Abstract
Cyclooxygenase-2 is frequently upregulated in epithelial tumors and contributes to poor outcomes in multiple malignancies. The COX-2 product prostaglandin E2 (PGE2) promotes tumor growth and metastasis by acting on a family of four G protein-coupled receptors (EP1–4). Using a novel small molecule EP4 antagonist (RQ-15986) and a syngeneic murine model of metastatic breast cancer, we determined the effect of EP4 blockade on innate immunity and tumor biology. Natural killer (NK)-cell functions are markedly depressed in mice bearing murine mammary tumor 66.1 or 410.4 cells owing to the actions of PGE2 on NK cell EP4 receptors. The EP4 agonist PGE1-OH inhibits NK functions in vitro, and this negative regulation is blocked by RQ-15986. Likewise, the treatment of tumor-bearing mice with RQ-15986 completely protected NK cells from the immunosuppressive effects of the tumor microenvironment in vivo. RQ-15986 also has direct effects on EP4 expressed by tumor cells, inhibiting the PGE2-mediated activation of adenylate cyclase and blocking PGE2-induced tumor cell migration. The pretreatment of tumor cells with a non-cytotoxic concentration of RQ-15986 inhibited lung colonization, a beneficial effect that was lost in mice depleted of NK cells. The oral administration of RQ-15986 inhibited the growth of tumor cells implanted into mammary glands and their spontaneous metastatic colonization to the lungs, resulting in improved survival. Our findings reveal that EP4 antagonism prevents tumor-mediated NK-cell immunosuppression and demonstrates the anti-metastatic activity of a novel EP4 antagonist. These observations support the investigation of EP4 antagonists in clinical trials.
Introduction
The cyclooxygenase-2 (COX-2) product prostaglandin E2 (PGE2) mediates tumor-associated immunosuppression. Natural killer (NK) cells, which play a critical role in the control of metastases, are inhibited by PGE2 acting on the cognate receptors EP2 and EP4.Citation1,Citation2 Previous studies from us and other laboratories support a key role for EP4 in promoting breast cancer metastasis,Citation3-Citation9 but until now there have been few effective EP4 antagonists available for preclinical and clinical investigations. Here, we describe a novel orally active EP4 antagonist, RQ-00015986 (hereafter abbreviated RQ-15986, CJ-042794).Citation10 In clinical trials, the RQ-15986-related compound RQ-07 has been shown to be safe and efficient against osteoarthritis-associated pain (unpublished observations) and is the only known EP4 antagonist available for clinical evaluation. The effects of RQ-15986 on innate immunity have not yet been investigated nor has its ability to inhibit tumor metastasis been determined. We now report that RQ-15986 restores the profound suppression of NK-cell functions that characterizes progressive tumor growth, an effect that is accompanied by marked antitumor and antimetastasis activity. These studies identify a potential new approach to prevent tumor-associated NK-cell suppression.
Results
A novel non-cytotoxic inhibitor of EP4
RaQualia Pharma has developed several high-affinity and highly selective EP4 antagonists.Citation10 RQ-15986 () potently inhibits PGE2-induced elevations in cAMP activity in HEK293 cells expressing murine EP4 (pA2 value of 8.7). In malignant cells derived from the mammary gland, EP4 is similarly coupled to adenylate cyclase activation.Citation6 We examined the ability of RQ-15986 to inhibit this response in malignant cells exposed to PGE2 or the EP4-specific agonist PGE1-OH (). Stimulation of murine 66.1 mammary tumor cells with 5 μM PGE2 or PGE1-OH for 15 min resulted in a 2.1- or 2-fold elevation in intracellular cAMP, respectively. These responses were significantly inhibited by the EP4 antagonists AH23848 or RQ-15986 in a dose dependent manner. The ability of RQ-15986 to inhibit PGE2-mediated adenylate cyclase activation was confirmed using 410.4 mammary tumor cells (). In these cells, PGE2 and PGE1-OH induced a 1.8- and 1.6-fold increase in cAMP levels, respectively. Both responses were inhibited by RQ-15986. Thus, like AH23848, RQ-15986 effectively antagonizes EP4 in mammary tumor cells.
Figure 1. RQ-15986, a noncytotoxic EP4 antagonist. (A) Chemical structure of RQ-00015986. (B) Murine 66.1 mammary tumor cells pretreated with 1 µM indomethacin for 24 h and transferred to complete media containing 100 µM IBMX were stimulated for 15 min with 5 µM PGE2 or PGE1-OH in the presence or absence of 0.5–3.0 µM AH23848 or RQ-15986 and intracellular cAMP levels were determined in triplicate cultures. (C) Murine 410.4 mammary tumor cells were treated as in (B) followed by the quantification of cAMP levels. *p < 0.05; **p < 0.01 (D) Proliferation of 66.1 cells in the presence of the indicated concentrations of RQ-15986 at 24 and 48 h.
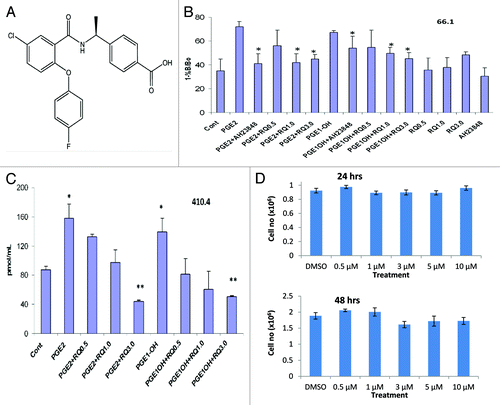
We then determined the effect of RQ-15986 on mammary tumor cell growth. To this aim, 66.1 cells were cultured in the presence of 0.5–10 μM RQ-15986 or vehicle and cell number was determined at 24 and 48 h. RQ-15986 did not affect the growth of tumor cells in vitro (). Likewise, the proliferation of 410.4 cells was not altered in the presence of RQ-15986 (data not shown).
RQ-15986 inhibits tumor growth and metastasis in vivo
The ability of malignant cells to migrate across an artificial membrane in a modified Boyden chamber is a surrogate measure of metastasis. PGE2 and PGE1-OH induced the migration of mammary tumor cells by 2.2- and 1.9-fold, respectively (). RQ-15986 was able to completely reverse PGE2 or PGE1-OH-induced tumor cell migration. Based on these encouraging results, we determined the ability of RQ-15986 to inhibit lung colonization by tumor cells in vivo. To this aim, 66.1 tumor cells were incubated with 3 μM RQ-15986 or vehicle for 15 min, washed and intravenously injected into syngenic BALB/cByJ female mice. Twenty-one days later, mice were euthanized and visceral metastases were enumerated. Mice injected with vehicle-treated tumor cells had an average of 49.6 ± 5 tumor colonies in the lungs, which were significantly less in mice injected with RQ-15986-treated tumor cells (, 15.2 ± 5, p < 0.003). Three of ten mice injected with vehicle-treated tumor cells also had grossly detectable tumor colonies on the heart, but 0/10 mice injected with RQ-15986-treated cells displayed cardiac metastases.
Figure 2. Antineoplastic and antimetastatic effects of RQ-15986. (A) Murine 66.1 mammary tumor cells labeled with calcein AM and placed in the upper well of a Boyden chamber containing 8 μm membranes coated with collagen I and fibronectin. Five µM PGE2 or PGE1-OH, 2% FBS and/or 3 µM RQ-15986 were placed in bottom chamber, and migration was assessed at 24 h. (B) Tumor cells were cultured in the presence of 3 µM RQ-15986 for 30 min were washed and injected (1 × 105 live cells) into the lateral tail vein of control BALB/cByJ female mice or mice depleted of NK cells with anti-asialo GM1 antibodies. All mice were euthanized on day 18–22 and soft tissues were examined for surface tumor colonies (10 mice/group). p < 0.003, vehicle-treated vs. RQ-15986-treated control mice. (C,D) Five × 105 66.1 tumor cells were subcutaneously injected proximal to the right abdominal mammary gland to BALB/cByJ female mice (day = 0, 10 mice/group). Beginning on day +1, mice were treated with 100 mg/Kg RQ-15986 twice a day by oral gavage for 21 d. Tumor diameters were monitored and mice were euthanized on an individual basis when tumors measured 18 mm in average diameter or earlier, if moribund (C), *p = 0.0004; **p < 0.0001, and surface lung tumor colonies were counted (D, p = 0.01). (E) Survival fraction plotted as number of surviving animals, i.e., not euthanized, at each time point. At the beginning of the experiment, there were 13 mice in the vehicle-treated group and 14 in the RQ-15986 treatment group (p = 0.0003).
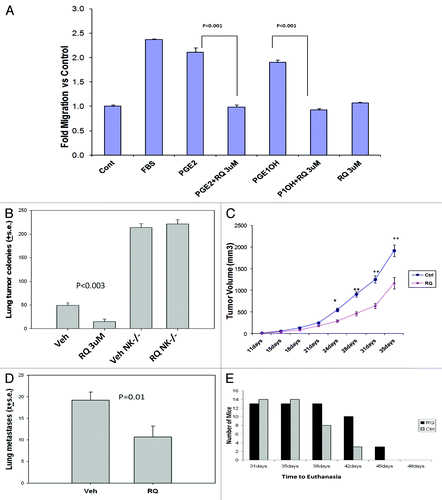
NK cells play an important role in controlling tumor dissemination. Vehicle-treated mammary tumor cells injected in BALB/cByJ mice depleted of NK cells produced more lung colonies (214 ± 8) than the same cells injected in control mice (), consistent with a protective role of endogenous NK cells. RQ-15986 was no longer able to limit the number of lung lesions (221.6 ± 9) in NK-depleted mice, indicating a critical role for NK cells in the mechanism by which EP4 antagonists inhibit metastasis. Sixty and 40% of NK-depleted mice displayed cardiac metastases after the injection of vehicle-treated vs. RQ-15986-treated tumor cells, respectively (data not shown).
To assess the effect of EP4 antagonism on tumor growth and spontaneous metastasis, 5 × 105 66.1 tumor cells were implanted proximal to the mammary gland of BALB/cByJ mice. Beginning on the next day, mice were treated twice a day with 100 mg/Kg RQ-15986 or vehicle for 21 d. Tumor volume was determined by caliper measurement and mice were euthanized when primary tumors achieved an average diameter of 18 mm, followed by the quantification of visceral metastases. Daily treatment with RQ-15986 resulted in an approximate 50% reduction in tumor volume (, which was significantly lower starting after day 21). Spontaneous metastases to the lungs were reduced by 44% (, 10.7 ± 2.5 vs. 19.2 ± 1.9, p = 0.01). Time to euthanasia (tumor > 18 mm) was also significantly extended by treatment with the EP4 antagonist (, p = 0.0003).
RQ-15986 rescues NK cells from immunosuppression
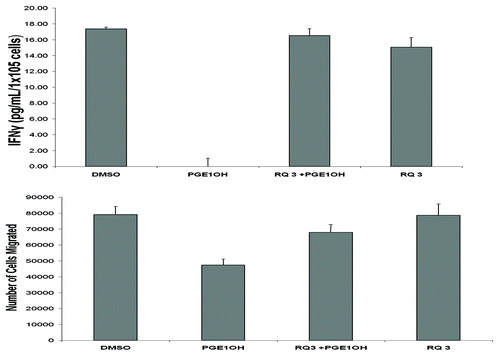
We have shown that NK cells are critical for the control of tumor dissemination ( and ref. Citation8) but NK-cell functions are profoundly suppressed in advanced malignancy. We assessed the capacity of RQ-15986 to rescue NK cells from PGE2-mediated immunosuppression. We first isolated NK-cell enriched populations from the spleens of normal BALB/cByJ mice by magnetic bead separation of DX5+CD3- cells, and examined the ability of RQ-15986 to protect NK-cell functions (migration, cytokine production) from PGE2-mediated inhibition. NK cells were pretreated with either vehicle or PGE1-OH for 30 min in the absence or presence of 3 μM RQ-15986. Then, NK cells were stimulated with 1,000 U/mL interleukin-2 (IL-2) for 18 h, after which conditioned culture media were harvested and assayed for interferon γ (IFNγ) levels by ELISA. The ability of NK cells to produce IFNγ upon IL-2 stimulation was completely suppressed in the presence of PGE1-OH (). RQ-15986 was able to completely protect NK cells from the immunosuppressive effects of PGE1-OH, restoring IFNγ production to the levels observed in control NK cells. We also determined the effect of PGE2 on NK-cell migration. The ability of NK cells to migrate in response to fetal bovine serum (FBS) was depressed in the presence of PGE1-OH, an inhibitory effect that was prevented by EP4 antagonism with RQ-15986 ().
Figure 3. Effects of RQ-15986 in NK cells. (A) Splenic NK cells isolated from normal mice as the DX5+CD3- cell population were treated with DMSO, 5 µM PGE1-OH and/or 3 µM RQ-15986 for 30 min followed by treatment with 1000 U/mL interleukin-2 (IL-2) for 18 h. Conditioned media were then assayed for interferon γ (IFNγ) levels by ELISA, in triplicate replicates. (B) NK cells were treated with 1 µM PGE1-OH for 10 min and allowed to migrate across the top of a microplate (3 μm pore size) for 3 h in response to fetal bovine serum (FBS).
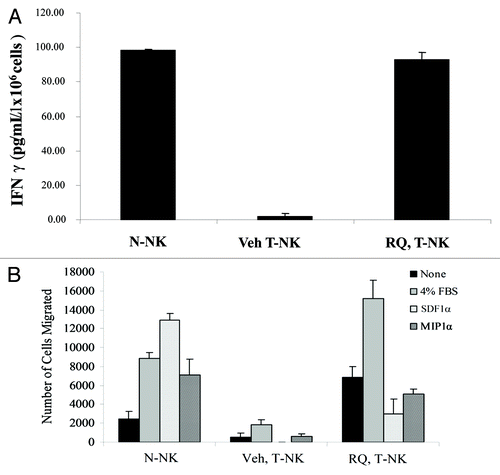
We then determined if RQ-15986 might protect NK cells from the functional inhibition that occurs in tumor-bearing mice. To this aim, we compared the functional activity of NK cells isolated from normal mice (N-NK cells) or 66.1 cell-derived tumor-bearing mice (T-NK cells) that either received vehicle or 100 mg/Kg RQ-15986 twice a day for 21 d. The ability of T-NK cells obtained from vehicle-treated mice to produce IFNγ was nearly absent as compared with that of N-NK cells (), indicating a profound suppression of NK cells by the tumor environment. Notably, RQ-15986 was able to reverse such a tumor-associated immune dysfunction. T-NK cells from RQ-15986-treated mice indeed produced IFNγ levels that were comparable to those generated by N-NK cells. Along similar lines, T-NK cells from vehicle-treated mice migrated poorly in response to FBS as well as upon chemotactic stimulation with the chemokines macrophage inflammatory protein 1α (MIP1α) and stromal cell derived factor 1α (SDF1α), compared with N-NK cells (). The administration of RQ-15986 to tumor-bearing mice rescued T-NK cells from this inhibition.
Figure 4. RQ-15986 rescues NK-cell functions in vivo. (A) NK cells were isolated from the spleens of normal mice (N-NK) or tumor-bearing mice (T-NK) that were treated with vehicle or 100 mg/Kg RQ-15986 twice a day by oral gavage for 21 d, and then stimulated in vitro with interleukin-2 (IL-2) for 18 h. Finally, interferon γ (IFNγ) secretion in culture media was assayed by ELISA. (B) N-NK and T-NK were isolated as in (A) and their migratory ability in response to 4% fetal bovine serum (FBS), 100 nM stromal cell derived factor 1α (SDF1α) and 100 nM macrophage inflammatory protein 1α (MIP1α) was determined.
Discussion
The suppression of NK-cell functions in patients with breast cancer is partially attributable to PGE2, which is generated in large amounts by the elevated activity of COX-2 that is common to many epithelial tumors.Citation11 PGE2 directly inhibits NK-cell functions primarily by binding EP4 receptors expressed on their surface.Citation1,Citation2 The results of clinical trials testing COX-2 inhibitors in cancer patients have been disappointing. The recent literature supports a potential benefit for targeting the COX-2 signaling pathway further downstream, at the level of individual EP receptors. However this branch of research has been hampered by the absence of clinically relevant EP4 antagonists. RQ-15986 is one of several high-affinity EP4 antagonists recently developed by RaQualia Pharma, Inc.Citation10 The RQ-15986-related compound RQ-07 has been investigated in four phase I studies in healthy subjects and two phase IIa trials in patients with osteoarthritis-associated pain and found to be safe and effective (unpublished observations). RQ-15986 has been shown to inhibit primary tumorigenesis in a model of inflammatory gastric cancer,Citation12 but the ability of this compound to modulate NK-cell functions and to inhibit tumor metastasis had not yet been investigated. Our findings provide support for the evaluation of the antineoplastic activity of novel EP4 antagonists in clinical trials.
Like many epithelial cancer cells, the mammary tumor cells used in this study produce significant levels of PGE2, both in vitro and in vivo. In our previous studies, we found cultured 66.1 cells to secrete 19.7 ± 3.4 ng/106 cells PGE2 in 48 h, while 410.4 cells released 18.2 ± 1.9 ng/106 cells PGE2.Citation13 In 66.1 and 410.4-cell derived tumor homogenates, PGE2 was quantified as 337 ± 70 and 321 ± 45 ng/g wet weight of tissue, respectively. In addition to the intrinsic COX-2 activity of mammary tumor cells, other cells in the tumor microenvironment including fibroblasts and myeloid cells can contribute to the increased production of PGE2.Citation14,Citation15 Experiments in which tumor cells were pre-treated with an EP4 antagonist point to a direct role for EP4 expression in the cell-intrinsic properties that contribute to their metastatic potential. Our previous findings also suggest that NK-mediated tumor cell lysis is enhanced in the presence of EP4 antagonists, a phenomenon that is mechanistically linked to a reduced expression of NK-inhibitory MHC Class I molecules.Citation8 Thus, EP4 antagonists inhibit tumor metastasis by multiple mechanisms, including reduced tumor cell migration, increased sensitivity to NK-mediated lysis and systemic protection of NK cells from PGE2-mediated immunosuppression.
NK-cell functions are profoundly inhibited in patients with advanced malignancies and in mammary tumor-bearing mice. Here, we have shown that the treatment of these mice with RQ-15986 leads to a nearly complete rescue of NK-cell functions. This is quite interesting because other non-prostaglandin tumor products also inhibit NK cells in vitro.Citation16 For example, COX-2 upregulates indoleamine-pyrrole 2,3-dioxygenase (IDO), a potent immunosuppressive mediator,Citation17,Citation18 implying that the COX-2/EP4 blockade may operate upstream of several other immunosuppressive mechanisms. These data are consistent with our finding demonstrating that neither RQ-15986, nor other EP4 antagonists nor an EP4-targeting short-hairpin RNA (shRNA) inhibit metastasis in the absence of NK cells.Citation8,Citation9
A consensus is growing on the notion that EP4 promotes multiple cancers of diverse histological origin.Citation19 EP4 is upregulated in colon adenomas/adenocarcinomas and promotes anchorage-independent growth and apoptosis resistance by a cAMP-dependent mechanism.Citation20,Citation21 Loss of miR-101 in colon cancer may explain the increased EP4 expression in this disease.Citation22 The EP4/CREB-mediated activation of ERK leads to downstream expression of early growth response gene-1 (EGR-1), which supports the proliferation of colon cancer cells.Citation23 In T-cell leukemia, EP4 promotes apoptosis resistance through a phosphoinositide-3-kinase (PI3K)/AKT pathway.Citation24 PGE2 acting on the EP4 receptor induces non-small cell lung carcinoma (NSCLC) cell growth through increased expression of an integrin-linked kinase.Citation25 Angiogenesis in prostate cancer is induced by PGE2 acting on EP2 and EP4 receptors.Citation26 EP4 is upregulated in castration-resistant hormone-naïve prostate cancer and an EP4 antagonist inhibits growth in two xenograft models of castration-resistant disease.Citation27 EP4 positively regulates the expression of aromatase, leading to estrogen synthesis and reduces the expression of BRCA1 in breast cancer.Citation28 In addition to affecting tumor cell proliferation, EP4 plays a key role in promoting tumor cell dissemination in breast, lung and colon cancer.Citation6,Citation7,Citation19,Citation29,Citation30 Both the pharmacologic antagonism of EP4 and EP4 gene silencing inhibits tumor cell invasion, migration and metastasis in preclinical models of invasive ductal carcinoma and inflammatory breast cancer.Citation3-Citation6 Thus, EP4 is a relevant target in multiple histologically diverse neoplasms.
Many of these studies, including our own, were performed using a commercially available EP4 antagonist, AH23848, a compound that is not compatible with clinical development. Here, we provide for the first time evidence that a novel family of compounds, developed by RaQualia Pharma may constitute effective EP4 antagonists in cancer. We employed RQ-15986 because it exhibits an excellent pharmacological profile in rodents. RQ-07, a similar compound, has been shown to be safe and efficient against osteoarthritis-associated pain in clinical trials. To our knowledge, RQ-07 is the only EP4 antagonist that is ready for evaluation as an antineoplastic agent in clinical trials.
The current study demonstrates that RQ-15986 is able to markedly inhibit breast cancer metastasis, likely due to both the protective effect of EP4 antagonism on critical NK-cell functions as well as direct effects on tumor cells. These findings add to the growing literature supporting a key role for EP4 in promoting tumor metastasis and provide a potentially novel approach to prevent cancer-associated immunosuppression. Our results support the further examination of EP4 as a new therapeutic target in multiple malignancies.
Materials and Methods
Cells
Highly tumorigenic and metastatic 66.1 and 410.4 cells were derived from a spontaneously occurring mammary adenocarcinoma in a BALB/cfC3H mouse. Cells were maintained as previously described.Citation2
Mice
BALB/cByJ female mice (Jackson Laboratory, 001026) were housed, cared for and used in strict accordance with the US. Department of Agriculture regulations and the NIH Health Guide for the Care and Use of Laboratory Animals.
In vivo studies
Five × 105 tumor cells were subcutaneously injected proximal to the right abdominal mammary gland of syngenic female mice (day = 0). On day +1–21, mice were treated twice a day with vehicle or 100 mg/Kg RQ-15986 by oral gavage. Tumor diameters were measured twice weekly and mice were euthanized on an individual basis when tumors measured 18 mm in average diameter or earlier, if moribund. Tumor volumes were calculated by the formula: (a × b2) × 0.5236, where a = longest diameter and b = perpendicular diameter. The lungs were weighed and surface tumor colonies quantified in a blinded fashion. Lung colonization was evaluated by intravenously injecting 1 × 105 viable tumor cells into the lateral tail vein of mice which were euthanized on day 18–22 post-transplantation or earlier, if moribund. For pretreatment protocols, tumor cells were cultured for 30 min in the presence of 3 μM RQ-15986, washed and injected into mice. To deplete NK cells, mice were injected with 20 μL rabbit anti-asialo GM1 ganglioside antibody (Wako Bioproducts, 986–10001) or control rabbit serum 1 d before and 3 d after tumor cell injection.
cAMP assays
Cells were pretreated with 1 μM indomethacin for 24 h and transferred to complete cell culture medium containing 100 μM IBMX (Sigma Chemical Co., 17018). Agonists or antagonists were added to cells and incubated for 15 min. Intracellular cAMP levels were determined using either the Amersham cAMP Biotrak EIA kit () or the the cAMP EIA system (Cayman Chemicals, 581–001) (). Software provided with the latter kit allowed for the conversion of binding data to pM cAMP.
NK-cell enrichment
An untouched population of NK cells derived from a splenic cell suspension obtained from normal BALB/cByJ or mice bearing 66.1 tumors was isolated by the NK Cell Isolation Kit (Miltenyi Biotec Inc., 130–090–864) as previously described.Citation1,Citation2
Migration assay
Tumor-cell and NK-cell migration in response to prostaglandins was assessed as previously described.Citation1,Citation2
ELISA
NK cells were pretreated with either vehicle, EP4 agonist or antagonist for 30 min, stimulated with 1000 U/mL IL-2 and, after 18 h, mouse IFNγ in culture supernatants was detected by ELISA Ready-SET-Go!® (eBioscience, 88–7914–29).
Statistical methods
The Student’s t-test was used to compare responses in cytokine, migration and cAMP studies. For animal studies, the nonparametric Wilcoxon test was applied. All tests were exact, two-sided and done at the p < 0.05 level of significance. Growth curves were analyzed using the linear mixed-effects models approach (LME). Comparison of the time to euthanasia between two groups was done using the general linear model approach (GLM). Statistical analyses were conducted in S-plus (TIBCO, v.8.2) and SAS (v.9.22).
| Abbreviations: | ||
| cAMP | = | cyclic adenosine monophosphate |
| COX-2 | = | cyclooxygenase 2 |
| EGR-1 | = | early growth response gene-1 |
| EP4 | = | prostaglandin E receptor 4 |
| FBS | = | fetal bovine serum |
| IBMX | = | 1-methyl-3-isobutylxanthine |
| IDO | = | indoleamine-pyrrole 2,3-dioxygenase |
| IFNγ | = | interferon γ |
| IL2 | = | interleukin 2 |
| MIP1α | = | macrophage inflammatory protein 1α |
| NK | = | natural killer |
| N-NK | = | NK from normal mice |
| NSCLC | = | non-small cell lung carcinoma |
| PGE2 | = | prostaglandin E2 |
| PI3K | = | phosphoinositide-3-kinase |
| SDF1α | = | stromal cell derived factor 1α |
| T-NK | = | NK from tumor-bearing mice |
Acknowledgments
We thank RaQualia Pharma, Inc. for the generous provision of RQ-15986 and scientific discussions. The authors received grant support from the US Department of Health and Human Services (CA120278), US Department of Veterans Affairs, US Department of Defense.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Holt D, Ma X, Kundu N, Fulton AM. Prostaglandin E(2) (PGE (2)) suppresses natural killer cell function primarily through the PGE(2) receptor EP4. Cancer Immunol Immunother 2011; 60:1577 - 86; http://dx.doi.org/10.1007/s00262-011-1064-9; PMID: 21681369
- Holt DM, Ma X, Kundu N, Collin PD, Fulton AM. Modulation of host natural killer cell functions in breast cancer via prostaglandin E2 receptors EP2 and EP4. J Immunother 2012; 35:179 - 88; http://dx.doi.org/10.1097/CJI.0b013e318247a5e9; PMID: 22306906
- Timoshenko AV, Xu G, Chakrabarti S, Lala PK, Chakraborty C. Role of prostaglandin E2 receptors in migration of murine and human breast cancer cells. Exp Cell Res 2003; 289:265 - 74; http://dx.doi.org/10.1016/S0014-4827(03)00269-6; PMID: 14499627
- Robertson FM, Simeone A-M, Mazumdar A, Shah AH, McMurray JS, Ghosh S, et al. Molecular and pharmacological blockade of the EP4 receptor selectively inhibits both proliferation and invasion of human inflammatory breast cancer cells. J Exp Ther Oncol 2008; 7:299 - 312; PMID: 19227010
- Robertson FM, Simeone A-M, Lucci A, McMurray JS, Ghosh S, Cristofanilli M. Differential regulation of the aggressive phenotype of inflammatory breast cancer cells by prostanoid receptors EP3 and EP4. Cancer 2010; 116:Suppl 2806 - 14; http://dx.doi.org/10.1002/cncr.25167; PMID: 20503412
- Ma X, Kundu N, Rifat S, Walser T, Fulton AM. Prostaglandin E receptor EP4 antagonism inhibits breast cancer metastasis. Cancer Res 2006; 66:2923 - 7; http://dx.doi.org/10.1158/0008-5472.CAN-05-4348; PMID: 16540639
- Fulton AM, Ma X, Kundu N. Targeting prostaglandin E EP receptors to inhibit metastasis. Cancer Res 2006; 66:9794 - 7; http://dx.doi.org/10.1158/0008-5472.CAN-06-2067; PMID: 17047037
- Kundu N, Ma X, Holt D, Goloubeva O, Ostrand-Rosenberg S, Fulton AM. Antagonism of the prostaglandin E receptor EP4 inhibits metastasis and enhances NK function. Breast Cancer Res Treat 2009; 117:235 - 42; http://dx.doi.org/10.1007/s10549-008-0180-5; PMID: 18792778
- Ma X, Kundu N, Collin PD, Goloubeva O, Fulton AM. Frondoside A inhibits breast cancer metastasis and antagonizes prostaglandin E receptors EP4 and EP2. Breast Cancer Res Treat 2012; 132:1001 - 8; http://dx.doi.org/10.1007/s10549-011-1675-z; PMID: 21761157
- Murase A, Taniguchi Y, Tonai-Kachi H, Nakao K, Takada J. In vitro pharmacological characterization of CJ-042794, a novel, potent, and selective prostaglandin EP(4) receptor antagonist. Life Sci 2008; 82:226 - 32; http://dx.doi.org/10.1016/j.lfs.2007.11.002; PMID: 18155068
- Mamessier E, Sylvain A, Thibult M-L, Houvenaeghel G, Jacquemier J, Castellano R, et al. Human breast cancer cells enhance self tolerance by promoting evasion from NK cell antitumor immunity. J Clin Invest 2011; 121:3609 - 22; http://dx.doi.org/10.1172/JCI45816; PMID: 21841316
- Oshima H, Hioki K, Popivanov K, Oguma K, Van Rooijen N, Ishikawa T-O, et al. Prostaglandin E2 signaling and bacterial infection recruit tumor-promoting macrophages to mouse gastric tumors. Gastroenterolology 2011; 140:596 - 607; http://dx.doi.org/10.1053/j.gastro.2010.11.007; PMID: 21070778
- Kundu N, Yang Q, Dorsey R, Fulton AM. Increased cyclooxygenase-2 (cox-2) expression and activity in a murine model of metastatic breast cancer. Int J Cancer 2001; 93:681 - 6; http://dx.doi.org/10.1002/ijc.1397; PMID: 11477578
- Balsamo M, Scordamaglia F, Pietra G, Manzini C, Cantoni C, Boitano M, et al. Melanoma-associated fibroblasts modulate NK cell phenotype and antitumor cytotoxicity. Proc Natl Acad Sci U S A 2009; 106:20847 - 52; http://dx.doi.org/10.1073/pnas.0906481106; PMID: 19934056
- Eruslanov E, Daurkin I, Ortiz J, Vieweg J, Kusmartsev S. Pivotal Advance: Tumor-mediated induction of myeloid-derived suppressor cells and M2-polarized macrophages by altering intracellular PGE₂ catabolism in myeloid cells. J Leukoc Biol 2010; 88:839 - 48; http://dx.doi.org/10.1189/jlb.1209821; PMID: 20587738
- Pietra G, Manzini C, Rivara S, Vitale M, Cantoni C, Petretto A, et al. Melanoma cells inhibit natural killer cell function by modulating the expression of activating receptors and cytolytic activity. Cancer Res 2012; 72:1407 - 15; http://dx.doi.org/10.1158/0008-5472.CAN-11-2544; PMID: 22258454
- Basu GD, Tinder TL, Bradley JM, Tu T, Hattrup CL, Pockaj BA, et al. Cyclooxygenase-2 inhibitor enhances the efficacy of a breast cancer vaccine: role of IDO. J Immunol 2006; 177:2391 - 402; PMID: 16888001
- Muller AJ, Prendergast GC. Indoleamine 2,3-dioxygenase in immune suppression and cancer. Curr Cancer Drug Targets 2007; 7:31 - 40; http://dx.doi.org/10.2174/156800907780006896; PMID: 17305476
- Reader JC, Holt D, Fulton AM. Prostaglandin E2 EP receptors as therapeutic targets in breast cancer. Cancer Metastasis Rev 2011; 30:449 - 63; http://dx.doi.org/10.1007/s10555-011-9303-2; PMID: 22002714
- Chell SD, Witherden IR, Dobson RR, Moorghen M, Herman AA, Qualtrough D, et al. Increased EP4 receptor expression in colorectal cancer progression promotes cell growth and anchorage independence. Cancer Res 2006; 66:3106 - 13; http://dx.doi.org/10.1158/0008-5472.CAN-05-3702; PMID: 16540660
- Hawcroft G, Ko CWS, Hull MA. Prostaglandin E2-EP4 receptor signalling promotes tumorigenic behaviour of HT-29 human colorectal cancer cells. Oncogene 2007; 26:3006 - 19; http://dx.doi.org/10.1038/sj.onc.1210113; PMID: 17130837
- Chandramouli A, Onyeagucha BC, Mercado-Pimentel ME, Stankova L, Shahin NA, LaFleur BJ, et al. MicroRNA-101 (miR-101) post-transcriptionally regulates the expression of EP4 receptor in colon cancers. Cancer Biol Ther 2012; 13:175 - 83; http://dx.doi.org/10.4161/cbt.13.3.18874; PMID: 22353936
- Cherukuri DP, Chen XBO, Goulet A-C, Young RN, Han Y, Heimark RL, et al. The EP4 receptor antagonist, L-161,982, blocks prostaglandin E2-induced signal transduction and cell proliferation in HCA-7 colon cancer cells. Exp Cell Res 2007; 313:2969 - 79; http://dx.doi.org/10.1016/j.yexcr.2007.06.004; PMID: 17631291
- George RJ, Sturmoski MA, Anant S, Houchen CW. EP4 mediates PGE2 dependent cell survival through the PI3 kinase/AKT pathway. Prostaglandins Other Lipid Mediat 2007; 83:112 - 20; http://dx.doi.org/10.1016/j.prostaglandins.2006.10.005; PMID: 17259077
- Zheng Y, Ritzenthaler JD, Sun X, Roman J, Han S-W. Prostaglandin E2 stimulates human lung carcinoma cell growth through induction of integrin-linked kinase: the involvement of EP4 and Sp1. Cancer Res 2009; 69:896 - 904; http://dx.doi.org/10.1158/0008-5472.CAN-08-2677; PMID: 19176380
- Jain S, Chakraborty G, Raja R, Kale S, Kundu GC. Prostaglandin E2 regulates tumor angiogenesis in prostate cancer. Cancer Res 2008; 68:7750 - 9; http://dx.doi.org/10.1158/0008-5472.CAN-07-6689; PMID: 18829529
- Terada N, Shimizu Y, Kamba T, Inoue T, Maeno A, Kobayashi T, et al. Identification of EP4 as a potential target for the treatment of castration-resistant prostate cancer using a novel xenograft model. Cancer Res 2010; 70:1606 - 15; http://dx.doi.org/10.1158/0008-5472.CAN-09-2984; PMID: 20145136
- Subbaramaiah K, Hudis C, Chang S-H, Hla T, Dannenberg AJ. EP2 and EP4 receptors regulate aromatase expression in human adipocytes and breast cancer cells. Evidence of a BRCA1 and p300 exchange. J Biol Chem 2008; 283:3433 - 44; http://dx.doi.org/10.1074/jbc.M705409200; PMID: 18083712
- Buchanan FG, Gorden DL, Matta P, Shi Q, Matrisian LM, DuBois RN. Role of β-arrestin 1 in the metastatic progression of colorectal cancer. Proc Natl Acad Sci U S A 2006; 103:1492 - 7; http://dx.doi.org/10.1073/pnas.0510562103; PMID: 16432186
- Yang L, Huang Y, Porta R, Yanagisawa K, Gonzalez A, Segi E, et al. Host and direct antitumor effects and profound reduction in tumor metastasis with selective EP4 receptor antagonism. Cancer Res 2006; 66:9665 - 72; http://dx.doi.org/10.1158/0008-5472.CAN-06-1271; PMID: 17018624