Abstract
Tumors arise and progress in immunocompetent hosts presumably by activating tolerance mechanisms critical for normal homeostasis. Host immune cells can mount anti-tumor responses by activation of Toll-like receptors (TLRs). However, emerging data suggests that molecules that negatively regulate TLRs are exploited by tumors to induce tolerance and mitigate the host immunosurveillance. Targeting these negative regulators can be a potential new immunotherapeutic strategy.
As an important extrinsic tumor suppressive mechanism, immune system can identify and destroy nascent tumor cells through a process known as cancer immunosurveillance. However, tumors still arise in immune-competent hosts, after progressing through three distinct phases of a process known as immunoediting.Citation1 In the first phase termed elimination, which also encompasses immunosurveillance, the innate and adaptive immune systems work together to detect the presence of a developing tumor and destroy it. Malignant cell variants that survive the elimination phase enter the equilibrium phase, in which the adaptive immune system prevents outgrowth of tumor and sculpts the immunogenicity of the malignant cells. During the escape phase, malignant cells acquire the ability to circumvent immune recognition and manifest into visible tumors. Tumors do so by exploiting several different tolerance mechanisms, which are also employed by the host to maintain the normal immune homeostasis.Citation1
Host immune cells can launch anti-tumor responses through activation of cell surface receptors, including toll-like receptors (TLRs), which are capable of sensing exogenous and endogenous danger signals. To strike an immunological balance between activation and inhibition and to avoid triggering inappropriate inflammatory responses, the immune system tightly regulates TLR signaling through multiple negative regulatory mechanisms.Citation2 Molecules that negatively regulate TLR signaling may be exploited by tumors to induce immune tolerance and mitigate host immunosurveillance. Recently, we demonstrated that tumor cells induce the expression of IRAK-M, a negative regulator of TLR signaling, in tumor associated macrophages, promoting an immunosuppressive M2 phenotype. Tumor cell induced IRAK-M is mediated by the cytokine TGFβ, which serves as a key mechanism by which lung tumors may circumvent anti-tumor responses of macrophages promoting tumor immunotolerance.Citation3 In this article, we discuss the implications, potential role in immunoediting and prospects for IRAK-M and other negative regulators of TLR signaling for potential therapeutic targeting in oncoimmunology.
TLR Signaling and Tolerance for Tumors
Toll like receptors are critical components of innate immunity and are broadly distributed on cells of the immune system. TLRs are evolutionarily conserved to recognize molecular patterns associated with pathogens (PAMPs) such as bacterial lipopolysaccharides (LPS), hypomethylated DNA, flagellin, dsRNA. In addition, molecular patterns associated with tissue damage (danger associated molecular patterns or DAMPs) including heat shock proteins, high mobility group box proteins and dsDNA can directly activate TLRs.Citation4 TLR signaling is initiated by dimerization of TLRs, forming homodimers or heterodimers. All TLRs, with the exception of TLR3, recruit and utilize the adaptor protein MyD88 for signaling upon receptor activation. This allows the recruitment and activation of a family of kinases, namely IRAKs (IL-1 receptor-associated kinases) 1, 2 and 4. IRAK-4 is initially recruited to the complex, becomes activated, and then phosphorylates IRAK-1. These kinases interact with MyD88 through the death domains common to both proteins, resulting in a cascade of interactions culminating in the activation of further downstream kinases, including inhibitor of NFκB (IκB) kinases (IKKs). Activation of IκB releases NFκB, allowing NFκB translocation to the nucleus to mediate an increase in inflammatory cytokine gene expression.Citation4,Citation5 The specificity and diversity of TLR function is conferred in part by the selective interaction with the adaptor molecules. For example, the adaptor MAL is vital for both TLR2 and TLR4 activation of NFκB, whereas, TLR3 uses the adaptor TRIF to induce interferon-β (IFNβ) synthesis and TLR4 uses both TRIF and TRAM to activate the IRF-3 signaling pathway.Citation5 There are other signaling pathways that contribute to TLR function, such as Jun N-terminal kinase (JNK) and the mitogen-activated protein kinases (MAPKs). The relationship and interaction between these various signaling pathways is a major subject of interest in TLR biology.
In the tumor microenvironment, the exact trigger(s) of TLR signaling in host immune cells is not known. However, the tumor microenvironment is rich in molecules that can potentially activate TLR signaling to trigger anti-tumor responses. This includes heat shock proteins, high mobility group proteins, double stranded DNA from necrotic tumor cells, and hyaluronic acid.Citation6,Citation7 Tumors may activate the same negative regulatory mechanisms that are critical for normal homeostasis of the immune system, and induce immune tolerance to cancer cells. Dynamic interactions between cancer cells and tumor associated host immune cells initiate and maintain tumor immune tolerance which eventually predominates and overcomes effective host immune response.Citation7,Citation8 The nature of the interactions between cancer cells and immune cells, and the molecular mechanisms underlying tumor-induced immune tolerance is poorly understood. Even though a number of molecules that negatively regulate TLR signaling has been identified and been shown to play an important role in both limiting excessive inflammation as well as immune tolerance, their role in tumor immunology has just started to emerge.
Negative Regulators of TLR Signaling
Negative regulators of TLRs can selectively inhibit one or more TLRs or target common components of the TLR-signaling pathway that function to block the effects of the entire TLR family (for a comprehensive review see ref. Citation2). The selective use of negative regulators by different TLRs, sometimes in a tissue specific manner, might therefore contribute to the functional specificity and diversity of TLR responses. In order to attenuate signaling events downstream of TLRs, at least three levels of negative regulation have so far been reported. These include extracellular decoy receptors to intracellular inhibitors, and membrane-bound suppressors ().Citation2 In addition, several miRNAs have been identified as negative regulators of TLR signaling.Citation9 However, functional studies establishing their roles are lacking and for that reason these molecules will not be addressed in this article. Moreover, while a number of TLR inhibitors have been shown to promote a tolerance phenotype in monocyte/macrophage populations under certain experimental conditions, only IRAK-M has been directly linked to macrophage tolerance within the tumor microenvironment.
Figure 1. Negative regulation of Toll-like receptor signaling. Toll-like receptor (TLR) signaling pathways are tightly regulated in multiple different ways by several endogenous regulators to prevent hyperactivation. To attenuate the signaling from TLRs, at least three levels of negative regulation have so far been reported. These range from intracellular inhibitors to extracellular decoy receptors, and membrane-bound suppressors.
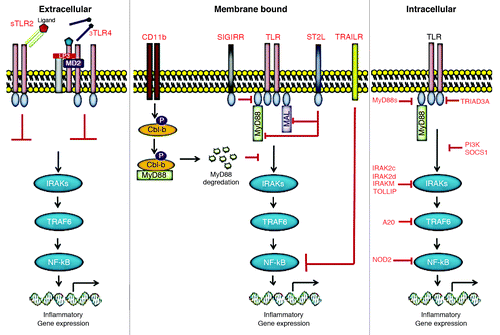
Extracellular decoy receptors
There are at least two soluble forms of TLRs (sTLRs) identified so far. These naturally produced decoy sTLRs may serve as first line of negative regulation to prevent overactivation of host response against microbial threat. Even though TLR4 and TLR2 are encoded by single distinct genes, multiple different mRNA products are detected for each in various mammalian hosts, including humans, indicating the presence of different isoforms. Recombinant form of sTLR4 inhibited LPS-induced NFκB activation and TNFα production by macrophages in vitro.Citation10 Similarly, sTLR2 isoforms, with molecular weights from 20–85 kDa, were detected in human milk and plasma, resulting from the post-translational modification of the transmembrane receptor.Citation11 Similarly, sTLR2 inhibited IL-8 and TNFα production by monocytes stimulated with the TLR2 agonist, bacterial lipopeptide. sTLR4 and sTLR2 may inhibit signaling by blocking the interaction between TLR4 or TLR-2 and other co-receptors, particularly MD2 and CD14 ().
Intracellular inhibitors
Intracellular molecules constitute the largest group of negative regulators that inhibit TLR signaling in both humans and mice. These molecules include short form of MyD88 (MyD88s), TRIAD3A, A20, NOD2, PI3K, SOCS1, TOLLIP, FADD, IRAK-2c, IRAK-2d and IRAK-M ().
Similar to the decoy sTLRs, an alternatively spliced MyD88s was identified that acts as a dominant negative inhibitor of MyD88. In contrast to the ubiquitous MyD88 expression, MyD88s was predominantly expressed in spleen and brain. MyD88s overexpression results in MyD88s-MyD88 heterodimers, rather than MyD88-MyD88 homodimers required for IRAK-4 recruitment. This prevents IRAK-1 phosphorylation by IRAK-4, resulting in the inhibition of MyD88-dependent signaling. Consequently, MyD88s inhibits LPS and IL-1 induced NFκB activation in vitro.Citation12,Citation13
TRIAD3A is a member of E3-ubiquitin ligases that promotes ubiquitylation and degradation of TLR4 and 9 but not TLR2. Overexpression of TRIAD3A reduced LPS- and CpG-induced NFκB activity, whereas knockdown of TRIAD3A enhances TLR expression and increases signaling in vitro.Citation14 Conversely, A20 is a deubiquitylating enzyme that blocks both MyD88-dependent and independent TLR signaling by deubiquitylating TRAF6, a common signaling component shared by all TLRs.Citation15
NOD2 is an intracellular pathogen pattern recognition receptor in the Nod-like receptor family that recognizes bacterial muramyl dipeptide (MDP). Interestingly, ligation of NOD2 has been shown to specifically inhibit TLR2-mediated signaling. NOD2-deficient mice demonstrated enhanced TH1 responses when stimulated with TLR2 ligands, but not other TLR ligands.Citation16 PI3K, a ubiquitously expressed signaling molecule that functions as an early event in many cellular responses, was also shown to inhibit TLR signaling. Unlike NOD2, PI3K-deficient mice show enhanced TH1 responses when stimulated with several TLR agonists, including TLR2, TLR4 and TLR9 ligands.Citation17 The mechanism(s) by which NOD2 and PI3K inhibit TLR signaling is not well defined, but both were shown to suppress NFκB activation downstream of TLR activation.Citation16,Citation17
An additional group of intracellular negative regulators are adaptor molecules that target IRAK-1. One such molecule, suppressor of cytokine signaling 1 (SOCS1), inhibits TLR4 and TLR9 signaling by suppressing IRAK-1 expression.Citation18 The Toll-interacting protein (TOLLIP) inhibits autophosphorylation of IRAK-1 to negatively regulate both TLR2 and TLR4 signaling.Citation19 The death receptor binding adaptor protein, FADD, impairs IRAK-1 interaction with MyD88 to block TLR2 and TLR4 mediated signaling in endothelial cells.Citation20 IRAK-2c and IRAK-2d are the alternatively spliced variants of IRAK-2 that lack death domains and have been shown to inhibit LPS-induced NFκB activation.Citation21 Lastly, IRAK-M (also referred to as IRAK-3) is the inactive kinase member of the IRAK family, one of the first identified and most studied negative regulators of TLR signaling.Citation22 A more detailed discussion on IRAK-M in the context of tumor immunosuppression will be discussed further.
Membrane-bound suppressors
Transmembrane proteins, such as ST2, SIGIRR, TRAILR and CD11b constitute another group of important negative regulators of TLR signaling. ST2, and SIGIRR are the two orphan receptors that block NFκB activation in response to IL-1, TLR4 and TLR9 ligands but not to TLR3 agonists.Citation23,Citation24 ST2 exists as two alternately spliced transmembrane forms (ST2L) expressed in hematopoietic cells, and a soluble form (sST2) that is present in both hematopoietic and non-hematopoietic cells. ST2-deficient mice were no different than wild-type mice in their response to endotoxin-induced shock, but failed to develop LPS tolerance.Citation23 This suggests that ST2 may plays an important role in regulating sustained TLR responses. ST2L suppresses IL-1 and TLR signaling by sequestering MyD88 and MAL through TIR domain. In addition, sST2 was shown to inhibit mRNA expression of TLR4 and TLR1 in response to LPS. SIGIRR is a transmembrane protein with a extracellular domain that interfers with TLR4, TLR5, and TLR9 ligand binding, and a long non-signaling intracellular domain that interfers with recruitment of IRAK-1 and TRAF6. Interestingly, SIGIRR is most highly expressed by epithelial cells, including in kidney, gut, and lung. SIGIRR deficient mice show exaggerated response to TLR4 and TLR9 activation and enhanced susceptibility to endotoxin shock.Citation24 Interestingly, mice deficient in the membrane-bound negative regulator, TRAILR, also showed enhanced viral clearance and increased cytokine production in response to TLR2, TLR3, and TLR4 but not to TLR9.Citation25 Another novel mechanism identified in macrophages is TLR-induced activation of CD11b, which inhibits TLR signaling by targeting MyD88 and TRIF for Cbl-b-mediated proteolytic degradation (). Consistent with other negative regulators, CD11b-deficient mice also show enhanced inflammatory cytokines in circulation and are susceptible to septic shock.Citation26
IRAK-M and Tumor-Induced Tolerance
IRAK-M is an intracellular negative regulator of TLR signaling and probably the only molecule to date investigated in the context of tumor immunology. It is an inactive kinase that antagonizes TLR signaling through protein-protein interactions preventing activation of IRAK-1. IRAK-M is regarded as a key negative regulator of TLR signaling in macrophages preventing excessive inflammatory responses.Citation22 Unfortunately, in pathologies such as sepsis and cancer, immune suppressive function of IRAK-M is exploited to evade host immune surveillance. Enhanced expression of IRAK-M was observed in alveolar macrophages during sepsis-induced immune suppressionCitation27 and in PBMCs isolated from patients with chronic myeloid leukemia.Citation28
We have shown that tumor associated macrophages (TAMs) express significantly higher levels of IRAK-M compared with the peritoneal macrophages (PEMs) from the same tumor bearing mice. Subcutaneous implantation of LLC cells in IRAK-M null mice resulted in a five-fold reduction in tumor growth, as compared with tumors in wild type (WT) animals. TAMs isolated from IRAK-M-deficient mice displayed a classically-activated M1 phenotype rather than M2 phenotype observed in TAMs from WT mice. Human lung cancer cells induced IRAK-M expression in human PBMCs when co-cultured together. Tumor cell-induced expression of IRAK-M was TGFβ dependent.Citation3 However, the mechanism by which TGFβ induced IRAK-M expression is not known. Bioinformatics analysis of human and mouse IRAK-M promoter identified Smad3 binding elements (unpublished observations). Ongoing studies in our lab will verify the direct binding of Smad3 to IRAK-M promoter and its functional significance in regulating TGFβ-induced IRAK-M expression. Importantly, enhanced IRAK-M expression in primary lung tumors correlated with poor survival in patients with lung cancer.Citation3 Collectively, our data demonstrated that TGFβ-dependent induction of IRAK-M expression is an important, clinically relevant mechanism by which tumors may circumvent anti-tumor responses of macrophages. Recent studies implicating a role for IRAK-M in regulating TLR responses in other cell types raising the question as to whether changes in macrophage phenotype in the IRAK-M deficient mice is solely responsible for inhibitory effects on tumor growth. Studies are underway in our laboratory to address this issue by using bone marrow chimera mouse models. The major deficiency in many of the earlier studies, including ours, was use of existing cell lines that are already immunoedited. To gain precise insights into the role of IRAK-M in the process of immunoediting, it is necessary to investigate by using a chemical or genetic model of carcinogenesis in an immunocompetent host. To this end we recently developed a mouse model by crossing the inducible K-ras mouse model of lung cancer into IRAK-M-deficient background (unpublished studies).
A Potential New Class of Therapeutic Targets for Oncoimmunology
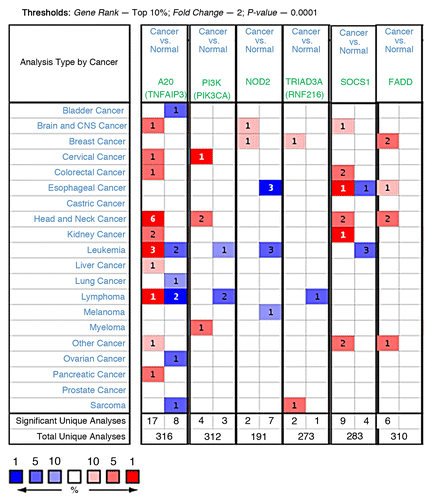
To develop more effective immunotherapies, immunologists must identify the cellular and molecular mechanisms that either eliminate or promote cancer development. For this reason, inhibitors of TLR signaling may be therapeutically useful in switching the nature of the tumor microenvironment from one of tumor-promoting inflammation to that of tumor-eliminating immunity. Surprisingly, with the exception of IRAK-M, the expression or the role of other TLR negative regulators has not been investigated in the context of tumor immunology. In a preliminary analysis, we assessed the mRNA expression of some of the negative regulators discussed above, in a spectrum of human cancers using Oncomine database (www.oncomine.com).Citation29 Interestingly, five out of six molecules assessed showed upregulation in the majority of unique analyses comparing cancer cells vs. normal tissue (). For example, A20 was upregulated (within top 10% with FC > 2 and p-value < 0.0001) in 17 unique analyses (sample sets) compared with its downregulation in 8 (). This shows a trend toward increased expression in tumors, where head and neck cancers stand out with upregulation in six unique analyses. This trend is consistent with our recent finding that high IRAK-M expression in tumors correlated with poor survival in patients with lung cancer.Citation3 By comparison, for NOD2 expression there was a reverse correlation with two up and seven down. It should be cautioned that the mRNA data in the oncomine data sets is derived from entire tumors, without any consideration or separation for their immune component. Therefore, it’s worth noting that these correlations may not truly represent a proof-of-principle but provides a rationale and starting point for further investigation. However, the lack-of or negative correlation in this analysis does not preclude a molecule from further investigation.
Figure 2. Analysis and visualization of A20, PI3K, NOD2, TRIAD3A, SOCS1 and FADD gene expressions in a spectrum of human cancers using Oncomine database. Each individual gene was analyzed separately for differential expression between cancers vs. normal by applying indicated thresholds and compiled together for visualization. Cell color is determined by the best gene rank percentile for the analyses within the cells. Red, upregulation; Blue, downregulation.
In light of emerging IRAK-M data, it’s reasonable to expect an important role for other negative regulators in conferring tumor tolerance. It is necessary to investigate the effect of each of these molecules on immunoediting, starting with molecules for which knockout mouse models are currently available. Given the TGFβ regulation of IRAK-M, it is important to explore the regulation of other negative regulators by TGFβ or other immunosuppressive cytokines in the tumor microenvironment. The tissue or cell type-specific expression of some regulators suggests that it would not be surprising to see tumor type specific mechanisms in operation. In summary, understanding the functional control of immune-suppressive networks, including the role of TLR signaling and their negative regulators, may offer new opportunities to shift the balance between tolerance and immunity. The identification and targeting such nodes and molecules will facilitate the elimination of tolerance phenomena in the tumor microenvironment and aid the development of effective cancer immunotherapeutic strategies.
| Abbreviations: | ||
| TLR | = | toll-like receptors |
| sTLR | = | soluble TLR |
| sMyD88 | = | short form of MyD88 |
| IRAK | = | interleukin-1 receptor-associated kinase |
| TGF | = | transforming growth factor |
| NFκB | = | nuclear factor kappa B |
| PAMP | = | pathogen associated molecular pattern |
| DAMP | = | danger associated molecular pattern |
| TNF | = | tumor necrosis factor |
| LPS | = | lipopolysacharide |
| NOD | = | nucleotide-binding oligomerization domain protein |
| SOCS | = | suppressor of cytokine signaling |
| TOLLIP | = | toll-interacting protein |
| FADD | = | Fas-associated death domain |
| TRAF | = | tumor necrosis factor associated factor |
| PI3K | = | phosphotidylinositol 3-kinase |
| SIGGIR | = | single immunoglobulin interleukin-1-related receptor |
| TRAILR | = | tumor necrosis factor-related apoptosis inducing ligand receptor |
| TRIF | = | toll/IL-1 receptor-domain containing adaptor protein inducing IFNβ |
| TRAM | = | TRIF-related adaptor molecule |
| LLC | = | Lewis lung carcinoma |
Acknowledgments
This research is funded by NIH/NCI (R01 CA132571-01), and American Cancer Society (RSG-CSM-116801) grants to V.G.K. We thank Dr. Ajay Kumar Reka for the art work in and Viritha Kaza for her help with the Oncomine analysis.
References
- Vesely MD, Kershaw MH, Schreiber RD, Smyth MJ. Natural innate and adaptive immunity to cancer. Annu Rev Immunol 2011; 29:235 - 71; http://dx.doi.org/10.1146/annurev-immunol-031210-101324; PMID: 21219185
- Liew FY, Xu D, Brint EK, O'Neill LA. Negative regulation of toll-like receptor-mediated immune responses. Nat Rev Immunol 2005; 5:446 - 58; http://dx.doi.org/10.1038/nri1630; PMID: 15928677
- Standiford TJ, Kuick R, Bhan U, Chen J, Newstead M, Keshamouni VG. TGF-beta-induced IRAK-M expression in tumor-associated macrophages regulates lung tumor growth. Oncogene 2011; 30:2475 - 84; http://dx.doi.org/10.1038/onc.2010.619; PMID: 21278795
- Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol 2004; 4:499 - 511; http://dx.doi.org/10.1038/nri1391; PMID: 15229469
- Akira S, Sato S. Toll-like receptors and their signaling mechanisms. Scand J Infect Dis 2003; 35:555 - 62; http://dx.doi.org/10.1080/00365540310015683; PMID: 14620134
- Benbaruch A. Inflammation-associated immune suppression in cancer: the roles played by cytokines, chemokines and additional mediators. Semin Cancer Biol 2006; 16:38 - 52; http://dx.doi.org/10.1016/j.semcancer.2005.07.006; PMID: 16139507
- Elgert KD, Alleva DG, Mullins DW. Tumor-induced immune dysfunction: the macrophage connection. J Leukoc Biol 1998; 64:275 - 90; PMID: 9738653
- Pardoll D. Does the immune system see tumors as foreign or self?. Annu Rev Immunol 2003; 21:807 - 39; http://dx.doi.org/10.1146/annurev.immunol.21.120601.141135; PMID: 12615893
- Nahid MA, Satoh M, Chan EK. MicroRNA in TLR signaling and endotoxin tolerance. Cell Mol Immunol 2011; 8:388 - 403; http://dx.doi.org/10.1038/cmi.2011.26; PMID: 21822296
- Iwami KI, Matsuguchi T, Masuda A, Kikuchi T, Musikacharoen T, Yoshikai Y. Cutting edge: naturally occurring soluble form of mouse Toll-like receptor 4 inhibits lipopolysaccharide signaling. J Immunol 2000; 165:6682 - 6; PMID: 11120784
- LeBouder E, Rey-Nores JE, Rushmere NK, Grigorov M, Lawn SD, Affolter M, et al. Soluble forms of Toll-like receptor (TLR)2 capable of modulating TLR2 signaling are present in human plasma and breast milk. J Immunol 2003; 171:6680 - 9; PMID: 14662871
- Burns K, Janssens S, Brissoni B, Olivos N, Beyaert R, Tschopp J. Inhibition of interleukin 1 receptor/Toll-like receptor signaling through the alternatively spliced, short form of MyD88 is due to its failure to recruit IRAK-4. J Exp Med 2003; 197:263 - 8; http://dx.doi.org/10.1084/jem.20021790; PMID: 12538665
- Janssens S, Burns K, Tschopp J, Beyaert R. Regulation of interleukin-1- and lipopolysaccharide-induced NF-kappaB activation by alternative splicing of MyD88. Curr Biol 2002; 12:467 - 71; http://dx.doi.org/10.1016/S0960-9822(02)00712-1; PMID: 11909531
- Chuang TH, Ulevitch RJ. Triad3A, an E3 ubiquitin-protein ligase regulating Toll-like receptors. Nat Immunol 2004; 5:495 - 502; http://dx.doi.org/10.1038/ni1066; PMID: 15107846
- Boone DL, Turer EE, Lee EG, Ahmad RC, Wheeler MT, Tsui C, et al. The ubiquitin-modifying enzyme A20 is required for termination of Toll-like receptor responses. Nat Immunol 2004; 5:1052 - 60; http://dx.doi.org/10.1038/ni1110; PMID: 15334086
- Watanabe T, Kitani A, Murray PJ, Strober W. NOD2 is a negative regulator of Toll-like receptor 2-mediated T helper type 1 responses. Nat Immunol 2004; 5:800 - 8; http://dx.doi.org/10.1038/ni1092; PMID: 15220916
- Fukao T, Tanabe M, Terauchi Y, Ota T, Matsuda S, Asano T, et al. PI3K-mediated negative feedback regulation of IL-12 production in DCs. Nat Immunol 2002; 3:875 - 81; http://dx.doi.org/10.1038/ni825; PMID: 12154357
- Kinjyo I, Hanada T, Inagaki-Ohara K, Mori H, Aki D, Ohishi M, et al. SOCS1/JAB is a negative regulator of LPS-induced macrophage activation. Immunity 2002; 17:583 - 91; http://dx.doi.org/10.1016/S1074-7613(02)00446-6; PMID: 12433365
- Zhang G, Ghosh S. Negative regulation of toll-like receptor-mediated signaling by Tollip. J Biol Chem 2002; 277:7059 - 65; http://dx.doi.org/10.1074/jbc.M109537200; PMID: 11751856
- Zhande R, Dauphinee SM, Thomas JA, Yamamoto M, Akira S, Karsan A. FADD negatively regulates lipopolysaccharide signaling by impairing interleukin-1 receptor-associated kinase 1-MyD88 interaction. Mol Cell Biol 2007; 27:7394 - 404; http://dx.doi.org/10.1128/MCB.00600-07; PMID: 17785432
- Hardy MP, O'Neill LA. The murine IRAK2 gene encodes four alternatively spliced isoforms, two of which are inhibitory. J Biol Chem 2004; 279:27699 - 708; http://dx.doi.org/10.1074/jbc.M403068200; PMID: 15082713
- Kobayashi K, Hernandez LD, Galan JE, Janeway CA Jr., Medzhitov R, Flavell RA. IRAK-M is a negative regulator of Toll-like receptor signaling. Cell 2002; 110:191 - 202; http://dx.doi.org/10.1016/S0092-8674(02)00827-9; PMID: 12150927
- Brint EK, Xu D, Liu H, Dunne A, McKenzie AN, O'Neill LA, et al. ST2 is an inhibitor of interleukin 1 receptor and Toll-like receptor 4 signaling and maintains endotoxin tolerance. Nat Immunol 2004; 5:373 - 9; http://dx.doi.org/10.1038/ni1050; PMID: 15004556
- Wald D, Qin J, Zhao Z, Qian Y, Naramura M, Tian L, et al. SIGIRR, a negative regulator of Toll-like receptor-interleukin 1 receptor signaling. Nat Immunol 2003; 4:920 - 7; http://dx.doi.org/10.1038/ni968; PMID: 12925853
- Diehl GE, Yue HH, Hsieh K, Kuang AA, Ho M, Morici LA, et al. TRAIL-R as a negative regulator of innate immune cell responses. Immunity 2004; 21:877 - 89; http://dx.doi.org/10.1016/j.immuni.2004.11.008; PMID: 15589175
- Han C, Jin J, Xu S, Liu H, Li N, Cao X. Integrin CD11b negatively regulates TLR-triggered inflammatory responses by activating Syk and promoting degradation of MyD88 and TRIF via Cbl-b. Nat Immunol 2010; 11:734 - 42; http://dx.doi.org/10.1038/ni.1908; PMID: 20639876
- Deng JC, Cheng G, Newstead MW, Zeng X, Kobayashi K, Flavell RA, et al. Sepsis-induced suppression of lung innate immunity is mediated by IRAK-M. J Clin Invest 2006; 116:2532 - 42; PMID: 16917541
- del Fresno C, Otero K, Gomez-Garcia L, Gonzalez-Leon MC, Soler-Ranger L, Fuentes-Prior P, et al. Tumor cells deactivate human monocytes by up-regulating IL-1 receptor associated kinase-M expression via CD44 and TLR4. J Immunol 2005; 174:3032 - 40; PMID: 15728517
- Rhodes DR, Yu J, Shanker K, Deshpande N, Varambally R, Ghosh D, et al. ONCOMINE: a cancer microarray database and integrated data-mining platform. Neoplasia 2004; 6:1 - 6; PMID: 15068665