
Abstract
Lamin A is a major component of the lamina, which creates a dynamic network underneath the nuclear envelope. Mutations in the lamin A gene (LMNA) cause severe genetic disorders, one of which is Hutchinson-Gilford progeria syndrome (HGPS), a disease triggered by a dominant mutant named progerin. Unlike the wild-type lamin A, whose farnesylated C-terminus is excised during post-translational processing, progerin retains its farnesyl tail and accumulates on the nuclear membrane, resulting in abnormal nuclear morphology during interphase. In addition, membrane-associated progerin forms visible cytoplasmic aggregates in mitosis. To examine the potential effects of cytoplasmic progerin, nuclear localization signal (NLS) deleted progerin and lamin A (PGΔNLS and LAΔNLS, respectively) have been constructed. We find that both ΔNLS mutants are farnesylated in the cytosol and associate with a sub-domain of the ER via their farnesyl tails. While the farnesylation on LAΔNLS can be gradually removed, which leads to its subsequent release from the ER into the cytoplasm, PGΔNLS remains permanently farnesylated and membrane-bounded. Moreover, both ΔNLS mutants dominantly affect emerin’s nuclear localization. These results reveal new insights into lamin A biogenesis and lamin A-emerin interaction.
Introduction
Lamin A, encoded by the LMNA gene, is a major component of the nuclear lamina in animal cells.Citation1-Citation4 As a type V intermediate filament, lamin A forms a dynamic network underneath the inner nuclear membrane (INM), providing mechanical support to the nuclear envelope.Citation5-Citation8 Besides the structural function, lamin A has been suggested to play essential roles in cell regulation, including chromatin organization, transcription, and apoptosis.Citation9-Citation15 These roles are at least partially accomplished by direct or indirect interactions with chromosomes and various nuclear regulators, including emerin, an integral protein of the INM.Citation16-Citation22
Similar to other intermediate filament proteins, lamin A contains a short globular N-terminal head domain, a central α-helical coiled-coil rod domain, and a long globular C-terminal tail domain.Citation23 In addition, between the central rod domain and C-terminal tail domain, lamin A has a nuclear localization signal sequence (NLS), which signals its nuclear residence.Citation23-Citation25 Moreover, a CaaX motif (C, cysteine; a, aliphatic amino acid; X, any amino acid) is located at the C-terminus of lamin A, with an exact sequence of CSIM (cysteine-serine-isoleucine-methionine).Citation26
It has been shown that proper processing of the CaaX motif is critical for membrane association, localization, and functionality of lamin A.Citation27-Citation31 After the DNA sequence is transcribed and translated into the lamin A precursor protein (prelamin A), the cysteine in the CSIM motif is farnesylated by a farnesyltransferase (FTase), followed by the removal of SIM by ZMPSTE24 and carboxylmethylation by Icmt. In the last step, the final 15 amino acids including the farnesylated C-terminus of prelamin A are excised by ZMPSTE24 to allow the release of mature lamin A from the INM.Citation2,Citation32-Citation36 ZMPSTE24 is an integral membrane zinc metalloprotease, which has a dual affinity to both the INM and the cytosolic ER membrane,Citation37-Citation39 and the INM has been shown to be the physiologically relevant compartment for prelamin A processing.Citation39
A wide range of human disorders known as laminopathies are associated with mutations within LMNA, among which Hutchinson–Gilford progeria syndrome (HGPS) has the most striking premature aging phenotypes.Citation10,Citation25,Citation40 HGPS is extremely rare, affecting 1 in 4–8 million live births. The patients appear normal at birth, but gradually show symptoms of accelerated aging after 12 mo, and often die of heart attacks or strokes in their early teens.Citation10 The culprit of HGPS is a lamin A mutant known as progerin which is caused by a de novo nucleotide substitution from C to T at position 1824 of LMNA. The mutation changes no amino acid (G608G), but induces a cryptic splicing donor site that generates a 150-nucleotide deletion on the mRNA sequence. The resulting progerin protein thus bears a 50-amino acid in-frame deletion that lacks the normal cleavage site of ZMPSTE24 for C-terminal farnesyl group release.Citation41-Citation43 Therefore, progerin permanently retains the farnesylated C-terminus and remains associated with the nuclear membrane, eliciting nuclear blebbings and other nuclear abnormalities in HGPS patient cells, including disrupted heterochromatin-lamin interactions and alterations in gene transcription.Citation11,Citation17,Citation44 Inhibiting farnesylation of progerin with farnesyltransferase inhibitors (FTIs) or mutating CSIM into non-farnesylable SSIM relocalizes progerin away from the nuclear envelope (NE) and alleviates the prominent nuclear phenotypes.Citation27,Citation29,Citation45,Citation46
Previously, we have reported that the anchorage of progerin to the INM disrupts the normal NE disassembly during mitosis, leading to an accumulation of progerin-membrane aggregates in mitosis.Citation44,Citation47 Importantly, there is a noticeable delay in the recruitment of progerin back to the nucleus at the end of mitosis.Citation44,Citation47 To investigate the possible effects of the cytoplasmic progerin, we created nuclear localization signal (NLS)-deleted progerin and lamin A (PGΔNLS and LAΔNLS, respectively). Analysis of these mutants has revealed new insights into lamin A processing and emerin targeting.
Results
Deletion of NLS directs lamin A and progerin to the ER
In the current study, the NLS sequence (AAAAAGCGCA AACTGGAG) was deleted from lamin A (LA) and progerin (PG) cDNA sequences using a PCR-mediated mutagenesis method.Citation48 These newly generated DNA segments were sequenced and sub-cloned into a pEGFP-C1 plasmid for expression (; Fig. S1). To examine the proteins’ sizes, we performed western blot analyses on transiently transfected HeLa cells with EGFP-LA, EGFP-PG, EGFP-LAΔNLS or EGFP-PGΔNLS plasmids. As expected, the sizes of EGFP-tagged ΔNLS mutants were slightly smaller than their NLS bearing counterparts, and the endogenous lamin A/C showed a consistent level across all transfected cell lines (). Untransfected HeLa cells were used as a control (CT, ).
Figure 1. Characterization of the NLS-deleted lamin A and progerin. (A) A schematic diagram of the generation of the NLS deletion mutants. Lamin A and progerin NLS deletion (LAΔNLS and PGΔNLS) were created via PCR and subcloned into the AscI and XbaI sites of the pEGFP-C1 plasmid. (B) Western Blot analysis. Protein samples were immunoblotted with antibodies of lamin A/C and β-actin. Non-transfected HeLa cells were used as a control (CT). (C) Confocal fluorescence images. HeLa cells transiently expressing EGFP-LA, EGFP-PG, EGFP-LAΔNLS or EGFP-PGΔNLS (green) were fixed and stained with anti-lamin B1 (red) by immunofluorescence at 24 h post transfection. DNA was stained with DAPI (blue). A representative cell under each condition is shown. Scale bar, 5 μm. (D) Confocal fluorescence images. HeLa cells transiently expressing EGFP-LAΔNLS or EGFP-PGΔNLS (green) were fixed and stained with anti-KDEL (a marker for ER, in red) or anti-GM130 (a marker for Golgi, in red). A representative cell under each condition is shown. Scale bar, 10 μm.
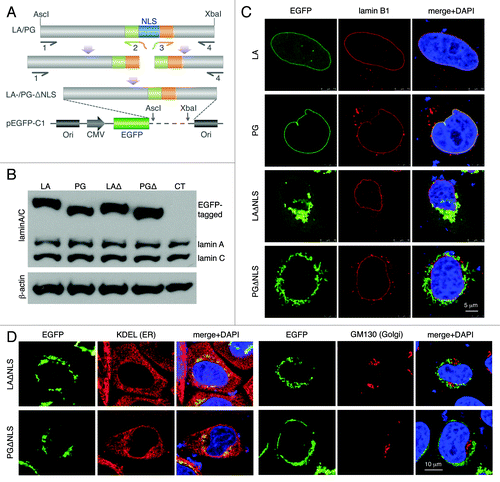
Next, we examined the cellular localization of EGFP-LAΔNLS and EGFP-PGΔNLS. We predicted that, without the NLS, neither of them could enter the nucleus. Indeed, 24 h post transfection, we found that the majority of the EGFP-LAΔNLS and EGFP-PGΔNLS stayed in the cytosol while EGFP-LA and EGFP-PG co-localized with lamin B underneath the INM (). Interestingly, we found that these cytosolic LAΔNLS and PGΔNLS were concentrated at specific locations. Moreover, time-course experiments revealed that in the EGFP-LAΔNLS transfected cells, diffuse cytoplasmic EGFP signals became detectable after 24 h post transfection, indicating that the EGFP-LAΔNLS accumulates gradually transformed into two distinct states with the passage of time: the insoluble state and the soluble cytoplasmic state. However, almost all PGΔNLS remained insoluble during the same time-course (Fig. S2).
To elucidate the cytosolic localization of these NLS mutants, we co-stained the EGFP-tagged NLS mutants with the anti-KDEL and anti-GM130 antibodies, markers for the ER and Golgi apparatus respectively.Citation49,Citation50 Microscopic analysis revealed that these mutant aggregates co-localized with a sub-domain of the ER while no overlaps were identified between LAΔNLS or PGΔNLS and the Golgi marker GM130 (). In addition, we observed a complete overlap between the signals of DsRed-LAΔNLS and EGFP-PGΔNLS at 24 h post transfection (Fig. S3), suggesting that both ΔNLS mutants localized to the same ER sub-domain. Consistently, FRAP experiments suggested a comparably slow motion of these ER-associated LAΔNLS and PGΔNLS (Fig. S4). In summary, we found that without the NLS, both lamin A and progerin immediately attached to a sub-domain of the ER after being synthesized.
The C-terminal farnesyl group tethers LAΔNLS and PGΔNLS to the ER membrane
To understand why LAΔNLS and PGΔNLS showed affinities to the ER, we hypothesized that both ΔNLS mutants were farnesylated at the C-terminus, which tethered these proteins to the ER membrane.
To test this hypothesis, we first determined the farnesylation status of LAΔNLS and PGΔNLS with a Click chemistry assay on transfected HeLa cells (see Methods). As expected,Citation10,Citation33 wild type mature lamin A was not farnesylated while progerin showed positive farnesylation signals due to its inability to be cleaved by ZMPSTE24 (). Notably, farnesylation signals were detected in both LAΔNLS and PGΔNLS, but the signal of LAΔNLS was significantly weaker compare with that of PGΔNLS (). We reason that the difference in farnesylation levels between LAΔNLS and PGΔNLS is likely caused by the cleavage of the C-terminal farnesyl group of LAΔNLS by the ER-associated ZMPSTE24, as ZMPSTE24 has been demonstrated to be a dually localized protein on both the ER membrane and the INM.Citation39 Supporting this notion, we observed a gradual increase of diffuse cytoplasmic LAΔNLS with time, which likely represented the cleaved form of LAΔNLS (Fig. S2).
Figure 2. C-terminal farnesyl group tethers LAΔNLS and PGΔNLS to the ER membrane. (A) Click chemistry analysis. HeLa cells were transfected with EGFP-LA, EGFP-PG, EGFP-LAΔNLS or EGFP-PGΔNLS and labeled with Click-iT farnesyl alcohol, followed by precipitation with GFP-Trap beads and detection with 647 Alkyne. Strong farnesylation signals appeared in PG and PGΔNLS (PGΔ) lanes, and weak but detectable farnsylation showed in LAΔNLS (LAΔ) lane. (B) Quantification of farnesylation levels in (A). The relative farnesylation level was calculated as the ratio of the farnesylation signal to the corresponding IP’ed protein signal. (C) Confocal fluorescence images of LAssimΔNLS and PGssimΔNLS. Immunofluorescence was performed 24 h after transfection. Confocal images show EGFP (green), lamin B1 (red), and DNA (blue). A representative cell under each condition is shown. Scale bar, 10 μm.
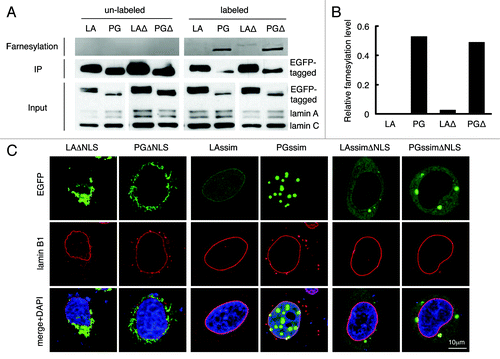
To further test this idea, we asked whether blocking the farnesylation of LAΔNLS and PGΔNLS could lead to dissociation from the ER membrane. It has been shown that farnesylation is abolished when the C-terminal sequence of CSIM on lamin A and progerin is mutated into SSIM.Citation29,Citation45,Citation51 Thus, we generated the SSIM-ΔNLS double mutants of lamin A and progerin (LAssimΔNLS and PGssimΔNLS). As expected, the SSIM mutation alone directed lamin A and progerin into the nucleoplasm ().Citation29,Citation45,Citation52,Citation53 Importantly, when the two features ΔNLS and SSIM were combined, the proteins were released from the ER into the cytoplasm (), validating the idea that the farnesyl groups on the C-termini of LAΔNLS and PGΔNLS tethered them to the ER membrane.
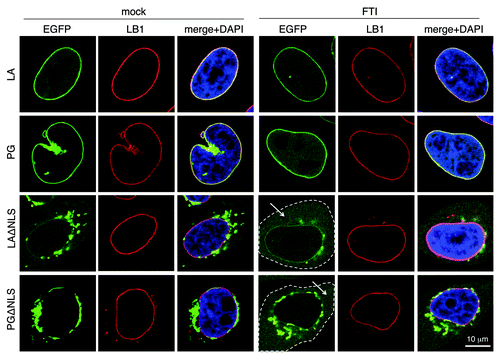
Additional support was obtained with a drug-treatment experiment using farnesyltransferase inhibitors (FTIs). When we blocked farnesylation with FTIs at 4 h post transfection for 16 h, we observed that the non-farnesylated LAΔNLS and PGΔNLS became soluble in the cytoplasm ().
Figure 3. The effect of FTI treatment resembles that of LAssimΔNLS. FTI treatment was performed on HeLa cells at 4 h post-transfection for 16 h. The cytoplasm of the ΔNLS mutants transfected cells with FTI treatment are outlined by dashed lines. Soluble cytoplasmic EGFP signals are pointed by arrows. Confocal images show EGFP (green), lamin B1 (red), and DNA (blue). A representative cell under each condition is shown. Scale bar, 10 μm.
Nuclear targeting of emerin is disrupted by LAΔNLS and PGΔNLS
It has been suggested that emerin localization is dependent on A-type lamins.Citation54 Thus, we investigated whether the distribution of emerin was altered by the ΔNLS mutants. In control LA and PG transfected HeLa cells, as expected, most of the endogenous emerin co-localized with LA or PG to the nuclear rim, outlining the shape of the nucleus (, first and third panels). However, in the ΔNLS mutant transfected cells, emerin became cytosolic and colocalized with the ΔNLS mutants to a sub-domain of the ER (, second and fourth panels), suggesting that emerin’s nuclear localization is dependent primarily on lamin A. Notably, emerin’s normal nuclear localization appeared to be more disrupted by PGΔNLS than by LAΔNLS, as the nuclear rim staining of emerin was almost absent in PGΔNLS transfected cells as it was still visible in LAΔNLS expressing cells (, second and fourth panels).
Figure 4. Disrupted emerin localization in cells expressing LAΔNLS or PGΔNLS. (A) Confocal fluorescence images. HeLa cells transiently expressing EGFP-LA, EGFP-PG, EGFP-LAΔNLS or EGFP-PGΔNLS (green) were fixed and stained with anti-emerin (red) at 24 h post transfection. A representative cell under each condition is shown. Scale bar, 10 μm. (B) IP with GFP-Trap®_A beads in the transfected HeLa cells. Un-transfected HeLa cells were used as a control (CT). (C) Relative intensity of emerin to EGFP-tagged proteins in each immunopricipitated sample. Band intensities were analyzed using ImageJ. Relative intensities were presented as the ratio of emerin to EGFP. Two biological duplicates were conducted. A representative experiment was shown.
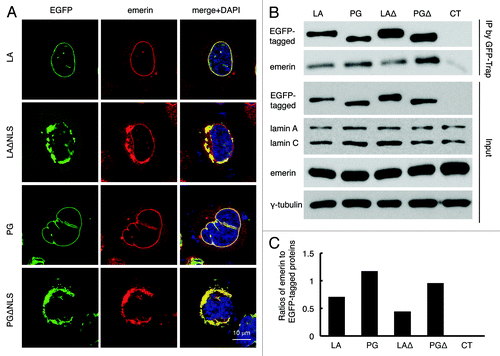
To determine the potential physical interactions between emerin and the ΔNLS mutants, an immunoprecipitation (IP) experiment was performed using GFP-Trap beads. Un-transfected HeLa cells were used as a control. We found that emerin co-precipitated with EGPF-LA, EGFP-PG, EGFP-LAΔNLS and EGFP-PGΔNLS (). Consistent with the microscopic observation that PGΔNLS more effectively sequestered emerin from the nucleus than LAΔNLS (), quantification revealed that the interaction of emerin with PGΔNLS was almost twice as strong as with LAΔNLS ().
Taken together, these data showed that the NLS deletion mutants interacted and sequestered emerin away from its normal INM localization. Our data suggest that emerin is targeted to the nucleus primarily through its interactions with lamin A, and there is a stronger affinity between emerin and progerin or PGΔNLS than emerin and lamin A or LAΔNLS.
Discussion
The processing of prelamin A by the INM and ER localized ZMPSTE24
ZMPSTE24, as one of the key players in the lamin A maturation process, is an integral membrane protein.Citation37-Citation39 The cytosolic face of the ER membrane was considered its primary residency until recently when Barrowman and colleagues clearly demonstrated that ZMPSTE24 was also localized to the INM, and that the nucleus was the physiologically relevant compartment where the C-terminal cleavage of prelamin A occurred.Citation39
In this study, we generate the cytoplasmic-resident lamin A mutant LAΔNLS. This mutant rapidly tethers to a sub-domain of the ER via its farnesyl tail after being synthesized on the ribosomes ( and ). Notably, we find that over a course of 72 h post transfection, the ER-associated LAΔNLS becomes gradually released into the cytoplasm (Fig. S2), which is likely to be resulted from the removal of the farnesylated C-terminus. In support of this notion, we detect a reduced level of farnesylation in LAΔNLS compared with PGΔNLS, and the double mutant LAssimΔNLS and PGssimΔNLS and FTI treatment experiments further support that the cytoplasmic soluble fraction of LAΔNLS is not farnesylated ( and ). Based on the previous finding that ZMPSTE24 is a dually localized enzyme,Citation39 we would like to suggest that the cleavage of LAΔNLS’s farnesylated tail is executed by the ER-associated ZMPSTE24.
Interestingly, the Click Chemistry labeling experiment reveals unexpected differences in enzymatic activities of the ER-associated and the INM-associated ZMPSTE24. As shown in , the processing of the wild-type lamin A is achieved in an extremely rapid manner on the INM, leading to no detection of the farnesylated prelamin A. In contrast, the processing of the ER-associated LAΔNLS by the ER-associated ZMPSTE24 appears to be much slower, which resulted in a clearly detectable fraction of the farnsylated LAΔNLS at 48 h post transfection.
Previously, Barrowman and colleagues have examined the ZMPSTE24 processing kinetics of a lamin A-tail construct that is fused with a large carrier protein HA-pyruvate kinase either with or without the NLS.Citation39 Without the NLS, the lamin A-tail construct produced a cytosolic protein.Citation39 Consistently with our observation, Barrowman and colleagues found that ZMPSTE24 was functional in both the INM and the ER locations.Citation39 However, they found that the rate of ZMPSTE24 processing of this lamin A fusion protein was quite similar in both locations.Citation39 The potential differences in the two studies may be caused by many variables including differential access of membrane-bound proteins vs. cytosolic proteins and differential enzymatic activity of ZMPSTE24 to LAΔNLS vs. Pyruvate kinase-lamin A tail fusion protein. Future studies, with controls of these variables, will be required to directly compare the processing kinetics of the ER and INM localized ZMPSTE24 to lamin A.
Emerin nuclear localization is disrupted by PGΔNLS
Mutations and aberrant targeting of emerin cause a number of diseases including muscular dystrophy, cardiomyopathy and Emery-Dreifuss muscular dystrophy, which is characterized by muscle weakening, contractures of major tendons and potentially lethal cardiac defects.Citation22,Citation55-Citation57 Emerin primarily localizes to the INM. Previously, Ostlund and colleagues suggested that the N-terminal nucleoplasmic domain of emerin was both necessary and sufficient for targeting emerin to the INM.Citation58 However, using SW13 cells that did not express lamin A, Vaughan et al. showed that the INM localization of emerin was dependent on the lamin A complex containing both lamins A and B.Citation54 In support of this argument, it has been shown biochemically that that emerin is in complexes with both A and B type lamins.Citation54,Citation59
In this study, we compartmentalize B type lamins and endogenous lamin A/C to the nucleus and LAΔNLS and PGΔNLS to the ER. With this geographic separation, we compare the effects of LAΔNLS, PGΔNLS, and endogenous lamins on emerin’s nuclear targeting. Our analyses reveal that at the presence of wildtype lamins A, B, and C, a large fraction of endogenous emerin is recruited to ER-localized (ΔNLS) lamin A or progerin (). This recruitment is stronger for ER-localized (ΔNLS) progerin, which appears to remove all endogenous emerin from the nuclear envelope (). In support, the IP experiment further suggests that emerin exhibits a stronger binding affinity to progerin or PGΔNLS than to lamin A or LAΔNLS, respectively (). It remains to be determined whether this phenotype is general to other INM proteins. Future work will focus on determining whether additional INM proteins, including the endogenous lamin A, are affected by the ER-localized progerin. Given the emerging roles of the emerin-lamin A complex in regulating muscle- and heart-specific gene expression,Citation60 we hope that these new insights gained from this study will promote a better understanding of gene misregulation in muscular dystrophy and cardiomyopathy.
Materials and Methods
Plasmid construction
Plasmids of pEGFP-C1-LAΔNLS, pEGFP-C1-PGΔNLS, pEGFP-C1-LASSIMΔNLS, and pEGFP-C1-PGSSIMΔNLS were constructed based on the pEGFP-C1 vector (Clontech). The NLS sequence (AAAAAGCGCA AACTGGAG) was removed from cDNA of Lamin A (LA), progerin (PG), LA-SSIM, and PG-SSIM by PCR splicing. Primers used were two targeting each ends of LMNA, LMNA 5F (5′-AGACCCCGTCC CAGCGGCGCGC-3′), and LMNA 3R (5′-GTCGACTCTA GATTACATGA TGCTGCAGTT CTG-3′), and two flanking NLS regions complementary each other, LMNA 5R (5′-TGCGGCTCTC AGTGGAGGTG ACGCTGCCC-3′) and LMNA 3F (5′-GGGCAGCGTC ACCTCCACTG AGAGCCGCA-3′). The 5′- and 3′- regions of LMNA were amplified using primer pairs “LMNA 5F + LMNA 5R” and “LMNA 3F + LMNA 3R” respectively, followed by a second amplification using the overlapping 5′- and 3′- fragments as templates to generate NLS-deleted sequences. The NLS deleted sequences were then sub-cloned into the AscI and XbaI sites of pEGFP-C1. A plasmid of pDsRed-monomer-C1-LA was created based on the pDsRed-monomer-C1 vector (Clontech). The full-length of lamin A, progerin, LAΔNLS or PGΔNLS was amplified using LMNA 5F and LMNA 3R, followed by sub-cloning into the BspEI and BamHI sites of pDsRed-monomer-C1.
Cell culture, transfection, and FTI treatment
HeLa cells were cultured in DMEM (Lonza) containing 10% heat-inactivated FBS (BenchMark) at 37 °C supplied with 5% CO2. Approximately 1.5 × 105 cells were seeded and incubated at 37 °C for one day, then transfected with 2 μg of the designated plasmids using FuGENE® 6 Transfection Reagent (Promega) following the manufacturer’s instructions. In the FTI treatment experiment, FTI (J&J) at a final concentration of 2 μM was added to culture media at 4 h after transfection for 16 h.
Antibodies
The antibodies used in western blotting analysis, immunofluorescence, and immunoprecipitation were: mouse-anti-human Lamin A/C (MAB3211, Millipore), goat-anti-Lamin A/C (N-18, Santa Cruz Biotechnology), goat- anti-Lamin B (sc-6217, Santa Cruz Biotechnology), mouse anti-β-Actin peroxidase conjugated (A3854, Sigma), mouse-anti-KDEL (ab12223, Abcam), mouse anti-GM130 (610822, BD Transduction Laboratories), rabbit-anti-emerin (ab14208–20, Abcam), mouse-anti-γ-tubulin (019K4794, Sigma).
Western blotting
Cell pellets were dissolved in Laemmli Sample Buffer containing 5% β-mercaptoethanol (Bio-Rad) to obtain whole cell lysates. Protein samples were then electrophoretically resolved on 10% SDS-PAGE gels and subsequently transferred onto nitrocellulose membranes (Bio-Rad) for primary and secondary antibodies detection. Bands were visualized by enhanced chemiluminescence (Pierce® ECL Western Blotting Substrate; Thermo SCIENTIFIC). Quantification was performed by ImageJ (National Institutes of Health, Bethesda).
Immunofluorescence microscopy
HeLa cells cultured on glass-bottom dishes were washed twice with tris-buffered saline (TBS) and fixed in 4% paraformaldehyde/phosphate buffered saline (PBS) for 20 min at room temperature. Subsequently, the cells were permeabilized with 0.5% Triton X-100 in PBS for 5 min at room temperature. After being washed twice with TBS, cells were blocked in 4% BSA/TBS for 1 h, and probed with the primary antibodies overnight at 4 °C. The cells were then washed five times with TBS, followed by secondary antibody incubation at room temperature for 1 h in the dark. Secondary antibodies used were Alexa Fluor® 594 donkey anti-rabbit IgG (Invitrogen), Alexa Fluor® 594 donkey anti-goat IgG (Invitrogen) and Alexa Fluor® 594 donkey anti-mouse IgG (Invitrogen). After being washed five times with TBS, the cells were stained with DAPI and mounted using VECTASHIELD® Mounting Medium with DAPI (H-1200, VECTOR). Immunofluorescence microscopy was performed on a Leica SP5 X Confocal Microscope (Leica Microsystems, Inc).
Immunoprecipitation (IP)
At 24 h after transfection, the transfected HeLa cell pellets were lysed in ice-cold 1% Triton buffer (1% Triton, 50 mM Tirs pH 7.4, 150 mM NaCl, 5 mM MgCl2, 1 X protease inhibitor cocktail [Roche]), and then centrifuged at 2700 g at 4 °C for 10 min to obtain supernatants. EGFP-tagged proteins were immunoprecipitated from the supernatants with GFP-Trap®_A beads (Chromoteck) according to manufacturer’s instructions. Both input supernatants and immunoprecipitates were then resolved on 10% SDS-PAGE gels and subsequently transferred onto nitrocellulose membranes (Bio-Rad) for staining with primary and secondary antibodies.
Click chemistry assay
At 15 h post transfection, the transfected HeLa cells with designated pEGFP-C1 based plasmids were incubated with Click-iT farnesyl alcohol azide (C10248, Invitrogen) for 14 h for labeling. Cell lysates were collected and immunoprecipitated using GFP-Trap®_A beads (Chromotek) according to the manufacturer’s instructions, followed by farnesyl detection using Alexa Fluor® 647, alkyne (A10278, Invitrogen). Protein samples were then separated with non-reducing 10% SDS-PAGE gels. After being fixed with methanol/acetic acid, the SDS-PAGE gels were scanned under a Typhoon imager.
Fluorescence recovery after photobleaching (FRAP) assay
HeLa cells transfected with designated constructs were grown on glass-bottom dishes and cultured at 37 °C prior to analysis. Photobleaching experiments were performed using a Leica SP5 X Confocal Microscope (Leica Microsystems, Inc). All procedures were done at 37 °C. Confocal images were taken every three seconds for the first 40 images and every ten seconds for the next 80 images. Quantification was conducted using Leica SP5 software.
Additional material
Download Zip (612.5 KB)Disclosure of Potential Conflicts of Interest
No potential conflict of interest was disclosed.
Acknowledgments
We thank members in Cao lab and Dr Norma Andrews for helpful discussion, as well as Amy Beaven in the imaging core and Kenneth class in the flow cytometry facility at the University of Maryland College Park for technical support. We also want to thank the two anonymous reviewers for their constructive comments that helped improve the quality of this manuscript. Funding: This work was supported by an NIH/NIA Grant R00AG029761 (K.C.) and by the New Scholar Award in Aging by the Ellison Medical Foundation (K.C.).
References
- Dechat T, Adam SA, Taimen P, Shimi T, Goldman RD. Nuclear lamins. Cold Spring Harb Perspect Biol 2010; 2:a000547; http://dx.doi.org/10.1101/cshperspect.a000547; PMID: 20826548
- Goldman RD, Gruenbaum Y, Moir RD, Shumaker DK, Spann TP. Nuclear lamins: building blocks of nuclear architecture. Genes Dev 2002; 16:533 - 47; http://dx.doi.org/10.1101/gad.960502; PMID: 11877373
- Broers JL, Ramaekers FC, Bonne G, Yaou RB, Hutchison CJ. Nuclear lamins: laminopathies and their role in premature ageing. Physiol Rev 2006; 86:967 - 1008; http://dx.doi.org/10.1152/physrev.00047.2005; PMID: 16816143
- Gordon LB, Cao K, Collins FS. Progeria: translational insights from cell biology. J Cell Biol 2012; 199:9 - 13; http://dx.doi.org/10.1083/jcb.201207072; PMID: 23027899
- Nikolova V, Leimena C, McMahon AC, Tan JC, Chandar S, Jogia D, Kesteven SH, Michalicek J, Otway R, Verheyen F, et al. Defects in nuclear structure and function promote dilated cardiomyopathy in lamin A/C-deficient mice. J Clin Invest 2004; 113:357 - 69; http://dx.doi.org/10.1172/JCI200419448; PMID: 14755333
- Worman HJ, Courvalin JC. How do mutations in lamins A and C cause disease?. J Clin Invest 2004; 113:349 - 51; http://dx.doi.org/10.1172/JCI20832; PMID: 14755330
- Dahl KN, Scaffidi P, Islam MF, Yodh AG, Wilson KL, Misteli T. Distinct structural and mechanical properties of the nuclear lamina in Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A 2006; 103:10271 - 6; http://dx.doi.org/10.1073/pnas.0601058103; PMID: 16801550
- Lammerding J, Schulze PC, Takahashi T, Kozlov S, Sullivan T, Kamm RD, Stewart CL, Lee RT. Lamin A/C deficiency causes defective nuclear mechanics and mechanotransduction. J Clin Invest 2004; 113:370 - 8; http://dx.doi.org/10.1172/JCI200419670; PMID: 14755334
- Shumaker DK, Dechat T, Kohlmaier A, Adam SA, Bozovsky MR, Erdos MR, Eriksson M, Goldman AE, Khuon S, Collins FS, et al. Mutant nuclear lamin A leads to progressive alterations of epigenetic control in premature aging. Proc Natl Acad Sci U S A 2006; 103:8703 - 8; http://dx.doi.org/10.1073/pnas.0602569103; PMID: 16738054
- Capell BC, Collins FS. Human laminopathies: nuclei gone genetically awry. Nat Rev Genet 2006; 7:940 - 52; http://dx.doi.org/10.1038/nrg1906; PMID: 17139325
- McCord RP, Nazario-Toole A, Zhang H, Chines PS, Zhan Y, Erdos MR, Collins FS, Dekker J, Cao K. Correlated alterations in genome organization, histone methylation, and DNA-lamin A/C interactions in Hutchinson-Gilford progeria syndrome. Genome Res 2013; 23:260 - 9; http://dx.doi.org/10.1101/gr.138032.112; PMID: 23152449
- Ly DH, Lockhart DJ, Lerner RA, Schultz PG. Mitotic misregulation and human aging. Science 2000; 287:2486 - 92; http://dx.doi.org/10.1126/science.287.5462.2486; PMID: 10741968
- Marji J, O’Donoghue SI, McClintock D, Satagopam VP, Schneider R, Ratner D, Worman HJ, Gordon LB, Djabali K. Defective lamin A-Rb signaling in Hutchinson-Gilford Progeria Syndrome and reversal by farnesyltransferase inhibition. PLoS One 2010; 5:e11132; http://dx.doi.org/10.1371/journal.pone.0011132; PMID: 20559568
- Csoka AB, English SB, Simkevich CP, Ginzinger DG, Butte AJ, Schatten GP, Rothman FG, Sedivy JM. Genome-scale expression profiling of Hutchinson-Gilford progeria syndrome reveals widespread transcriptional misregulation leading to mesodermal/mesenchymal defects and accelerated atherosclerosis. Aging Cell 2004; 3:235 - 43; http://dx.doi.org/10.1111/j.1474-9728.2004.00105.x; PMID: 15268757
- Halaschek-Wiener J, Brooks-Wilson A. Progeria of stem cells: stem cell exhaustion in Hutchinson-Gilford progeria syndrome. J Gerontol A Biol Sci Med Sci 2007; 62:3 - 8; http://dx.doi.org/10.1093/gerona/62.1.3; PMID: 17301031
- Wilson KL, Foisner R. Lamin-binding Proteins. Cold Spring Harb Perspect Biol 2010; 2:a000554; http://dx.doi.org/10.1101/cshperspect.a000554; PMID: 20452940
- Goldman RD, Shumaker DK, Erdos MR, Eriksson M, Goldman AE, Gordon LB, Gruenbaum Y, Khuon S, Mendez M, Varga R, et al. Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A 2004; 101:8963 - 8; http://dx.doi.org/10.1073/pnas.0402943101; PMID: 15184648
- Kubben N, Voncken JW, Demmers J, Calis C, van Almen G, Pinto Y, Misteli T. Identification of differential protein interactors of lamin A and progerin. Nucleus 2010; 1:513 - 25; PMID: 21327095
- Zhang H, Kieckhaefer JE, Cao K. Mouse models of laminopathies. Aging Cell 2013; 12:2 - 10; http://dx.doi.org/10.1111/acel.12021; PMID: 23095062
- Berk JM, Maitra S, Dawdy AW, Shabanowitz J, Hunt DF, Wilson KL. O-Linked β-N-acetylglucosamine (O-GlcNAc) regulates emerin binding to barrier to autointegration factor (BAF) in a chromatin- and lamin B-enriched “niche”. J Biol Chem 2013; 288:30192 - 209; http://dx.doi.org/10.1074/jbc.M113.503060; PMID: 24014020
- Gruenbaum Y, Lee KK, Liu J, Cohen M, Wilson KL. The expression, lamin-dependent localization and RNAi depletion phenotype for emerin in C. elegans. J Cell Sci 2002; 115:923 - 9; PMID: 11870211
- Holaska JM, Wilson KL. Multiple roles for emerin: implications for Emery-Dreifuss muscular dystrophy. Anat Rec A Discov Mol Cell Evol Biol 2006; 288:676 - 80; http://dx.doi.org/10.1002/ar.a.20334; PMID: 16761279
- Stuurman N, Heins S, Aebi U. Nuclear lamins: their structure, assembly, and interactions. J Struct Biol 1998; 122:42 - 66; http://dx.doi.org/10.1006/jsbi.1998.3987; PMID: 9724605
- Vlcek S, Foisner R. Lamins and lamin-associated proteins in aging and disease. Curr Opin Cell Biol 2007; 19:298 - 304; http://dx.doi.org/10.1016/j.ceb.2007.04.001; PMID: 17466505
- Eriksson JE, Dechat T, Grin B, Helfand B, Mendez M, Pallari HM, Goldman RD. Introducing intermediate filaments: from discovery to disease. J Clin Invest 2009; 119:1763 - 71; http://dx.doi.org/10.1172/JCI38339; PMID: 19587451
- Holtz D, Tanaka RA, Hartwig J, McKeon F. The CaaX motif of lamin A functions in conjunction with the nuclear localization signal to target assembly to the nuclear envelope. Cell 1989; 59:969 - 77; http://dx.doi.org/10.1016/0092-8674(89)90753-8; PMID: 2557160
- Yang SH, Bergo MO, Toth JI, Qiao X, Hu Y, Sandoval S, Meta M, Bendale P, Gelb MH, Young SG, et al. Blocking protein farnesyltransferase improves nuclear blebbing in mouse fibroblasts with a targeted Hutchinson-Gilford progeria syndrome mutation. Proc Natl Acad Sci U S A 2005; 102:10291 - 6; http://dx.doi.org/10.1073/pnas.0504641102; PMID: 16014412
- Shumaker DK, Lopez-Soler RI, Adam SA, Herrmann H, Moir RD, Spann TP, Goldman RD. Functions and dysfunctions of the nuclear lamin Ig-fold domain in nuclear assembly, growth, and Emery-Dreifuss muscular dystrophy. Proc Natl Acad Sci U S A 2005; 102:15494 - 9; http://dx.doi.org/10.1073/pnas.0507612102; PMID: 16227433
- Capell BC, Erdos MR, Madigan JP, Fiordalisi JJ, Varga R, Conneely KN, Gordon LB, Der CJ, Cox AD, Collins FS. Inhibiting farnesylation of progerin prevents the characteristic nuclear blebbing of Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A 2005; 102:12879 - 84; http://dx.doi.org/10.1073/pnas.0506001102; PMID: 16129833
- Gruenbaum Y, Margalit A, Goldman RD, Shumaker DK, Wilson KL. The nuclear lamina comes of age. Nat Rev Mol Cell Biol 2005; 6:21 - 31; http://dx.doi.org/10.1038/nrm1550; PMID: 15688064
- Gelb MH, Brunsveld L, Hrycyna CA, Michaelis S, Tamanoi F, Van Voorhis WC, Waldmann H. Therapeutic intervention based on protein prenylation and associated modifications. Nat Chem Biol 2006; 2:518 - 28; http://dx.doi.org/10.1038/nchembio818; PMID: 16983387
- Hennekes H, Nigg EA. The role of isoprenylation in membrane attachment of nuclear lamins. A single point mutation prevents proteolytic cleavage of the lamin A precursor and confers membrane binding properties. J Cell Sci 1994; 107:1019 - 29; PMID: 8056827
- Sinensky M, Fantle K, Trujillo M, McLain T, Kupfer A, Dalton M. The processing pathway of prelamin A. J Cell Sci 1994; 107:61 - 7; PMID: 8175923
- Boyartchuk VL, Ashby MN, Rine J. Modulation of Ras and a-factor function by carboxyl-terminal proteolysis. Science 1997; 275:1796 - 800; http://dx.doi.org/10.1126/science.275.5307.1796; PMID: 9065405
- Dai Q, Choy E, Chiu V, Romano J, Slivka SR, Steitz SA, Michaelis S, Philips MR. Mammalian prenylcysteine carboxyl methyltransferase is in the endoplasmic reticulum. J Biol Chem 1998; 273:15030 - 4; http://dx.doi.org/10.1074/jbc.273.24.15030; PMID: 9614111
- Wright LP, Philips MR. Thematic review series: lipid posttranslational modifications. CAAX modification and membrane targeting of Ras. J Lipid Res 2006; 47:883 - 91; http://dx.doi.org/10.1194/jlr.R600004-JLR200; PMID: 16543601
- Bergo MO, Gavino B, Ross J, Schmidt WK, Hong C, Kendall LV, Mohr A, Meta M, Genant H, Jiang Y, et al. Zmpste24 deficiency in mice causes spontaneous bone fractures, muscle weakness, and a prelamin A processing defect. Proc Natl Acad Sci U S A 2002; 99:13049 - 54; http://dx.doi.org/10.1073/pnas.192460799; PMID: 12235369
- Barrowman J, Hamblet C, Kane MS, Michaelis S. Requirements for efficient proteolytic cleavage of prelamin A by ZMPSTE24. PLoS One 2012; 7:e32120; http://dx.doi.org/10.1371/journal.pone.0032120; PMID: 22355414
- Barrowman J, Hamblet C, George CM, Michaelis S. Analysis of prelamin A biogenesis reveals the nucleus to be a CaaX processing compartment. Mol Biol Cell 2008; 19:5398 - 408; http://dx.doi.org/10.1091/mbc.E08-07-0704; PMID: 18923140
- Vlcek S, Foisner R. A-type lamin networks in light of laminopathic diseases. Biochim Biophys Acta 2007; 1773:661 - 74; http://dx.doi.org/10.1016/j.bbamcr.2006.07.002; PMID: 16934891
- Eriksson M, Brown WT, Gordon LB, Glynn MW, Singer J, Scott L, Erdos MR, Robbins CM, Moses TY, Berglund P, et al. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature 2003; 423:293 - 8; http://dx.doi.org/10.1038/nature01629; PMID: 12714972
- De Sandre-Giovannoli A, Bernard R, Cau P, Navarro C, Amiel J, Boccaccio I, Lyonnet S, Stewart CL, Munnich A, Le Merrer M, et al. Lamin a truncation in Hutchinson-Gilford progeria. Science 2003; 300:2055; http://dx.doi.org/10.1126/science.1084125; PMID: 12702809
- D’Apice MR, Tenconi R, Mammi I, van den Ende J, Novelli G. Paternal origin of LMNA mutations in Hutchinson-Gilford progeria. Clin Genet 2004; 65:52 - 4; http://dx.doi.org/10.1111/j..2004.00181.x; PMID: 15032975
- Cao K, Capell BC, Erdos MR, Djabali K, Collins FS. A lamin A protein isoform overexpressed in Hutchinson-Gilford progeria syndrome interferes with mitosis in progeria and normal cells. Proc Natl Acad Sci U S A 2007; 104:4949 - 54; http://dx.doi.org/10.1073/pnas.0611640104; PMID: 17360355
- Yang SH, Chang SY, Ren S, Wang Y, Andres DA, Spielmann HP, Fong LG, Young SG. Absence of progeria-like disease phenotypes in knock-in mice expressing a non-farnesylated version of progerin. Hum Mol Genet 2011; 20:436 - 44; http://dx.doi.org/10.1093/hmg/ddq490; PMID: 21088111
- Toth JI, Yang SH, Qiao X, Beigneux AP, Gelb MH, Moulson CL, Miner JH, Young SG, Fong LG. Blocking protein farnesyltransferase improves nuclear shape in fibroblasts from humans with progeroid syndromes. Proc Natl Acad Sci U S A 2005; 102:12873 - 8; http://dx.doi.org/10.1073/pnas.0505767102; PMID: 16129834
- Dechat T, Shimi T, Adam SA, Rusinol AE, Andres DA, Spielmann HP, Sinensky MS, Goldman RD. Alterations in mitosis and cell cycle progression caused by a mutant lamin A known to accelerate human aging. Proc Natl Acad Sci U S A 2007; 104:4955 - 60; http://dx.doi.org/10.1073/pnas.0700854104; PMID: 17360326
- Lee J, Lee HJ, Shin MK, Ryu WS. Versatile PCR-mediated insertion or deletion mutagenesis. Biotechniques 2004; 36:398 - 400; PMID: 15038153
- Nakamura N, Rabouille C, Watson R, Nilsson T, Hui N, Slusarewicz P, Kreis TE, Warren G. Characterization of a cis-Golgi matrix protein, GM130. J Cell Biol 1995; 131:1715 - 26; http://dx.doi.org/10.1083/jcb.131.6.1715; PMID: 8557739
- Munro S, Pelham HR. A C-terminal signal prevents secretion of luminal ER proteins. Cell 1987; 48:899 - 907; http://dx.doi.org/10.1016/0092-8674(87)90086-9; PMID: 3545499
- Yang SH, Meta M, Qiao X, Frost D, Bauch J, Coffinier C, Majumdar S, Bergo MO, Young SG, Fong LG. A farnesyltransferase inhibitor improves disease phenotypes in mice with a Hutchinson-Gilford progeria syndrome mutation. J Clin Invest 2006; 116:2115 - 21; http://dx.doi.org/10.1172/JCI28968; PMID: 16862216
- Yang SH, Andres DA, Spielmann HP, Young SG, Fong LG. Progerin elicits disease phenotypes of progeria in mice whether or not it is farnesylated. J Clin Invest 2008; 118:3291 - 300; http://dx.doi.org/10.1172/JCI35876; PMID: 18769635
- Capell BC, Olive M, Erdos MR, Cao K, Faddah DA, Tavarez UL, Conneely KN, Qu X, San H, Ganesh SK, et al. A farnesyltransferase inhibitor prevents both the onset and late progression of cardiovascular disease in a progeria mouse model. Proc Natl Acad Sci U S A 2008; 105:15902 - 7; http://dx.doi.org/10.1073/pnas.0807840105; PMID: 18838683
- Vaughan A, Alvarez-Reyes M, Bridger JM, Broers JL, Ramaekers FC, Wehnert M, Morris GE, Hutchison CJ, Whitfield WGF. Both emerin and lamin C depend on lamin A for localization at the nuclear envelope. J Cell Sci 2001; 114:2577 - 90; PMID: 11683386
- Astejada MN, Goto K, Nagano A, Ura S, Noguchi S, Nonaka I, Nishino I, Hayashi YK. Emerinopathy and laminopathy clinical, pathological and molecular features of muscular dystrophy with nuclear envelopathy in Japan. Acta Myol 2007; 26:159 - 64; PMID: 18646565
- Lee KK, Haraguchi T, Lee RS, Koujin T, Hiraoka Y, Wilson KL. Distinct functional domains in emerin bind lamin A and DNA-bridging protein BAF. J Cell Sci 2001; 114:4567 - 73; PMID: 11792821
- Mislow JM, Holaska JM, Kim MS, Lee KK, Segura-Totten M, Wilson KL, McNally EM. Nesprin-1alpha self-associates and binds directly to emerin and lamin A in vitro. FEBS Lett 2002; 525:135 - 40; http://dx.doi.org/10.1016/S0014-5793(02)03105-8; PMID: 12163176
- Ostlund C, Ellenberg J, Hallberg E, Lippincott-Schwartz J, Worman HJ. Intracellular trafficking of emerin, the Emery-Dreifuss muscular dystrophy protein. J Cell Sci 1999; 112:1709 - 19; PMID: 10318763
- Clements L, Manilal S, Love DR, Morris GE. Direct interaction between emerin and lamin A. Biochem Biophys Res Commun 2000; 267:709 - 14; http://dx.doi.org/10.1006/bbrc.1999.2023; PMID: 10673356
- Ho CY, Jaalouk DE, Vartiainen MK, Lammerding J. Lamin A/C and emerin regulate MKL1-SRF activity by modulating actin dynamics. Nature 2013; 497:507 - 11; http://dx.doi.org/10.1038/nature12105; PMID: 23644458