Abstract
The discovery of a stem cell population in human neoplasias has given a new impulse to the study of the origins of cancer. The tissue compartment in which transformation first occurs likely comprises stem cells, since these cells need to consolidate the short-term and long-term requisites of tissue renewal. Because of their unique role, stem cells have a combination of characteristics that makes them susceptible to genetic damage, transformation, and tumor initiation. One type of genetic damage in particular, chromosomal instability, might affect the stem cell compartment, because it induces an ongoing cycle of DNA damage and alters cellular programming. Here, we will discuss some of the recently described links between SC, chromosomal instability, and carcinogenesis, and outline some of the consequences for oncoimmunology.
Keywords: :
Introduction
The complexity of malignant neoplasias can be appreciated with the recent identification of an intra-tumoral hierarchy and cancer stem cells (CSC) in different types of cancer. In the CSC hypothesis, the terms initiation and propagation are used to assign a function to cell types identified inside established tumors, but these terms do not necessarily refer to the cells from which the tumor originates. A few decades ago, the possibility that cancer arises from a dedifferentiation of mature cells still was under discussion.Citation1 The characteristics of cancer described in the last years indicate that neoplasias can be considered a true stem cell (SC) disease.Citation2 Whereas initial CSC theories proposed that “primitive” (embryonic) cells could migrate throughout the body, the identification of tissue-specific (somatic) SC has provided a more satisfactory explanation for the origin of cancer.Citation1
Somatic SC have to adjust their proliferation to the need for renewal of the tissue in which they reside. In addition, SC need to safeguard their own health and genetic integrity, in order to carry out their task during the entire life of the organism. SC divide asymmetrically, so each SC produces a new SC and a cell destined for differentation termed progenitor.Citation3 Since the genetic integrity of cells is largely determined by the number of divisions,Citation4 the number of SC divisions is kept as low as possible; progenitor cells can undergo several divisions which amplify the cell population destined for differentiation and tissue renewal.Citation4
In addition to a control over the number of divisions, genetic integrity itself is monitored.Citation5 Chromosome damage, for example radiation-induced double strand breaks, inhibits replication of all cell types including SC.Citation6 In order to prevent genetic defects, chromosomal damage of a big enough magnitude will cause the affected cells to enter in apoptosis.Citation5,Citation6 Apoptotic control in SC however cannot be too strict, since this might eliminate the self-renewing population and endanger tissue maintenance in the future. SC thus have the difficult task to balance the level of proliferation for short-term and long-term tissue stability.
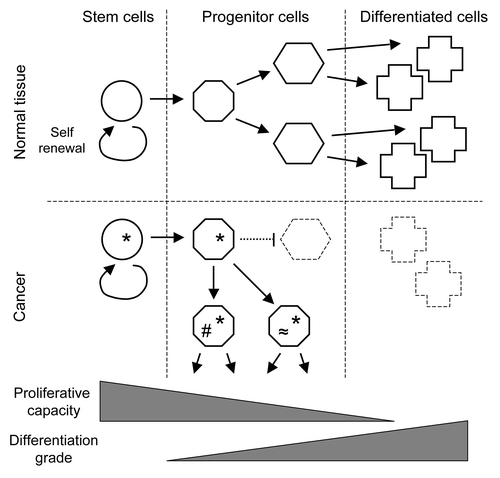
Most models that propose a SC basis for cancer include a partial blockage of differentiation; an imbalance results when the rate of proliferation exceeds the rate of differentiation and cell loss. If this situation persist, the transient accumulation of stem or progenitor cells might facilitate the acquisition of genetic changes, cause a further loss of control over proliferation, and ultimately lead to cancer (). Although the CSC theory is well on its way of being generally accepted, many details still are a matter of debate. In addition, work on integration of the CSC theory with other aspects of oncology, for example genetics and immunology, is just starting. Nonetheless, combining information from different theories might illustrate the problems SC encounter when preserving the equilibrium between short-term and long-term tissue requisites. Here, we will discuss possible connections between chromosomal instability and the SC theory of cancer, and briefly outline some of the consequences for oncoimmunology.
Figure 1. The role of stem cells in cancer. Whereas normal tissue homeostasis requires the coordination of the proliferation of stem and progenitor cells with their differentiation (above), cancer-inducing defects (*) are thought to inhibit stem cell differentiation (below). The resulting accumulation of progenitor cells with a residual capacity to proliferate allows for additional mutations (#, ≈), which promote further tumor growth and heterogeneity. Theories that propose the dedifferentiation of fully differentiated cells have lost importance in the last years.
Chromosomal Instability and Gene Dosage
Most sporadic tumors undergo a combination of numerical and structural changes. This combination of genetic defects is termed chromosomal instability (CIN), and is found in approximately 85% of non-hereditary carcinomas.Citation7,Citation8 Although CIN is most prominent in solid tumors, CIN has also been detected in leukemias recently.Citation9 Other types of genomic instability, such as microsattelite instability and the recently described chromothripsis,Citation10 are found only in a small percentage of clinical tumor samples. Even though CIN represents the most common form of genetic alterations in human cancer, the role of CIN in tumorigenesis has been a matter of debate for a long time. In contrast to basepair mutations in oncogenes and tumor suppressor genes, which can directly influence the activity of growth-regulating pathways,Citation11 the gain or loss of large chromosome fragments does not seem to stimulate cell replication. Moreover, since the relation between aneuploidy (involving whole chromosomes) and segmental alterations (dependent on chromosome breakage) has remained obscure until recently. Many oncologists therefore regarded CIN as a passenger phenotype for a long time.Citation8
The aspect of CIN that is studied in most detail, aneuploidy, can be induced experimentally in tissue culture cells, for example through a transient block of mitosis by spindle poisons. In the resulting mixed diploid and aneuploid population, the diploid cells outgrow their aneuploid counterparts, indicating that aneuploidy itself slows the proliferation rate of cells.Citation12,Citation13 In humans, additional copies of small chromosomes can be tolerated but is associated with developmental alterations such as Down or Edwards syndrome.Citation14,Citation15 Copy number changes of large chromosomes however are not compatible with mammalian development. The growth-inhibiting effect of chromosome number changes is further illustrated by gene mutations that induce aneuploidy; homozygous deletion of genes that control the mitotic checkpoint results in embryonic lethality in mice.Citation16,Citation17 Whereas treatments or mutations that induce chromosome copy number changes usually have a antiproliferative and therefore tumor suppressing effect, aneuploidy also can promote tumorigenesis. Haploinsufficiency of spindle checkpoint genes (the same genes that cause lethality when homozygously deleted) results in increased tumor susceptibility,Citation18,Citation19 and aneuploidy is detected before intragenic mutations in experimentally induced tumors.Citation20 Also mathematical models, which do not take into account potential effects on cellular viability, indicate that aneuploidy is an initiating step in tumorigenesis.Citation21
The other aspect of CIN is a combination of translocations, duplications, and deletions, known together as segmental chromosome defects. Until recently, the origin of segmental defects remained unknown. Under experimental conditions, chromosome breaks can be induced by stalling replication forks or imposing telomere erosion.Citation22,Citation23 However, these mechanisms give rise to their own particular kind of genetic alteration, which are detected only in a small proportion of tumor samples.Citation24,Citation25 Furthermore, the dominant theory of telomere erosionCitation23 is at odds with the CSC theory because telomere length distribution in a tissue places the most protected telomeres in SC.Citation26 Also in clinical tumors, telomere erosion probably plays an minor role.Citation27 Recent studies show that aneuploidy and segmental defects occur together in the majority of tumors, and probably are different aspects of a single phenomenon.Citation28 Like aneuploidy, segmental defects can be caused by mitotic spindle defects.Citation29 Furthermore, the observation that the most frequent alteration in CIN tumors are gains and losses of whole chromosomes and chromosome arms supports the idea that spindle defects cause the different genetic defects in chromosomal instability;Citation30 spindle defects not only result in missegregation of whole chromosomes (resulting in aneuploidy), but also generate enough force to shear centromeric DNA during mitosis (a phenomenon termed centromere fission) and cause chromosome breakage.Citation29 Chromosome breaks play an important role in tumor initiation and progression, because they are an obligatory intermediate in the breakage-fusion-bridge cycles that endow cancer cells with their genetic plasticity.Citation27 The localization of spindle-induced chromosome breaks, adjacent to the centromere, has profound effects on the fusion products that are formed in CIN tumors. Just as any break in the DNA, the cell will try to repair centromeric breaks as soon as possible. Rapid repair of breaks seems especially important in the SC compartment, in order to prevent loss of individual SC and to maintain the capacity for tissue renewal.Citation31 For this reason, at least some SC have a high activity of the non-homologous end-joining repair system.Citation32 Although this strategy prevents the accumulation of externally induced breaks, it also catalyzes the fusion of chromosome arms to the ends of receptor chromosomes.Citation33 This combination of the preferential repair pathway in SC and centromere fission is reflected in the number of whole arm translocations, which are one of the most common genetic alteration in carcinomas.Citation27 The fusion of chromosome arms can generate highly unstable dicentric chromosomes,Citation27 which produce random breaks and trigger breakage-fusion-bride cycles.Citation34 The fusion of chromosome arms to the end of receptor chromosomes can therefore promote ongoing instability and theoretically spread through the entire genome, which provides a mechanism of genetic plasticity in tumor progression.
Irrespective of the origin of the segmental defects and aneuploidy in CIN, the gain and loss of large genomic fragments is accompanied by dosage effects due to copy number changes in active genes (protein- and micro RNA-encoding genes). The overall effect of gene dosage on cell proliferation appears to be inhibitory; whole arm translocations of small chromosome arms, bearing fewer genes, are more frequent than those of large chromosome arms that harbor many genes.Citation24 Even the presence of an oncogene is not sufficient to break the trend of gene dosage, although it favors local translocations.Citation24,Citation25 The mechanisms by which gene dosage disturb cell proliferation are not exactly known; surplus protein produced in aneuploid cells apparently is degraded, because aneuploid cells are more sensitive to autophagy and protease inhibitors than diploid cells,Citation35 and augmented proteasomal protein degradation increases tolerance for aneuploidy in yeast.Citation36 Since excess proteins not incorporated in multiprotein complexes are especially susceptible to degradation, proteolysis has an important buffering effect on subunit ratios in protein complexes and stabilization of gene regulatory networks. Also feedback loops that control expression levels of subunits in multiprotein complexes contribute to the stability of regulatory networks.Citation37 When the various compensatory mechanisms cannot control gene dosage, a p53-dependent pathway is activated, resulting in cell cycle arrest and apoptosis.Citation38,Citation39 In conclusion, the gains and losses of large chromosomal segments characteristic for CIN do not directly favor an increased growth rate, but instead seem to be a burden for proliferating cancer cells.
In most tumors, aneuploidy is not a simple acquired genetic state but an ongoing process. Even though genes involved in control of the mitotic checkpoint rarely are mutated in cancer, CIN evolves from relatively simple patterns in low grade tumors to hyperploidy and complex translocations in high grade tumors.Citation30 The level of segmental defects and number of aneuploid chromosomes both correlate with the level of malignancy. The exact mechanisms that drive cancer cells to ever greater instability and malignancy are not exactly known, but a likely scenario is spill over of gene dosage on pathways that control cell proliferation and apoptosis. Gains and losses of whole chromosomes or large segments not only introduce copy number changes of genes, which can be partly buffered by regulatory pathways and protein turnover, but also changes the levels of miRNAs that affect the stability of large groups of mRNAs.Citation40 In contrast to intragenic mutations which generally affect a single regulatory network, aneuploidy-linked gene dosage has a general effect on cell physiology.
Do SC Tolerate CIN?
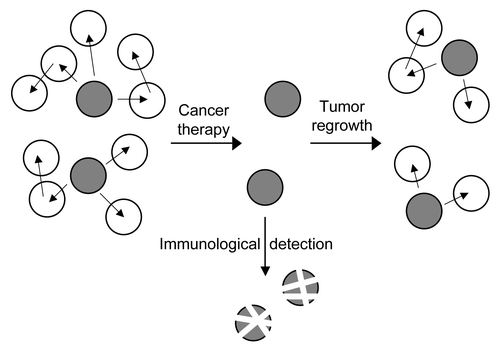
Because chromosomal instability itself reduces cellular fitness, the large percentage of tumors presenting this type of genetic defect seems paradoxical. Still, the most widely accpedted models include CSC that are more resistant to genetic damage than the surrounding tumor cells (). Whereas established tumors like have acquired tolerance for the gene dosage effects associated with aneuploidy, the initial steps in tumorigenesis however involve cells that yet have to acquire tolerance. Thus, the cells in which cancer first develops probably proliferate even in the presence of a low level of CIN, thereby allowing a certain gene dosage effect. This initial gene dosage promotes an adaptive response in the cell, and an ever increasing tolerance for gene dosage could drive a parallel increase in chromosomal instability, resistance to apoptosis, dedifferentiation, and malignancy. Since the tumor initiating cells in cancer are thought to belong to the SC compartment, could these cells have an intrinsic tolerance for chromosomal instability? Although the CSC theory implies that a normal SC acquires the initial genetic defects that trigger tumorigenesis, no data showing transformation of a SC in vivo is available yet.
Figure 2. CSC and cancer therapy. In heterogeneous primary tumors, more differentiated cells (white) have been shown to be susceptible to treatment, such as radiation or chemotherapy. CSC (gray) however are more resistant to currently used treatments, and might be responsible for tumor recurrence and ongoing chromosomal instability if not eliminated efficiently. Immunological strategies that directly target the CSC that are more resistant to classical therapies might thus help to ensure complete eradication of cancer cells.
Since the signals that connect gene dosage cell cycle arrest and apoptosis depend on p53,Citation38,Citation39 signaling of other pathways through p53 might yield clues regarding the resistance of SC against gene dosage. Notwithstanding the general view that p53 has an important role in apoptotic signaling, some studies report a protective effect of p53; whereas homozygous p53 mutation in hematopoietic SC increases resistance against high levels of radiation, p53 proficient hematopoietic SC are more resistant to low levels of radiation.Citation41 Also in a model for colon cancer, p53 positive cells were more resistant to the aneuploidy-inducing compound paclitaxel.Citation42 These data suggest that p53 allows for low level DNA damage without inducing apoptosis.
Although intuition dictates that the SC need most protection against genetic damage, the role of p53 in the SC compartment has remained unclear until recently. The asymmetric divisions that establish functionally different daughter cells are governed by the p53-stabilizing protein Numb.Citation43 Numb distributes unevenly over the daughter cells brought about by asymmetric divisions, but the first studies did not show if the protein was enriched in the self-renewing or differentiating cells.Citation44 Thus, also the faith of p53 in the two daughter cells remained unknown. Only recently, Numb was shown to accumulate in one of the daughter cells together with the SC marker CD133,Citation45 indicating that p53 is stabilized in the self-renewing population. In clinical tumors, expression of CD133 is indicative of a high resistance to radiationCitation46 and chemotherapy,Citation47 corroborating the hypothesis of resistant SC. The way in which p53 protects SC against genetic damage apparently involves the induction of a transient dormant state termed quiescence.Citation48 In contrast to other modes of cell cycle arrest, the correct stimuli can induce a rapid exit from quiescence and thereby allow SC to respond to the needs of the surrounding tissue.
An important effect of CIN, gene dosage, induces apoptosis through the p53-dependent pathways;Citation38,Citation39 since p53 protects against low levels of DNA damage through quiescence, it might also protect against some effects of gene dosage. Just as high doses of DNA damage, however, cells cannot be protected against high levels of gene dosage. Even though larger chromosomes bear larger centromeric regions and thus suffer more easily from whole arm translocations, large scale analysis of tumor samples has shown that smaller chromosome arms are involved in more translocations than bigger chromosome arms.Citation24 Probably, apoptotic pathways other than p53 respond to gene dosage, as forced differentiation induces apoptosis irrespective of p53 status.Citation49-Citation51 The tolerance for chromosome damage and gene dosage thus are determined by a combination of SC characteristics, some of which remain to be discovered.
SC Induction of Immune Tolerance
Although a regulatory ability has only recently been identified for SC, there is now ample evidence that SC can act as immunosuppressors. The precise mechanism by which these cells suppress the immune system remains to be elucidated, but they are thought to participate in several pathways: (1) in prevention of T cell activation and proliferation; (2) in apoptosis induction in type Th1 cells; (3) in inhibition of proinflammatory cytokine secretion.Citation52
SC-mediated immunosuppression was observed in a rat model of experimental autoimmune encephalomyelitis in which transplanted neural SC elicited an anti-inflammatory response, reducing brain inflammation and disease severity.Citation53 It is thought this is achieved by the triggering of neural SC-induced T cell apoptosis, which shifted the inflammatory process in the brain toward a more favorable Th2 response. In vitro studies demonstrated that mesenchymal SC are also immunosuppressive. Again in in vivo experiments, mesenchymal SC induced peripheral tolerance and migrated to injured tissues, where they inhibited release of proinflammatory cytokines and promoted the survival of damaged cells, preventing type 1 diabetes, graft-vs.-host disease, collagen-induced arthritis, and multiple sclerosis.Citation54 Hematopoietic SC also appear to have the ability to sense and respond to situations such as tissue damage, infection, and migration to sites of immune privilege, although in a distinct manner that involves recognition of the hematopoietic SC niche, its proximity to regulatory T cells, and tolerance induction.Citation55 These cells were shown to prevent diabetes in NOD mice,Citation56 and are currently undergoing evaluation in clinical trials. Umbilical cord blood SC facilitated the generation of regulatory T cells, thereby reversing hyperglycemia in NOD mice.
Despite limitations in our current knowledge, allo- and xenogeneic mesenchymal SC have been used experimentally to evade host immune response and to trigger immunosuppression, thus showing privilege in preventing rejection. This behavior might be explained by their ability to migrate rapidly to a damaged organ, contributing to clearance of damage following the release of immunomodulatory molecules.Citation54
Concluding Remarks
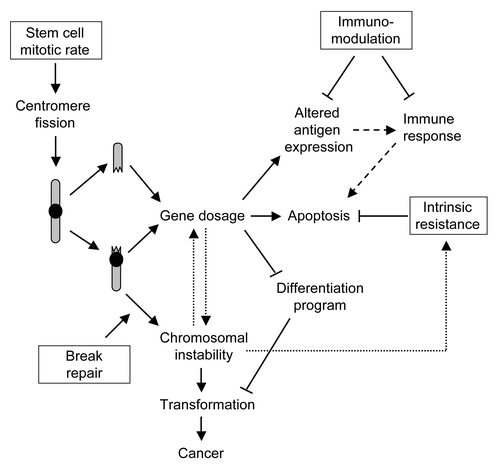
In the SC model, asymmetric divisions allocate one daughter cell for self renewal, and the other for differentiation and tissue repair. The maintenance of tissues by SC usually includes an intermediate population of progenitor cells, which remain some proliferative capacity until differentiation is complete.Citation2 Since centromere fission comprises two arms of a chromosome that are connected to opposite spindle poles and segregated into different daughter cells,Citation27 a single centromere fission causes a genetic alteration in a cell dedicated to self renewal and in a cell destined for differentiation. In practice, it might be difficult (and unnecessary) to distinguish between a “true” SC and a progenitor cell blocked early in differentiation. Although the segregation of chromosome arms into two daughter cells renders the question of stem or progenitor cells less relevant, it favors the theory of a blocked differentiation over the dedifferentiation of mature cells. In order to perform their function in tissue renewal, SC have a unique combination of high proliferative capacity, high tolerance for gene dosage, and high break repair activity (). SC also show immune-suppresive effects, which target antigen presentation by dendritic cells, natural killer-cell cytotoxicity, the activation of neutrophils and the acquired immune response by inducing T-cell peripheral tolerance, in addition to a potent tissue-protective effect through the release of anti-inflammatory, anti-apoptotic and trophic molecules. Together with the ability of SC to evade immune response and to regulate effector functions of innate immune cells,Citation57 the genetic plasticity of CSC raises serious dificulties in developing appropriate therapeutical tools.
Figure 3. Stem cell characteristics favor tumor development. A combination of stem cell properties (boxes), essential for their role in tissue renewal, contributes to the long-term accumulation of cells bearing genetic defects (arrows). A sustained mitotic rate in combination with highly active break repair and resistance to apoptosis are important to avoid stem cell depletion. These factors however promote chromosomal instability after accidental breakage and fusion of centromere-containing chromosome arms, resulting in the survival of genetically imbalanced cells. Elimination of cancer cells by the immune system (dashed arrows) might be ineffective as a consequence of stem cell immunosuppressive capacity. Once chromosomal instability is initiated, it promotes a self-amplifying cycle of gene dosage and reduced apoptosis (dotted arrows) through the duplication and deletion of genomic segments.
Even though the basic principles of CSC are only just becoming clear, the consequences of the CSC theory are for related fields of study are evident. Since SC are a likely origin of cancer, CSC represent a distinct cellular population within tumors. The identification of stemness markers such as CD133Citation45,Citation46 might help to develop combined treatments to eradicate the cells that are more resistant to chemo- and radiotherapy. Already, some data are available on drugs that target chromosomal instability and gene dosage.Citation35 Specific targeting of these therapeutic compounds to CSC might increase efficiency and reduce secondary effects.Citation58 The immunosuppressive effects of SC however add an additional dimension to the CSC theory, which might present a mayor challenge for oncoimmunologists. In conclusion, linking the CSC theory to other disciplines can help to develop useful strategies for the combat of cancer.
| Abbreviations: | ||
| CIN | = | chromosomal instability |
| CSC | = | cancer stem cells |
| SC | = | stem cells |
Acknowledgments
The authors thank Catherine Mark for editorial assistance. This work is financed by grants PS09/00572 and RD08/0075/0010 (Fondo de Investigación en Salud), BFU 2009-08395-E, SAF2010-21205, and PIB2010BZ-00564 (Ministerio de Ciencia y Investigación). KvW is supported by a JAE-doc fellowship from the Spanish National Research Council (CSIC). The Department of Immunology and Oncology was founded and is supported by the CSIC and by Pfizer.
References
- Sell S. Cellular origin of cancer: dedifferentiation or stem cell maturation arrest?. Environ Health Perspect 1993; 101:Suppl 5 15 - 26; PMID: 7516873
- Sell S. Stem cell origin of cancer and differentiation therapy. Crit Rev Oncol Hematol 2004; 51:1 - 28; http://dx.doi.org/10.1016/j.critrevonc.2004.04.007; PMID: 15207251
- Bergmann A, Steller H. Apoptosis, stem cells, and tissue regeneration. Sci Signal 2010; 3:re8; http://dx.doi.org/10.1126/scisignal.3145re8; PMID: 20978240
- Ruzankina Y, Asare A, Brown EJ. Replicative stress, stem cells and aging. Mech Ageing Dev 2008; 129:460 - 6; http://dx.doi.org/10.1016/j.mad.2008.03.009; PMID: 18462780
- Canman CE, Chen CY, Lee MH, Kastan MB. DNA damage responses: p53 induction, cell cycle perturbations, and apoptosis. Cold Spring Harb Symp Quant Biol 1994; 59:277 - 86; PMID: 7587079
- Park Y, Gerson SL. DNA repair defects in stem cell function and aging. Annu Rev Med 2005; 56:495 - 508; http://dx.doi.org/10.1146/annurev.med.56.082103.104546; PMID: 15660524
- Draviam VM, Xie S, Sorger PK. Chromosome segregation and genomic stability. Curr Opin Genet Dev 2004; 14:120 - 5; http://dx.doi.org/10.1016/j.gde.2004.02.007; PMID: 15196457
- Pihan G, Doxsey SJ. Mutations and aneuploidy: co-conspirators in cancer?. Cancer Cell 2003; 4:89 - 94; http://dx.doi.org/10.1016/S1535-6108(03)00195-8; PMID: 12957283
- Talamo A, Chalandon Y, Marazzi A, Jotterand M. Clonal heterogeneity and chromosomal instability at disease presentation in high hyperdiploid acute lymphoblastic leukemia. Cancer Genet Cytogenet 2010; 203:209 - 14; http://dx.doi.org/10.1016/j.cancergencyto.2010.09.005; PMID: 21156235
- Stephens PJ, Greenman CD, Fu B, Yang F, Bignell GR, Mudie LJ, et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 2011; 144:27 - 40; http://dx.doi.org/10.1016/j.cell.2010.11.055; PMID: 21215367
- Doppler W, Jaggi R, Groner B. Induction of v-mos and activated Ha-ras oncogene expression in quiescent NIH 3T3 cells causes intracellular alkalinisation and cell-cycle progression. Gene 1987; 54:147 - 53; http://dx.doi.org/10.1016/0378-1119(87)90357-X; PMID: 3038697
- Thompson SL, Compton DA. Examining the link between chromosomal instability and aneuploidy in human cells. J Cell Biol 2008; 180:665 - 72; http://dx.doi.org/10.1083/jcb.200712029; PMID: 18283116
- Williams BR, Prabhu VR, Hunter KE, Glazier CM, Whittaker CA, Housman DE, et al. Aneuploidy affects proliferation and spontaneous immortalization in mammalian cells. Science 2008; 322:703 - 9; http://dx.doi.org/10.1126/science.1160058; PMID: 18974345
- Carr DH. The chromosome abnormality in mongolism. Can Med Assoc J 1962; 87:490 - 5; PMID: 13876803
- Tolksdorf M, Lehmann W, Hansen HG, Wiedemann HR. Edwards Syndrome with Unusual Chromosome Findings. Z Kinderheilkd 1965; 93:55 - 63; http://dx.doi.org/10.1007/BF00438567; PMID: 14322784
- Burds AA, Lutum AS, Sorger PK. Generating chromosome instability through the simultaneous deletion of Mad2 and p53. Proc Natl Acad Sci USA 2005; 102:11296 - 301; http://dx.doi.org/10.1073/pnas.0505053102; PMID: 16055552
- Kalitsis P, Earle E, Fowler KJ, Choo KH. Bub3 gene disruption in mice reveals essential mitotic spindle checkpoint function during early embryogenesis. Genes Dev 2000; 14:2277 - 82; http://dx.doi.org/10.1101/gad.827500; PMID: 10995385
- Dai W, Wang Q, Liu T, Swamy M, Fang Y, Xie S, et al. Slippage of mitotic arrest and enhanced tumor development in mice with BubR1 haploinsufficiency. Cancer Res 2004; 64:440 - 5; http://dx.doi.org/10.1158/0008-5472.CAN-03-3119; PMID: 14744753
- Weaver BA, Cleveland DW. Aneuploidy: instigator and inhibitor of tumorigenesis. Cancer Res 2007; 67:10103 - 5; http://dx.doi.org/10.1158/0008-5472.CAN-07-2266; PMID: 17974949
- Duesberg P, Li R, Rasnick D, Rausch C, Willer A, Kraemer A, et al. Aneuploidy precedes and segregates with chemical carcinogenesis. Cancer Genet Cytogenet 2000; 119:83 - 93; http://dx.doi.org/10.1016/S0165-4608(99)00236-8; PMID: 10867141
- Nowak MA, Komarova NL, Sengupta A, Jallepalli PV, Shih Ie M, Vogelstein B, et al. The role of chromosomal instability in tumor initiation. Proc Natl Acad Sci USA 2002; 99:16226 - 31; http://dx.doi.org/10.1073/pnas.202617399; PMID: 12446840
- Arlt MF, Casper AM, Glover TW. Common fragile sites. Cytogenet Genome Res 2003; 100:92 - 100; http://dx.doi.org/10.1159/000072843; PMID: 14526169
- Artandi SE, DePinho RA. Telomeres and telomerase in cancer. Carcinogenesis 2010; 31:9 - 18; http://dx.doi.org/10.1093/carcin/bgp268; PMID: 19887512
- Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, et al. The landscape of somatic copy-number alteration across human cancers. Nature 2010; 463:899 - 905; http://dx.doi.org/10.1038/nature08822; PMID: 20164920
- Bignell GR, Greenman CD, Davies H, Butler AP, Edkins S, Andrews JM, et al. Signatures of mutation and selection in the cancer genome. Nature 2010; 463:893 - 8; http://dx.doi.org/10.1038/nature08768; PMID: 20164919
- Flores I, Canela A, Vera E, Tejera A, Cotsarelis G, Blasco MA. The longest telomeres: a general signature of adult stem cell compartments. Genes Dev 2008; 22:654 - 67; http://dx.doi.org/10.1101/gad.451008; PMID: 18283121
- Martínez-A C, van Wely KH. Centromere fission, not telomere erosion, triggers chromosomal instability in human carcinomas. Carcinogenesis 2011; 32:796 - 803; http://dx.doi.org/10.1093/carcin/bgr069; PMID: 21478459
- Geigl JB, Obenauf AC, Schwarzbraun T, Speicher MR. Defining 'chromosomal instability'. Trends Genet 2008; 24:64 - 9; http://dx.doi.org/10.1016/j.tig.2007.11.006; PMID: 18192061
- Guerrero AA, Cano Gamero M, Trachana V, Futterer A, Pacios-Bras C, Panadero Diaz-Concha N, et al. Centromere-localized breaks indicate the generation of DNA damage by the mitotic spindle. Proc Natl Acad Sci USA 2010; 107:4159 - 64; http://dx.doi.org/10.1073/pnas.0912143106; PMID: 20142474
- Martínez-A C, van Wely KH. Are aneuploidy and chromosome breakage caused by a CINgle mechanism?. Cell Cycle 2010; 9:2275 - 80; http://dx.doi.org/10.4161/cc.9.12.11865; PMID: 20519949
- Mathews LA, Cabarcas SM, Farrar WL. DNA repair: the culprit for tumor-initiating cell survival?. Cancer Metastasis Rev 2011; 30:185 - 97; http://dx.doi.org/10.1007/s10555-011-9277-0; PMID: 21340664
- Sotiropoulou PA, Candi A, Mascre G, De Clercq S, Youssef KK, Lapouge G, et al. Bcl-2 and accelerated DNA repair mediates resistance of hair follicle bulge stem cells to DNA-damage-induced cell death. Nat Cell Biol 2010; 12:572 - 82; http://dx.doi.org/10.1038/ncb2059; PMID: 20473297
- Guerrero AA, Martinez AC, van Wely KH. Merotelic attachments and non-homologous end joining are the basis of chromosomal instability. Cell Div 2010; 5:13; http://dx.doi.org/10.1186/1747-1028-5-13; PMID: 20478024
- McClintock B. The Stability of Broken Ends of Chromosomes in Zea Mays. Genetics 1941; 26:234 - 82; PMID: 17247004
- Tang YC, Williams BR, Siegel JJ, Amon A. Identification of aneuploidy-selective antiproliferation compounds. Cell 2011; 144:499 - 512; http://dx.doi.org/10.1016/j.cell.2011.01.017; PMID: 21315436
- Torres EM, Dephoure N, Panneerselvam A, Tucker CM, Whittaker CA, Gygi SP, et al. Identification of aneuploidy-tolerating mutations. Cell 2010; 143:71 - 83; http://dx.doi.org/10.1016/j.cell.2010.08.038; PMID: 20850176
- Bekaert M, Conant GC. Transcriptional robustness and protein interactions are associated in yeast. BMC Syst Biol 2011; 5:62; http://dx.doi.org/10.1186/1752-0509-5-62; PMID: 21545728
- Li M, Fang X, Baker DJ, Guo L, Gao X, Wei Z, et al. The ATM-p53 pathway suppresses aneuploidy-induced tumorigenesis. Proc Natl Acad Sci USA 2010; 107:14188 - 93; http://dx.doi.org/10.1073/pnas.1005960107; PMID: 20663956
- Thompson SL, Compton DA. Proliferation of aneuploid human cells is limited by a p53-dependent mechanism. J Cell Biol 2010; 188:369 - 81; http://dx.doi.org/10.1083/jcb.200905057; PMID: 20123995
- Chaubey A, Karanti S, Rai D, Oh T, Adhvaryu SG, Aguiar RC. MicroRNAs and deletion of the derivative chromosome 9 in chronic myeloid leukemia. Leukemia 2009; 23:186 - 8; http://dx.doi.org/10.1038/leu.2008.154; PMID: 18548097
- Hirabayashi Y, Matsuda M, Matumura T, Mitsui H, Sasaki H, Tukada T, et al. The p53-deficient hemopoietic stem cells: their resistance to radiation-apoptosis, but lasted transiently. Leukemia 1997; 11:Suppl 3 489 - 92; PMID: 9209435
- Rakovitch E, Mellado W, Hall EJ, Pandita TK, Sawant S, Geard CR. Paclitaxel sensitivity correlates with p53 status and DNA fragmentation, but not G2/M accumulation. Int J Radiat Oncol Biol Phys 1999; 44:1119 - 24; http://dx.doi.org/10.1016/S0360-3016(99)00109-1; PMID: 10421546
- Colaluca IN, Tosoni D, Nuciforo P, Senic-Matuglia F, Galimberti V, Viale G, et al. NUMB controls p53 tumour suppressor activity. Nature 2008; 451:76 - 80; http://dx.doi.org/10.1038/nature06412; PMID: 18172499
- Pece S, Confalonieri S, R Romano P, Di Fiore PP. . NUMB-ing down cancer by more than just a NOTCH. Biochim Biophys Acta2011; 1815:26-43.
- Lathia JD, Hitomi M, Gallagher J, Gadani SP, Adkins J, Vasanji A, et al. Distribution of CD133 reveals glioma stem cells self-renew through symmetric and asymmetric cell divisions. Cell Death Dis 2011; 2:e200; http://dx.doi.org/10.1038/cddis.2011.80; PMID: 21881602
- He J, Shan Z, Li L, Liu F, Liu Z, Song M, et al. Expression of glioma stem cell marker CD133 and O6-methylguanine-DNA methyltransferase is associated with resistance to radiotherapy in gliomas. Oncol Rep 2011; 26:1305 - 13; PMID: 21769436
- Suetsugu A, Osawa Y, Nagaki M, Moriwaki H, Saji S, Bouvet M, et al. Simultaneous color-coded imaging to distinguish cancer “stem-like” and non-stem cells in the same tumor. J Cell Biochem 2010; 111:1035 - 41; http://dx.doi.org/10.1002/jcb.22792; PMID: 20672309
- Li L, Bhatia R. Stem cell quiescence. Clin Cancer Res 2011; 17:4936 - 41; http://dx.doi.org/10.1158/1078-0432.CCR-10-1499; PMID: 21593194
- Wallington LA, Bunce CM, Durham J, Brown G. Particular combinations of signals, by retinoic acid and 1 alpha, 25 dihydroxyvitamin D3, promote apoptosis of HL60 cells. Leukemia 1995; 9:1185 - 90; PMID: 7630194
- Lee MK, Hande MP, Sabapathy K. Ectopic mTERT expression in mouse embryonic stem cells does not affect differentiation but confers resistance to differentiation- and stress-induced p53-dependent apoptosis. J Cell Sci 2005; 118:819 - 29; http://dx.doi.org/10.1242/jcs.01673; PMID: 15687103
- Campos B, Wan F, Farhadi M, Ernst A, Zeppernick F, Tagscherer KE, et al. Differentiation therapy exerts antitumor effects on stem-like glioma cells. Clin Cancer Res 2010; 16:2715 - 28; http://dx.doi.org/10.1158/1078-0432.CCR-09-1800; PMID: 20442299
- Jones BJ, McTaggart SJ. Immunosuppression by mesenchymal stromal cells: from culture to clinic. Exp Hematol 2008; 36:733 - 41; http://dx.doi.org/10.1016/j.exphem.2008.03.006; PMID: 18474304
- Einstein O, Ben-Hur T. The changing face of neural stem cell therapy in neurologic diseases. Arch Neurol 2008; 65:452 - 6; http://dx.doi.org/10.1001/archneur.65.4.452; PMID: 18413466
- Uccelli A, Moretta L, Pistoia V. Mesenchymal stem cells in health and disease. Nat Rev Immunol 2008; 8:726 - 36; http://dx.doi.org/10.1038/nri2395; PMID: 19172693
- Massberg S, Schaerli P, Knezevic-Maramica I, Kollnberger M, Tubo N, Moseman EA, et al. Immunosurveillance by hematopoietic progenitor cells trafficking through blood, lymph, and peripheral tissues. Cell 2007; 131:994 - 1008; http://dx.doi.org/10.1016/j.cell.2007.09.047; PMID: 18045540
- Fiorina P, Voltarelli J, Zavazava N. . Immunological Applications of Stem Cells in Type 1 Diabetes. Endocr Rev2011.
- Marigo I, Dazzi F.. The immunomodulatory properties of mesenchymal stem cells. Semin Immunopathol2011.
- Damek-Poprawa M, Volgina A, Korostoff J, Sollecito TP, Brose MS, O'Malley BW Jr., et al. Targeted inhibition of CD133+ cells in oral cancer cell lines. J Dent Res 2011; 90:638 - 45; http://dx.doi.org/10.1177/0022034510393511; PMID: 21220361