:2055; http://dx.doi.org/10.1126/science.1084125; PMID: 12702809
- Eriksson M, Brown WT, Gordon LB, Glynn MW, Singer J, Scott L, Erdos MR, Robbins CM, Moses TY, Berglund P, et al. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature 2003; 423:293 - 8; http://dx.doi.org/10.1038/nature01629; PMID: 12714972
- Stewart CL, Kozlov S, Fong LG, Young SG. Mouse models of the laminopathies. Exp Cell Res 2007; 313:2144 - 56; http://dx.doi.org/10.1016/j.yexcr.2007.03.026; PMID: 17493612
- Zhang H, Kieckhaefer JE, Cao K. Mouse models of laminopathies. Aging Cell 2013; 12:2 - 10; http://dx.doi.org/10.1111/acel.12021; PMID: 23095062
- Sullivan T, Escalante-Alcalde D, Bhatt H, Anver M, Bhat N, Nagashima K, Stewart CL, Burke B. Loss of A-type lamin expression compromises nuclear envelope integrity leading to muscular dystrophy. J Cell Biol 1999; 147:913 - 20; http://dx.doi.org/10.1083/jcb.147.5.913; PMID: 10579712
- Jahn D, Schramm S, Schnölzer M, Heilmann CJ, de Koster CG, Schütz W, Benavente R, Alsheimer M. A truncated lamin A in the Lmna -/- mouse line: implications for the understanding of laminopathies. Nucleus 2012; 3:463 - 74; http://dx.doi.org/10.4161/nucl.21676; PMID: 22895093
- Osorio FG, Bárcena C, Soria-Valles C, Ramsay AJ, de Carlos F, Cobo J, Fueyo A, Freije JM, López-Otín C. Nuclear lamina defects cause ATM-dependent NF-κB activation and link accelerated aging to a systemic inflammatory response. Genes Dev 2012; 26:2311 - 24; http://dx.doi.org/10.1101/gad.197954.112; PMID: 23019125
- Pendás AM, Zhou Z, Cadiñanos J, Freije JM, Wang J, Hultenby K, Astudillo A, Wernerson A, Rodríguez F, Tryggvason K, et al. Defective prelamin A processing and muscular and adipocyte alterations in Zmpste24 metalloproteinase-deficient mice. Nat Genet 2002; 31:94 - 9; PMID: 11923874
- Mounkes LC, Kozlov S, Hernandez L, Sullivan T, Stewart CL. A progeroid syndrome in mice is caused by defects in A-type lamins. Nature 2003; 423:298 - 301; http://dx.doi.org/10.1038/nature01631; PMID: 12748643
- Hernandez L, Roux KJ, Wong ES, Mounkes LC, Mutalif R, Navasankari R, Rai B, Cool S, Jeong JW, Wang H, et al. Functional coupling between the extracellular matrix and nuclear lamina by Wnt signaling in progeria. Dev Cell 2010; 19:413 - 25; http://dx.doi.org/10.1016/j.devcel.2010.08.013; PMID: 20833363
- Shumaker DK, Dechat T, Kohlmaier A, Adam SA, Bozovsky MR, Erdos MR, Eriksson M, Goldman AE, Khuon S, Collins FS, et al. Mutant nuclear lamin A leads to progressive alterations of epigenetic control in premature aging. Proc Natl Acad Sci U S A 2006; 103:8703 - 8; http://dx.doi.org/10.1073/pnas.0602569103; PMID: 16738054
- Liu B, Wang J, Chan KM, Tjia WM, Deng W, Guan X, Huang JD, Li KM, Chau PY, Chen DJ, et al. Genomic instability in laminopathy-based premature aging. Nat Med 2005; 11:780 - 5; http://dx.doi.org/10.1038/nm1266; PMID: 15980864
- Viteri G, Chung YW, Stadtman ER. Effect of progerin on the accumulation of oxidized proteins in fibroblasts from Hutchinson Gilford progeria patients. Mech Ageing Dev 2010; 131:2 - 8; http://dx.doi.org/10.1016/j.mad.2009.11.006; PMID: 19958786
- Scaffidi P, Misteli T. Lamin A-dependent nuclear defects in human aging. Science 2006; 312:1059 - 63; http://dx.doi.org/10.1126/science.1127168; PMID: 16645051
- Gonzalez-Suarez I, Redwood AB, Gonzalo S. Loss of A-type lamins and genomic instability. Cell Cycle 2009; 8:3860 - 5; http://dx.doi.org/10.4161/cc.8.23.10092; PMID: 19901537
- Gonzalez-Suarez I, Redwood AB, Grotsky DA, Neumann MA, Cheng EH, Stewart CL, Dusso A, Gonzalo S. A new pathway that regulates 53BP1 stability implicates cathepsin L and vitamin D in DNA repair. EMBO J 2011; 30:3383 - 96; http://dx.doi.org/10.1038/emboj.2011.225; PMID: 21750527
- Gonzalez-Suarez I, Redwood AB, Perkins SM, Vermolen B, Lichtensztejin D, Grotsky DA, Morgado-Palacin L, Gapud EJ, Sleckman BP, Sullivan T, et al. Novel roles for A-type lamins in telomere biology and the DNA damage response pathway. EMBO J 2009; 28:2414 - 27; http://dx.doi.org/10.1038/emboj.2009.196; PMID: 19629036
- Redwood AB, Perkins SM, Vanderwaal RP, Feng Z, Biehl KJ, Gonzalez-Suarez I, Morgado-Palacin L, Shi W, Sage J, Roti-Roti JL, et al. A dual role for A-type lamins in DNA double-strand break repair. Cell Cycle 2011; 10:2549 - 60; http://dx.doi.org/10.4161/cc.10.15.16531; PMID: 21701264
- Gonzalo S, Blasco MA. Role of Rb family in the epigenetic definition of chromatin. Cell Cycle 2005; 4:752 - 5; http://dx.doi.org/10.4161/cc.4.6.1720; PMID: 15908781
- Gonzalo S, García-Cao M, Fraga MF, Schotta G, Peters AH, Cotter SE, Eguía R, Dean DC, Esteller M, Jenuwein T, et al. Role of the RB1 family in stabilizing histone methylation at constitutive heterochromatin. Nat Cell Biol 2005; 7:420 - 8; http://dx.doi.org/10.1038/ncb1235; PMID: 15750587
- Solovei I, Wang AS, Thanisch K, Schmidt CS, Krebs S, Zwerger M, Cohen TV, Devys D, Foisner R, Peichl L, et al. LBR and lamin A/C sequentially tether peripheral heterochromatin and inversely regulate differentiation. Cell 2013; 152:584 - 98; http://dx.doi.org/10.1016/j.cell.2013.01.009; PMID: 23374351
- Johnson BR, Nitta RT, Frock RL, Mounkes L, Barbie DA, Stewart CL, Harlow E, Kennedy BK. A-type lamins regulate retinoblastoma protein function by promoting subnuclear localization and preventing proteasomal degradation. Proc Natl Acad Sci U S A 2004; 101:9677 - 82; http://dx.doi.org/10.1073/pnas.0403250101; PMID: 15210943
- Nitta RT, Jameson SA, Kudlow BA, Conlan LA, Kennedy BK. Stabilization of the retinoblastoma protein by A-type nuclear lamins is required for INK4A-mediated cell cycle arrest. Mol Cell Biol 2006; 26:5360 - 72; http://dx.doi.org/10.1128/MCB.02464-05; PMID: 16809772
- Liu B, Zhou Z. Lamin A/C, laminopathies and premature ageing. Histol Histopathol 2008; 23:747 - 63; PMID: 18366013
- Pegoraro G, Kubben N, Wickert U, Göhler H, Hoffmann K, Misteli T. Ageing-related chromatin defects through loss of the NURD complex. Nat Cell Biol 2009; 11:1261 - 7; http://dx.doi.org/10.1038/ncb1971; PMID: 19734887
- Prokocimer M, Barkan R, Gruenbaum Y. Hutchinson-Gilford progeria syndrome through the lens of transcription. Aging Cell 2013; 12:533 - 43; http://dx.doi.org/10.1111/acel.12070; PMID: 23496208
- Blasco MA, Lee HW, Hande MP, Samper E, Lansdorp PM, DePinho RA, Greider CW. Telomere shortening and tumor formation by mouse cells lacking telomerase RNA. Cell 1997; 91:25 - 34; http://dx.doi.org/10.1016/S0092-8674(01)80006-4; PMID: 9335332
- Herrera E, Samper E, Blasco MA. Telomere shortening in mTR-/- embryos is associated with failure to close the neural tube. EMBO J 1999; 18:1172 - 81; http://dx.doi.org/10.1093/emboj/18.5.1172; PMID: 10064584
- García-Cao M, O’Sullivan R, Peters AH, Jenuwein T, Blasco MA. Epigenetic regulation of telomere length in mammalian cells by the Suv39h1 and Suv39h2 histone methyltransferases. Nat Genet 2004; 36:94 - 9; http://dx.doi.org/10.1038/ng1278; PMID: 14702045